

Abstract
Wild type (wt) activated protein C (APC) and cytoprotective-selective APC variants such as 3K3A-APC (< 10% anticoagulant but normal cytoprotective activity) are neuroprotective in murine focal ischemic stroke models. Here we compared the neuroprotective effects of the anticoagulant-selective E149A-APC variant (> 3-fold increased anticoagulant activity but defective cytoprotective activities) to those of the cytoprotective-selective 5A-APC variant (< 10 % anticoagulant activity). After transient distal middle cerebral artery occlusion, mice received vehicle, E149A-APC or 5A-APC at 0.2 mg/kg at 4 h after stroke. Treatment with 5A-APC was neuroprotective, as it improved performance on forelimb use asymmetry test and foot fault test (P < 0.05), reduced by 48% and 50% the infarct and edema volumes, respectively (P < 0.05), and was not associated with an increased risk of bleeding as indicated by normal hemoglobin levels in the ischemic brain at day 7. In contrast, E149A-APC treatment worsened neurological outcome determined by foot fault tests and forelimb use asymmetry tests, and increased significantly by 44% and 60% infarct and edema volume, respectively (P < 0.05). At 7 days after treatment, E149A-APC compared to vehicle or 5A-APC notably increased by ~5-fold the hemoglobin level in the ischemic hemisphere suggesting it provoked significant intracerebral bleeding. Thus, the enhanced anticoagulant activity of E149A-APC increased post-ischemic accumulation of neurotoxic erythrocyte-derived hemoglobin which likely worsened the neurological and neuropathological outcome after stroke. Our data emphasize that APC’s cytoprotective activities, but not its anticoagulant activity, are key for APC neuroprotection after transient ischemic stroke.
Keywords: stroke, activated protein C, mouse, ischemia
INTRODUCTION
Activated protein C (APC) is the active trypsin-like, serine protease derived from its zymogen form, protein C, which is a normal component of blood [1,2]. APC exerts multiple activities, including antithrombotic activities by downregulating thrombin generation due to proteolysis of clotting factors Va and VIIIa and cytoprotective activities involving APC-initiated cell signaling. The cytoprotective cell-signaling effects involve multiple cell receptors. APC is a homeostatic blood protease that beneficially reduces morbidity and mortality in a wide variety of preclinical injury models, including ischemic stroke and other brain traumas [1–4]. Some initial insights into which of APC’s many activities might be most important for its neuroprotective actions came from protein engineering of APC variants with greatly reduced anticoagulant activity but apparently normal cytoprotective activities, such as in the APC mutants known as 3K3A-APC and 5A-APC in which either 3 or 5 positively charged residues were replaced by Ala residues [5–7]. These cytoprotective-selective APC mutants demonstrated normal neuroprotection [3,8–10], implying that APC’s cell signaling actions were the primary activities responsible for APC’s neuroprotective activities.
APC’s cell signaling actions involve multiple receptors, including but not limited to endothelial cell protein C receptor (EPCR) and protease activated receptors 1 and 3 (PAR1 and PAR3) [1–3]. The mystery of how thrombin’s activation of PAR1 could be proinflammatory and vasculo-disruptive when APC activation of PAR1 could be anti-inflammatory and vasculoprotective was solved when studies showed that thrombin cleaves PAR1 at Arg41 whereas APC cleaves PAR1 at Arg46, causing biased signaling via PAR1 [11]. In contrast to cytoprotective-selective APC variants like 5A-APC, an anticoagulant-selective APC variant, E149A-APC, was engineered to have greatly reduced anti-inflammatory and anti-apoptotic activities (< 10 % of normal) but greatly increased anticoagulant activity (> 3-fold normal) [12]. When compared to 5A-APC or wt-APC, this E149A-APC mutant failed to reduce endotoxin-induced mortality in mice [12], implicating a primary role for APC’s robust cytoprotective activities in reducing sepsis-related mortality. When the remarkable ability of wt APC to reduce death caused by total body radiation of mice was recently discovered, both the E149A-APC variant and wt-APC reduced mortality whereas the 5A-APC variant failed to reduce radiation-induced death [13]. These striking findings for the in vivo mortality-reducing actions of the anticoagulant-selective E149A-APC mutant in response to lethal total body radiation stimulated us to ascertain this mutant’s neuroprotective activity compared to the well described beneficial effects of cytoprotective-selective 5A-APC [3,8–10]. Here we show that treatment of mice with E149A-APC following ischemic brain injury is actually neurotoxic, supporting the concept that one or more of APC’s cytoprotective actions that is not provided by the E149A-APC variant are key for APC’s neuroprotection after ischemic brain injury.
