

Abstract
The development of sarcomatous component (SC) in testicular germ cell tumor (GCT) is an uncommon phenomenon. We searched our surgical pathology files from 1985 to 2007 and identified 33 cases of testicular GCTs with SC. The average age of patients was 31 years. All patients underwent radical orchiectomy, which demonstrated a GCT in all patients except for 3 patients who had received neoadjuvant chemotherapy. All testicular GCTs contained a teratomatous component. The GCTs were pure teratomas in 3 cases, and were mixed GCTs in the other cases. The SC was observed in primary testicular tumor (n = 19), in metastasis (n = 11), or in both primary testicular tumor and metastasis (n=3). The average percentage of the SC in the primary testicular GCT was 32% (range, 5% to 99%). The most common histologic type of SC was rhabdomyosarcoma (n = 24), followed by high-grade unclassified sarcoma (n = 5), rhabdomyosarcoma admixed with high-grade unclassified sarcoma (n = 2), angiosarcoma (n = 1), and low-grade myxoid sarcoma (n = 1). Clinical follow-up information was available for 27 patients. Of the 13 patients whose SC was limited to the testicular GCT, 2 died of GCT not otherwise specified (NOS) at 37 and 68 months, respectively; and 11 patients were free of disease at a mean of 46 months. Of the 14 patients with a SC in the metastasis, 7 patients died of GCT NOS at a mean of 95 months, and 7 patients were free of disease at a mean of 104 months. These results suggest that patients with a SC confined to the primary testicular GCT may not have a higher risk of mortality than those at a comparable stage without a SC. However, patients with a SC in the metastasis have an increased risk of mortality.
Keywords: testicular germ cell tumor, sarcomatous component, rhabdomyosarcoma
Introduction
Germ cell tumor (GCT) of the testis is the most common tumor affecting men 15 to 35 years old. Each year in the United States, 8,090 new cases of testicular GCT are diagnosed.11 Remarkably, testicular GCT is one of the most curable tumors with only 380 cancer-related deaths reported annually in the US. With multiple therapeutic modalities including surgery, radiation and chemotherapy available for patients, the five-year survival rate of testicular GCT patients is now reaching 96%.11 Even for patients with metastatic GCT at initial presentation, over 80% of these patients can still be cured with appropriate therapy regimen.2
Testicular GCT demonstrates a wide spectrum of differentiation. Common histologic types of testicular GCT include seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma and teratoma.6 More than half of testicular GCTs are composed of more than one histologic type.17 Different histologic types of GCT show distinctive clinical behaviors. For example, most seminomas are clinically confined to the testis and respond well to radiation therapy, whereas the majority of nonseminomas present with metastatic disease and respond well to chemotherapy.20 Therefore, the accurate histologic classification of testicular GCTs is crucial to disease management.
GCTs may develop a somatic (or non-germ cell) malignant component. Although this is relatively common in mediastinal GCTs, it is rare in testicular GCTs, accounting for 3-6% of GCTs with a teratomatous component.1, 24 Among secondary somatic malignancies in GCTs, sarcomatous components (SC) of various histologic types are the most commonly observed.14, 18 There have been limited studies on this phenomenon and most studies have been single-case reports. Furthermore, some series studies included GCTs of mixed origins of the testis, ovary, retroperitoneum and intracranial cavity,3,14,18 which are known to have different clinical outcomes.8,9 For example, mediastinal non-seminomatous GCTs are a highly aggressive disease with a 3-year mortality of up to 72% despite therapy.16 In contrast, testicular GCTs carry a 5-year mortality of less than 5%.11 Furthermore, limited studies also suggest that the development of SC in GCTs of different orgins have various impacts on the patient's clinical outocome.14, 18 In the current study, we evaluated the clinical and pathologic features of SC in GCTs that arose exclusively in the tesitis.
Materials and Methods
With approval from the Institutional Review Board of The University of Texas M. D. Anderson Cancer Center (Houston, TX), we retrospectively searched our surgical pathology report files for patients who presented with testicular GCT with SC during the period from January 1, 1985 to December 31, 2007. Our inclusion criterion was an expansile sarcomatous tumor growth of at least one low-power field (4 × objective) replacing the GCT component.25 Cases with scattered clusters of atypical spindle cells mixed with GCT were considered atypical stromal proliferations and excluded from the analysis, as were cases of nonsarcomatous somatic transformation (eg, carcinomas and hematopoietic malignancies). We identified 33 cases of testicular GCT with SC in the primary or metastatic tumor. None of the 33 patients had any other documented malignancy before the testicular GCT.