Materials and methods
Reagents
5A-APC and E149A-APC were characterized and produced as described previously [7,12,14].
Animals
Procedures were approved by the Institutional Animal Care and Use Committee. Male C57Bl6 mice (22–26 g; Jackson Laboratory, Bar Harbor, Maine) were anaesthetized with 100 mg per kg body weight intraperitoneal ketamine and 10 mg per kg body weight xylazine. Rectal temperature was maintained between 36.5 and 37.0°C using a feedback-controlled heating system.
Transient distal middle cerebral artery occlusion
Transient distal middle cerebral artery occlusion (MCAO) was performed using a modified technique [15,16]. In brief, the left common carotid artery was isolated through a neck incision and ligated using a 5–0 silk (Roboz surgical instrument Co.). A skin incision was made between the left orbit and tragus, the temporal muscle was retracted laterally. A 2-mm burr hole was made with a high-speed micro-drill through the outer surface of the semitranslucent skull, just over the visually identified middle cerebral artery (MCA). The dura was carefully opened and the MCA was ligated with a 10–0 nylon suture (S & T AG, Neuhausen, Switzerland). Interruption of blood flow was confirmed under the microscope. The right common carotid arteries were transiently occluded for 20 min and then released. The MCA remained occluded for 60 min, at which time the blood flow to the MCA territory was restored by removing the suture. The wound was sutured and rectal temperature was controlled until mice regained full consciousness.
Treatment with APC variants
Mice were randomly assigned to the vehicle-treated group, E149A-APC-treated group, 5A-APC-treated group and sham-operated control group. E149A-APC, 5A-APC and vehicle were administered via the tail vein 4 h after stroke. E149A-APC and 5A-APC at 0.2 mg/kg were given intravenously 50% as a bolus and 50% as a 60 min infusion. Vehicle-treated group received saline.
Functional tests
The following behavioral tests were performed in mice at 0, 1 and 7 days after stroke: (i) forelimb use asymmetry test, for sensorimotor activity [17,18]; and (ii) foot-fault test, for locomotor assessment [19–21].
Neuropathological Analysis
The mice were sacrificed at 7 days after MCAO and the brains were removed and rapidly frozen in CO2 snow. Brains were cut into serial 20 μm cryostat sections. Every 10th section was stained with cresyl-violet and the lesion area determined using an image analysis system (Image J, Bethesda, MD, USA). On these sections, the areas of infarction, brain swelling and infarct volume were determined, as previously described [17,22].
Hemoglobin Assay
Hemoglobin levels were determined by a spectrophotometric assay using Drabkin’s reagent (Sigma) [23,24].
Protease Activated Receptor 1 Cleavage Assays
Secretory alkaline phosphatase (SEAP)-PAR1 constructs were made and transfected into HEK293 cells and were used for studies of the ability of APC to cleave SEAP-PAR1 at Arg41 or Arg46 as described [11]. The SEAP reporter is an N-terminal extension on PAR1 such that cleavage at either Arg41 or Arg46 can release the SEAP reporter enzyme into the cell supernatant following cleavage. When either Arg is replaced by Gln, then cleavage can be ascribed to a unique Arg residue.
Statistics
Data are presented as mean ± standard error of the mean. One-way analysis of variance (ANOVA) followed by Tukey post hoc test was used to determine statistically significant differences. P < 0.05 was considered statistically significant.