We reviewed patient records at M. D. Anderson Cancer Center for demographic, clinical, and outcome information. Clinical stage at presentation was determined by reviewing pathologic, radiologic, and clinical findings and using staging criteria from the American Joint Committee on Cancer as follows: stage I tumors were confined to the testis, epididymis, or spermatic cord; stage II tumors metastasized locally to the retroperitoneal lymph nodes below the diaphragm; and stage III tumors metastasized outside the retroperitoneal lymph nodes or above the diaphragm.7 Treatment and follow-up data were recorded. Survival times were measured from diagnosis to death or last follow-up.
All 33 patients had undergone radical orchiectomy; 22 patients had undergone retroperitoneal lymph node dissection (RPLND); and 9 had undergone resection of distant metastatic tumors. All specimens were adequately sampled for histologic examination. We reviewed the hematoxylin and eosin stained slides. The reviewed parameters included the histologic classification of the GCT and SCs components and the percentage of each in the overall tumor volume. In selective cases, immunohistochemical staining was performed using the avidin–biotin-peroxidase complex method in a Dako AutoStainer (Dako, Carpinteria, CA). The primary antibodies used were antibodies to desmin (Dako, 1:100 dilution) and CD34 (Dako, 1:50 dilution).
Results
Clinical Presentation and Therapy
The average age of patients was 31 years (range, 16 to 55 y). Clinical stage, treatment, and follow-up data were available for 27 patients (Table 1). At presentation, 7 patients had stage I disease confined to the testis, and 20 patients developed metastasis. In the latter group, 9 patients had stage II disease with metastases to the retroperitoneal lymph nodes below the diaphragm, and 11 patients had stage III disease with metastases to the lungs, liver, mediastinum, or neck. All the patients underwent radical orchiectomy, and 22 patients received retroperitoneal lymph node dissection (RPLND). In addition, 21 patients received cisplatin-based chemotherapy regimens, BEP (bleomycin, etoposide, and cisplatin) and ITP (paclitaxel, ifosfamide, and cisplatin).
Table 1. Clinical and Pathologic Features of 33 Cases of Testicular Germ Cell Tumor with Sarcomatous Transformation*.
| CaseNo. | Age(y) | Stage† | Orchiectomy Histology | Metastasis Histology | Therapy | Follow-up (mo) | Status |
|---|---|---|---|---|---|---|---|
| 1 | 38 | I | T, RHGS (80) | N/A | O, R | 5 | FOD |
| 2 | 44 | I | T, EC, S | T, RMS (90) | O, R | 19 | AWD |
| 3 | 34 | I | T, RMS (99) | N/A | O, R, | 12 | FOD |
| 4 | 21 | I | T, EC, YS, RMS (40) | CC | O, R, C | 50 | FOD |
| 5 | 18 | I | T, CC, EC, YS, RMS (25) | N/A | O, C | 68 | FOD |
| 6 | 24 | I | T, EC, S, YS, RMS (5) | EC | O | 68 | DOD |
| 7 | 28 | I | T, EC, S | T, HGUS (95) | O | 71 | DOD |
| 8 | 34 | II | T, S, RMS (70) | T | O, R | 36 | FOD |
| 9 | 30 | II | T, S, RMS (5) | T | O, R, C | 12 | FOD |
| 10 | 37 | II | T, EC, YS, RMS (60) | T | O, R, C | 91 | FOD |
| 11 | 44 | II | T, EC, S, YS | HGUS (100) | O, R, C | 168 | DOD |
| 12 | 16 | II | T, EC, YS | YS, RMS (80) | O, R, C, | 20 | DOD |
| 13 | 39 | II | T, RMS (5) | T, YS, RMS (50) | O, R, C | 9 | FOD |
| 14 | 40 | II | Fibrosis | EC, RMS (10) | O, R, C | 185 | FOD |
| 15 | 24 | II | T, YS, LGMS (5) | LGMS (100) | O, R, C | 240 | FOD |
| 16 | 17 | II | T, CC, EC, ANG (5) | T, ANG (5) | O, R, C | 28 | FOD |
| 17 | 30 | III | T, S, RMS (15) | T | O, R, C | 81 | FOD |
| 18 | 30 | III | T, EC, YS, RMS (60) | T | O, R, C | 79 | FOD |