Results
To define the neuroprotective activity of E149A-APC, a murine ischemia reperfusion injury model was used. After transient distal MCAO, mice received vehicle, E149A-APC or 5A-APC at 0.2 mg/kg at 4 h after stroke (given intravenously 50% as a bolus and 50% as a 60 min infusion). Within 7 days of treatment, E149A-APC compared to vehicle notably increased by ~5-fold the hemoglobin level in the ischemic hemisphere (p < 0.01) suggesting significant intracerebral bleeding (Fig. 1). In the absence of MCAO, neither E149A-APC nor 5A-APC had any effect on hemoglobin leakage (Fig. 1). Compared to vehicle control, E149A-APC worsened neurological outcome determined by foot fault tests and forelimb use asymmetry tests, and increased significantly by 44% and 60% infarct and edema volume, respectively (P < 0.05) (Figs. 2a and 2b). In contrast, compared to vehicle control, treatment with 5A-APC improved performance on forelimb use asymmetry test and foot fault test (P < 0.05) (Figs. 2a and 2b), reduced by 48% and 50% the infarct and edema volumes, respectively (P < 0.05) (Figs. 3a and 3b), and was not associated with an increased risk of bleeding as indicated by normal hemoglobin levels in the ischemic brain (Fig. 1).
Fig. 1. Effects of E149A-APC and 5A-APC on post-ischemic hemoglobin levels after transient distal MCAO.
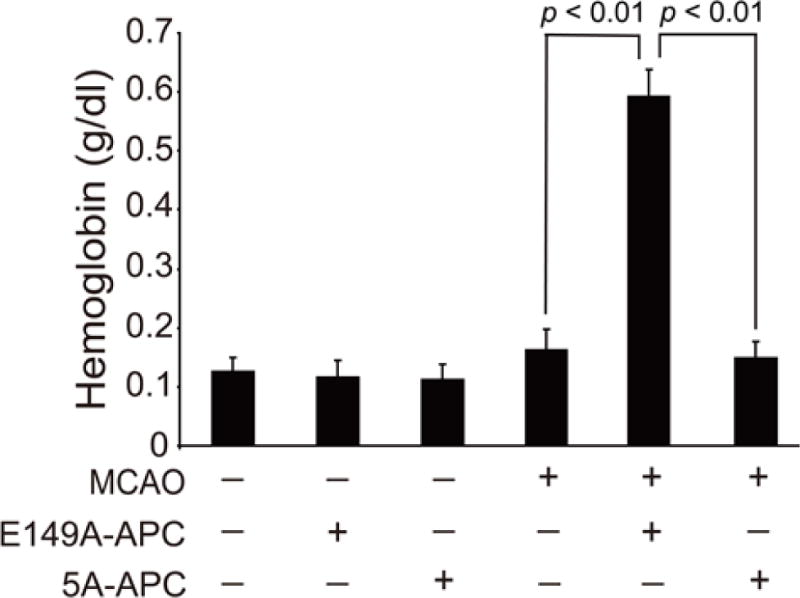
Vehicle, E149A-APC (0.2 mg/kg) and 5A-APC (0.2 mg/kg) were administered via tail vein 4 h after transient distal MCAO. Hemoglobin levels in the ischemic hemisphere of animals subjected to MCAO or in brains of control animals that did not have MCAO but received APC mutants or vehicle were determined at 7 days after stroke. Mean ± SEM, n = 5 mice per group.
Fig. 2. Effects of E149A-APC and 5A-APC on behavior after transient distal MCAO.
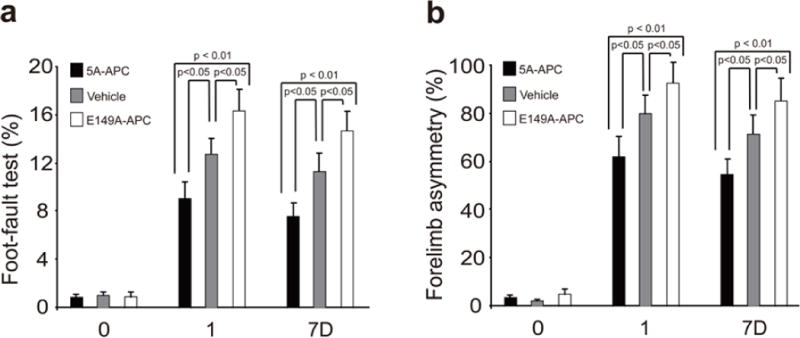
Vehicle, E149A-APC (0.2 mg/kg) and 5A-APC (0.2 mg/kg) were administered via tail vein 4 h after transient distal MCAO. Foot-fault test (a) and forelimb asymmetry test (b) were applied prior to MCAO (“0”), after one Day (“1”) or after 7 days (“7D”). Mean ± SEM, n = 5 mice per group. P values were as indicated.