| 19 | 27 | III | T, EC, S, YS, RMS (5) | CC | O, R, C | 69 | FOD |
| 20 | 28 | III | T, EC | T, RMS (5) | O, R, C | 70 | DOD |
| 21 | 21 | III | T, EC, YS | T, RMS (10) | O, R, C | 81 | FOD |
| 22 | 32 | III | T, CC, EC, RMS (20) | T | O, R, C | 23 | FOD |
| 23 | 55 | III | T, S, YS, RHGS (50) | N/A | O, C | 37 | DOD |
| 24 | 43 | III | T, EC, S | T, RMS (90) | O, R, C | 60 | DOD |
| 25 | 17 | III | T, EC, S, YS | RMS (100) | O, R, C | 87 | FOD |
| 26 | 31 | III | Fibrosis | HGUS (100) | O, R, C | 132 | DOD |
| 27 | 34 | III | Fibrosis | T, HGUS (70) | O, R, C | 146 | DOD |
| 28 | 26 | N/A | T, S, RMS (15) | N/A | N/A | N/A | N/A |
| 29 | 21 | N/A | T, EC, YS, RMS (80) | N/A | N/A | N/A | N/A |
| 30 | 33 | N/A | T, EC, RMS (35) | N/A | N/A | N/A | N/A |
| 31 | 52 | N/A | T, EC, S, YS, RMS (20) | N/A | N/A | N/A | N/A |
| 32 | 22 | N/A | T, EC, YS, HGUS (5) | N/A | N/A | N/A | N/A |
| 33 | 29 | N/A | T, EC, S, RMS (N/A**) | N/A | N/A | N/A | N/A |
ANG, angiosarcoma; AWD, alive with disease; C, chemotherapy; CC, choriocarcinoma; DOD, died of disease; EC, embryonal carcinoma; FOD; free of disease; HGUS, high-grade unclassified sarcoma; LGMS, low-grade myxoid sarcoma; N/A, not available; O, orchiectomy; R, retroperitoneal lymph node dissection; RHGS, rhabdomyosarcoma admixed with high-grade unclassified sarcoma; RMS, rhabdomyosarcoma; S, seminoma; T, teratoma; YS, yolk sac tumor.
Type of sarcomatous component appears in bold and percentage of sarcomatous component in parenthesis.
Clinical stage determined at initial diagnosis.
Patient received neoadjuvant chemotherapy before orchiectomy.
The percentage of SC could not be assessed in this case, because only representative (not all) slides were available for review.
Pathologic Features
Our review of the orchiectomy specimens revealed a GCT in 30 cases. In 3 cases, GCT was not present in the orchiectomy specimen, and all the 3 patients had received neoadjuvant chemotherapy before orchiectomy. A teratomatous component was present in all 30 primary GCTs (range of tumor volume, 5% to 100%; average, 34%). Teratoma was the only GCT type in 3 cases. In 27 primary tumors, the teratoma was admixed with other GCT components, including embryonal carcinoma (n = 21), yolk sac tumor (n = 15), seminoma (n = 15), and choriocarcinoma (n = 3).
The SC was observed in the primary testicular tumor (n = 19), in a metastasis (n = 11), or in both the primary testicular tumor and metastasis (n=3). The average percentage of SC in the primary testicular tumor was 32.1% (range, 5% to 99%). In 24 cases, the SC was composed of rhabdomyosarcoma with rhabdomyoblasts at various stages of myogenesis. The primitive rhabdomyoblasts showed oval nuclei with scant amphophilic cytoplasm and expressed desmin, which was demonstrated by immunohistochemical staining (Fig. 1). The more differentiated rhabdomyoblasts were elongated with abundant eosinophilic cytoplasm (Fig. 2). In 5 cases, the SC was composed of atypical spindle cells that exhibited no specific histologic differentiation after comprehensive immunohistochemical analysis. The tumors in these cases were considered high-grade unclassified sarcoma (Fig. 3). In 2 cases, the tumor showed high-grade unclassified sarcoma admixed with rhabdomyosarcoma. In 1 case, the SC was composed of low-grade spindle cells in a myxoid background and was classified as low-grade unclassified myxoid sarcoma (Fig. 4). In 1 case, the SC formed irregular vascular spaces lined with atypical cells positive for the endothelial marker CD34, which was consistent with an angiosarcoma (Fig. 5).