Fig. 3. Effects of E149A-APC and 5A-APC on infarct and edema volumes after transient distal MCAO.
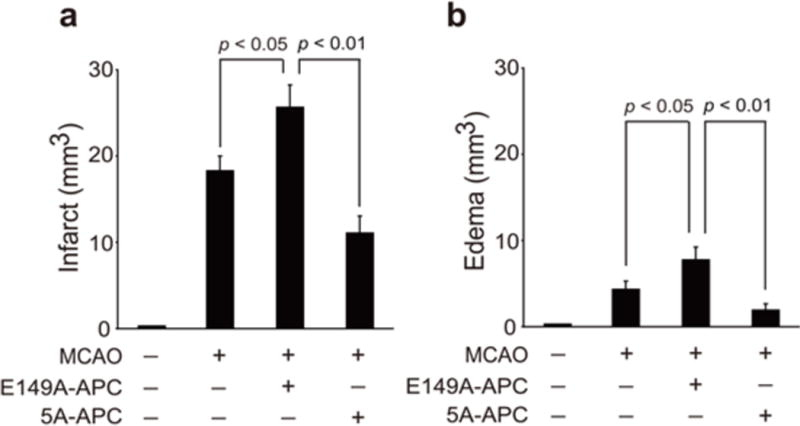
Vehicle, E149A-APC (0.2 mg/kg) and 5A-APC (0.2 mg/kg) were administered via tail vein 4 h after transient distal MCAO. Infarct volume (a) and edema volume (b) were determined 7 days after stroke. Mean ± SEM, n = 5 mice per group. P values were as indicated.
The ability of E149A-APC to cleave PAR1 at Arg46, which generates a novel cytoprotective N-terminal peptide sequence [11], was studied using three SEAP-PAR1 constructs which contained the normal wt Arg41 and Arg46 residues or a R41Q or a R46Q mutation. Based on dose-response comparisons, E149A-APC cleaved each SEAP-PAR1 construct with the same efficacy as the control wt APC (Fig. 4). Specifically, E149A-APC cleaved PAR1 at Arg46 as well as did wt APC. For the negative control, the active site Ser360Ala-APC mutant which lacks all proteolytic activity did not cleave any SEAP-PAR1 constructs (Fig. 4).
Fig. 4. E149A-APC cleaves PAR1 normally at Arg46.
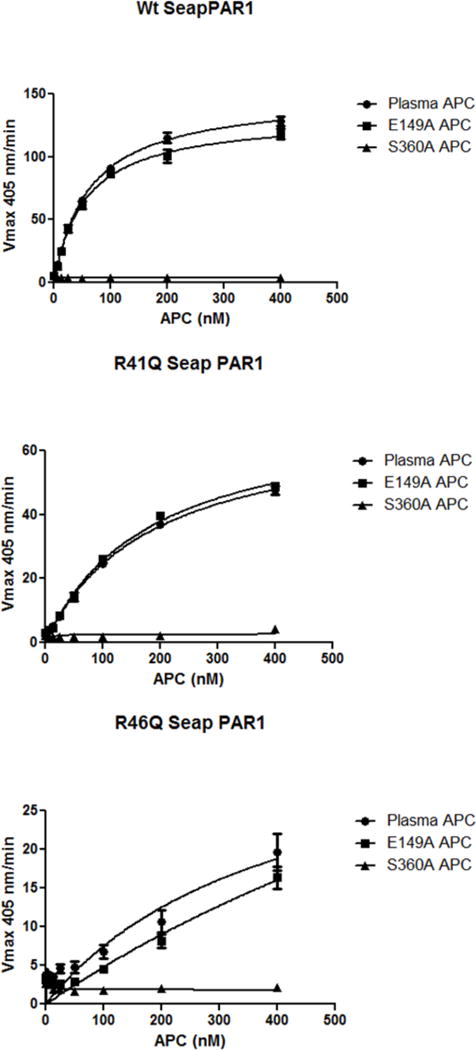
Concentration-response curves are shown for the cleavage of wt-SEAP-PAR1 (upper panel) and of the R41Q mutant (middle panel) or of the R46Q mutant (lower panel) by human APC species. Curves are seen for wt-plasma-derived APC (circles), anticoagulant-selective E149A-APC variant (squares), and enzymatically dead, S360A-APC (triangles). The ordinate denotes SEAP activity (Vmax 405 nm/min) that was released into the supernatant from SEAP-PAR1-transfected cells caused by APC’s cleavage of the PAR1 N-terminal polypeptide tail. The upper panel reported cleavage at either Arg41 or Arg46 (wt-PAR1 containing Arg41 and Arg46) while the middle and lower panels reported cleavage at Arg46 (R41Q indicating PAR1 has Gln41) or at Arg41 (R46Q indicating PAR1 has Gln46), respectively. See Methods section for details.