FIGURE 1.

Primitive rhabdomyosarcoma in a testicular GCT. (A) Rhabdomyosarcoma was adjacent to teratoma with chondroid differentiation. (B) The rhabdomyoblasts were stellate with scant amphophilic cytoplasm and oval nuclei. (C) Rhabdomyoblasts expressed desmin on immunohistochemical staining.
FIGURE 2.

Well differentiated rhabdomyosarcoma in a testicular GCT. The rhabdomyoblasts were elongated with abundant eosinophilic cytoplasm.
FIGURE 3.

High-grade unclassified sarcoma in a testicular GCT.
FIGURE 4.

Low-grade myxoid sarcoma in a testicular GCT.
FIGURE 5.
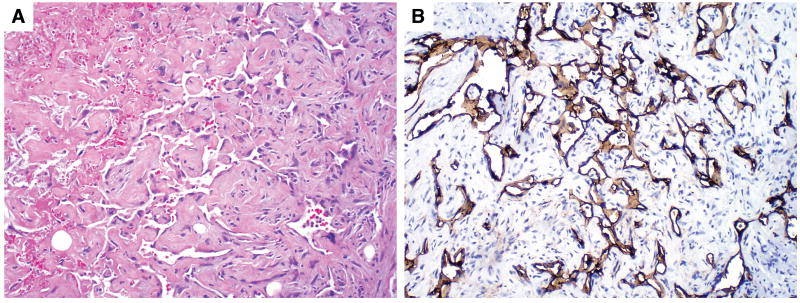
Angiosarcoma in a testicular GCT. (A) The irregular vascular spaces were lined by atypical endothelial cells. (B) The atypical endothelial cells were highlighted by immunohistochemical staining with CD34.
Clinical Follow-up
Follow-up information was available for 27 patients for a mean of 75 months (range, 5 to 240 months). Of the 13 patients who had the SC only in the testicular GCT, 2 died of GCT not otherwise specified (NOS) at 37 and 68 months, respectively; one patient who died did not have a SC in the metastasis; and 11 patients were alive and free of disease at a mean of 46 months (range, 5 to 81 months). Of the 14 patients who developed a SC in the metastasis, including 3 patients who also had a SC in the testicular GCT, 7 patients died of GCT NOS at a mean of 95 months (range, 20 to 168 months); 6 patients were alive and free of disease at a mean of 118 months (range, 9 to 185 months); and 1 patient was alive with disease at 19 months.
Discussion
The development of a secondary somatic (or non-germ cell) malignant component in GCT is a well-known phenomenon.1,24 This phenomenon has been observed in GCTs of various origins including those of the testis, ovary, mediastinum and intracranial cavity.10,14,18,19,21 Histologically, the secondary component may resemble a somatic malignancy derived from any of the three germinal layers. Sarcomas are the most common somatic malignancies observed in GCTs, while other somatic malignancies, such as carcinomas and hematologic malignancies, have been also reported.3,18 Among a variety of sarcomas reported in GCTs, rhabdomyosarcoma is by far the most common.3,18 In our study, rhabdomyosarcoma was present in 26 of 33 (79%) cases of GCTs that arose in the testis. In 24 cases the SC was entirely rhabdomyosarcoma, and in 2 cases the SC was composed of rhabdomyosarcoma admixed with high-grade unclassified sarcoma.
There are several hypotheses regarding the origin of SC in GCT. Because SC tends to occur in GCTs that contain a teratomatous component, the SC may arise from “malignant transformation” of teratomatous mesenchyma.18,24 The extremely close association of the SC with teratomas has led to the term, “teratoma with a secondary malignant transformation,” to describe this phenomenon.18 However, SCs have also been reported in GCTs without a teratomatous component. True et al23 reported 5 cases of spermatocytic seminoma with SC, in which no teratomatous component was present. Malagón et al14 also reported the development of rhabdomyosarcoma in a retroperitoneal GCT composed of pure seminoma. These results suggest that the development of a SC in a GCT may use other mechanisms, such as aberrant differentiation of primitive germ cells and dedifferentiation of blastomatous stroma in yolk sac tumor.14,23 In our study, a teratoma component was present in all primary testicular GCTs, but only one testicular GCT was composed of pure teratoma. The other 29 GCTs were composed of a teratomatous component admixed with other GCT components, such as seminoma, embryonal carcinoma, yolk sac tumor and choriocarcinoma. Therefore, we prefer to classify these testicular tumors as GCT (state type or types) with a secondary somatic malignancy (state type).