Discussion
Protein engineering using site-directed mutagenesis is a powerful tool to decipher the molecular properties that are responsible for the biologic activities of agents, such as APC, which manifest multiple activities derived from interactions with multiple molecular partners. Comparisons of two APC variants, cytoprotective-selective 5A-APC and anticoagulant-selective E149A-APC, for their neuroprotection show that the former reduced infarct and edema volumes and improved neurologic outcome. The implications of these observations extend our previous studies of recombinant human and murine 3K3A-APC [8–10] and further establish that cytoprotective selective-APC variants form the basis for second generation biologics that offer neuroprotective activities [3]. This emphasis on the protective properties of APC for its neuroprotective benefits mirrors the implications from studies of APC variants in murine sepsis models [14,25,26] or in murine amyotrophic lateral sclerosis models [27] or acute traumatic brain injury models [28] which show that cytoprotective actions provide the primary actions that reduce mortality or promote recovery from serious injury.
The failure of E149A-APC to provide neuroprotection in mice mirrors its failure to reduce mortality in murine endotoxemia [12]. This is not due to a failure to cleave PAR1 specifically at Arg46 which is a recently discovered mechanism that helps explain some of APC’s cytoprotective actions [11]. The E149A variant was previously shown to lack anti-inflammatory and anti-apoptotic activities when tested using cultured cells [12], but the loss of these activities is unlikely to explain the toxicities observed here. The enhanced anticoagulant activity of E149A-APC increased post-ischemic accumulation of neurotoxic erythrocyte-derived hemoglobin which likely worsened the neurological and neuropathological outcome after stroke. In mice, following controlled cortical impact, treatment with wt APC but not 3K3A-APCcaused excess intracerebral bleeding [28]. Another possibility to account for toxicity in the current study is that infusion of nonprotective E149A-APC may competitively block the beneficial actions of the protective endogenous protein C pathways involving EPCR, thereby exacerbating the injury due to ischemia reperfusion. It was shown that infusion of proteins, such as inactivated-APCi or inactivated-factor VIIai, that compete with endogenous protein C for binding to EPCR can actually increase the plasma levels of protein C by two-to three-fold [29]. Moreover, if endogenous neuroprotective APC which is known to be generated in response to cerebral ischemia [30] needs to bind simultaneously to two or more receptors on target cells and if the E149A mutation interferes with binding to only one of those targeted receptors, then this mutant could be toxic by blocking endogenous APC protection. It is also possible that the Glu to Ala mutation promotes off-target interactions that result in neurotoxicities in this injury model.
This study indicates that one or more properties of APC that is strongly influenced by mutation of Glu149 is relevant for APC’s neuroprotective actions. A protein C gene (PROC) mutation involving deletion of Lys150, the residue following Glu149, is associated with increased risk for ischemic stroke in the Han Chinese population (Odds Ratio = 2.56, 95 % CI 1.45–4.52, p = 0.001) [31]. Plasma from a homozygous carrier of the Lys150 deletion had 26 % protein C anticoagulant activity, but no information is available about the cytoprotective activities of the Lys150del-APC molecule [31]. APC residues 142 to 155 in the light chain comprise a functional exosite in APC that is important for APC’s anticoagulant activity [12,32]. Keeping in mind the hydrophilic residues 142 to 155 that likely mediate binding of substrates and receptors, there remains much to be deciphered about the mechanisms for APC’s beneficial or toxic actions in different preclinical injury models in the course of translational research on the protein C pathways.
Acknowledgments
Support was provided, in part, by NIH grants HL063290 (BVZ), HL031950 and HL052246 (JHG), and HL104165 (LOM). We thank Dr. Jose Fernandez and Ms. Xiao Xu for helpful assistance with this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authorship
Y. Wang and R. Sinha conducted experiments. L.O. Mosnier and J.H. Griffin provided unique reagents. Y. Wang, B.V. Zlokovic and J.H. Griffin wrote the manuscript.
Disclosures
LO Mosnier, JH Griffin, and BV Zlokovic are inventors for subject matters related to cytoprotective, neuroprotective APC variants. JH Griffin is a consultant for ZZBiotech LLC. BV Zlokovic is the scientific founder of ZZBiotech LLC. Y Wang and R Sinha have nothing to declare.
References
- 1.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 2.Griffin JH, Zlokovic BV, Mosnier LO. Protein C anticoagulant and cytoprotective pathways. Int J Hematol. 2012;95:333–345. doi: 10.1007/s12185-012-1059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011;34:198–209. doi: 10.1016/j.tins.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffin JH, Fernández JA, Mosnier LO, Liu D, Cheng T, Guo H, Zlokovic BV. The promise of protein C. Blood Cells Mol Dis. 2006;36:211–216. doi: 10.1016/j.bcmd.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 5.Gale AJ, Tsavaler A, Griffin JH. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J Biol Chem. 2002;277:28836–28840. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- 6.Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104:1740–1744. doi: 10.1182/blood-2004-01-0110. [DOI] [PubMed] [Google Scholar]
- 7.Mosnier LO, Yang XV, Griffin JH. Activated protein C mutant with minimal anticoagulant activity, normal cytoprotective activity, and preservation of thrombin activated fibrinolysis inhibitor-dependent cytoprotective functions. J Biol Chem. 2007;282:33022–33033. doi: 10.1074/jbc.M705824200. [DOI] [PubMed] [Google Scholar]
- 8.Guo H, Singh I, Wang Y, Deane R, Barrett T, Fernández JA, Chow N, Griffin JH, Zlokovic BV. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. 2009;29:1119–1130. doi: 10.1111/j.1460-9568.2009.06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Guo H, Wang Y, Singh I, Liu D, Fernández JA, Griffin JH, Chow N, Zlokovic BV. Species-dependent neuroprotection by activated protein C mutants with reduced anticoagulant activity. J Neurochem. 2009;109:116–124. doi: 10.1111/j.1471-4159.2009.05921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Zhang Z, Chow N, Davis TP, Griffin JH, Chopp M, Zlokovic BV. An activated protein C analog with reduced anticoagulant activity extends the therapeutic window of tissue plasminogen activator for ischemic stroke in rodents. Stroke. 2012;43:2444–2449. doi: 10.1161/STROKEAHA.112.658997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012;120:5237–5246. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosnier LO, Zampolli A, Kerschen EJ, Schuepbach RA, Banerjee Y, Fernández JA, Yang XV, Riewald M, Weiler H, Ruggeri ZM, Griffin JH. Hyperantithrombotic, noncytoprotective Glu149Ala-activated protein C mutant. Blood. 2009;113:5970–5978. doi: 10.1182/blood-2008-10-183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geiger H, Pawar SA, Kerschen EJ, Nattamai KJ, Hernandez I, Liang HP, Fernández JA, Cancelas JA, Ryan MA, Kustikova O, Schambach A, Fu Q, Wang J, Fink LM, Petersen KU, Zhou D, Griffin JH, Baum C, Weiler H, Hauer-Jensen M. Pharmacological targeting of the thrombomodulin-activated protein C pathway mitigates radiation toxicity. Nat Med. 2012;18:1123–1129. doi: 10.1038/nm.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, Castellino FJ, Mackman N, Griffin JH, Weiler H. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, Cairns B, Tumas D, Gerlai R, Williams SP, van Lookeren Campagne M, Ferrara N. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. doi: 10.1172/JCI8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoang S, Liauw J, Choi M, Guzman RG, Steinberg GK. Netrin-4 enhances angiogenesis and neurologic outcome after cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:385–397. doi: 10.1038/jcbfm.2008.128. [DOI] [PubMed] [Google Scholar]
- 17.Zlokovic BV, Zhang C, Liu D, Fernandez JA, Griffin JH, Chopp M. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. 2005;58:474–477. doi: 10.1002/ana.20602. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Thiyagarajan M, Chow N, Singh I, Guo H, Davis TP, Zlokovic BV. Differential neuroprotection and risk for bleeding from activated protein C with varying degrees of anticoagulant activity. Stroke. 2009;40:1864–1869. doi: 10.1161/STROKEAHA.108.536680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 20.Gong Y, Hua Y, Keep RF, Hoff JT, Xi G. Intracerebral hemorrhage: effects of aging on brain edema and neurological deficits. Stroke. 2004;35:2571–2575. doi: 10.1161/01.STR.0000145485.67827.d0. [DOI] [PubMed] [Google Scholar]
- 21.Liu D, Cheng T, Guo H, Fernandez JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein. C Nat Med. 2004;10:1379–1383. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Kilic E, Kilic U, Weber B, Bassetti CL, Marti HH, Hermann DM. VEGF overexpression induces post-ischaemic neuroprotection, but facilitates haemodynamic steal phenomena. Brain. 2005;128:52–63. doi: 10.1093/brain/awh325. [DOI] [PubMed] [Google Scholar]
- 23.Choudhri TF, Hoh BL, Solomon RA, Connolly ES, Jr, Pinsky DJ. Use of a spectrophotometric hemoglobin assay to objectively quantify intracerebral hemorrhage in mice. Stroke. 1997;28:2296–2302. doi: 10.1161/01.str.28.11.2296. [DOI] [PubMed] [Google Scholar]
- 24.Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernandez JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- 25.Kerschen E, Hernandez I, Zogg M, Jia S, Hessner MJ, Fernandez JA, Griffin JH, Huettner CS, Castellino FJ, Weiler H. Activated protein C targets CD8+ dendritic cells to reduce the mortality of endotoxemia in mice. J Clin Invest. 2010;120:3167–3178. doi: 10.1172/JCI42629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bir N, Lafargue M, Howard M, Goolaerts A, Roux J, Carles M, Cohen MJ, Iles KE, Fernández JA, Griffin JH, Pittet JF. Cytoprotective-selective activated protein C attenuates Pseudomonas aeruginosa-induced lung injury in mice. Am J Respir Cell Mol Biol. 2011;45:632–641. doi: 10.1165/rcmb.2010-0397OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N, Thiyagarajan M, Deane R, Fernandez JA, Lane S, Zlokovic AB, Liu T, Griffin JH, Chow N, Castellino FJ, Stojanovic K, Cleveland DW, Zlokovic BV. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. 2009;119:3437–3449. doi: 10.1172/JCI38476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker CT, Marky AH, Petraglia AL, Ali T, Chow N, Zlokovic BV. Activated protein C analog with reduced anticoagulant activity improves functional recovery and reduces bleeding risk following controlled cortical impact. Brain Res. 2010;6:125–131. doi: 10.1016/j.brainres.2010.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sen P, Clark CA, Gopalakrishnan R, Hedner U, Esmon CT, Pendurthi UR, Rao LV. Factor VIIa binding to endothelial cell protein C receptor: differences between mouse and human systems. Thromb Haemost. 2012;107:951–961. doi: 10.1160/TH11-09-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macko RF, Killewich LA, Fernández JA, Cox DK, Gruber A, Griffin JH. Brain-specific protein C activation during carotid artery occlusion in humans. Stroke. 1999;30:542–545. doi: 10.1161/01.str.30.3.542. [DOI] [PubMed] [Google Scholar]
- 31.Lu X, Tang L, Xu K, Ma J, Guo T, Mei H, Yang R, Yu J, Wang Q, Yang Y, Jian X, Hu Y. Novel association of a PROC variant with ischemic stroke in a Chinese Han population. Hum Genet. 2013;132:69–77. doi: 10.1007/s00439-012-1225-8. [DOI] [PubMed] [Google Scholar]
- 32.Mesters RM, Heeb MJ, Griffin JH. A novel exosite in the light chain of human activated protein C essential for interaction with blood coagulation factor Va. Biochemistry. 1993;32:12656–12663. doi: 10.1021/bi00210a014. [DOI] [PubMed] [Google Scholar]