Cytogenetic studies have provided some insight into the origin of SCs in GCTs. To investigate the clonality of SCs in GCTs, Motzer et al18 studied the cytogenetic features of secondary somatic components in 12 cases of GCT. They found the presence of isochromosome 12p in 10 cases and a deletion of chromosome 12q in 1 case. Similarly, Korski et al13 also reported that isochromosome 12p was present in metastatic rhabdomyosarcoma which developed from a testicular GCT. Since the majority of testicular GCTs are also characterized by isochromosome 12p,22 these findings support the germ cell clonal origin of the SC in GCT.
Although SC may develop in a primary testicular GCT or in a metastasis, the result of our study indicates that only the presence of a SC in a metastasis is associated with an increased risk for mortality. In our study, 50% of the patients with a SC in the metastasis died of disease. In contrast, only 15% of the patients with a SC in the primary testicular GCT died of disease, despite that 64% of the patients had metastatic disease. Because advanced (or metastatic) testicular GCTs carry a 5-year mortality of 16-18%,9 the patients whose SC was confined to the primary testicular GCT did not have a higher risk of mortality than those at a comparable stage but who lacked a SC in the testicular GCT. Furthermore, the cause of death in these patients may not be exclusively due to the SC, as several patients had conventional GCT components in the metastasis that were also resistant to chemotherapy. One patient who died did not have a SC in the metastasis, although there was a SC in his primary testicular tumor. In addition, Ahmed et al 1 also reported similar findings in a study of 17 cases of testicular teratoma with SC. They found that all 5 patients with a SC in the primary testicular teratoma were free of disease, whereas 7 of 12 patients with a SC in the metastasis died of disease. Thus, their findings also support that only the presence of a SC in a metastasis is associated with an increased risk for mortality.
Although testicular GCT is a highly curable disease, the development of a SC presents a challenge in treatment.4,5 Surgical resection can provide substantial benefits to the patients, but only if the tumor is localized and resectable. Cisplatin-based chemotherapy is highly effective for GCTs, but not successful in those GCTs with a SC because the SC is generally resistant to cisplatin-based chemotherapy. Recently Donadio et al4 reported that GCT patients with a single histologic type of SC responded well to specific chemotherapy when the treatment choice was based on the histology of the SC. For example, GCTs with rhabdomyosarcoma respond well to a doxorubicin-based regimen, and GCTs with adenocarcinoma benefit from a 5-fluorouracil-based regimen. Therefore, the accurate classification of SC in GCT is important to the type of chemotherapy used in these patients.
The primary testicular GCT may differ histologically from that of the metastatic tumor.15 In our study, of the 22 patients with a SC in the primary testicular GCT, 12 underwent resection for metastasis, but only 3 patient's metastasis contained the SC. Similarly, 11 patients did not have a SC in the primary testicular tumor, but developed it in the metastasis. Considering that all GCT components, including the SC, are derived from totipotential germ cells, this histologic discrepancy between the primary and metastatic tumors is not entirely surprising.12,17 Alternatively, the failure to detect a SC in the primary testicular GCT may be partly due to insufficient sampling of the specimens. Another reason for this discrepancy may be due to the poor response of the SC to the cisplatin-based chemotherapy, which would allow selective proliferation and metastasis of the SC over GCT components.
Because the presence of SCs in testicular GCTs is associated with a poor prognosis and patients may benefit from a sarcoma-specific chemotherapy regimen, it is important to differentiate SC from atypical stromal proliferations in the GCTs. Focal areas of atypical stroma are common in GCTs, especially those with teratoma or yolk sac tumor. In such cases, the spindle cells usually lack marked cytologic atypia, are admixed with other GCT components, and do not form an expansile nodule. Ulbright et al25 have proposed at least half to a whole low-power field (4 × objective) of involvement as the minimum requirement for SC, which usually forms an expansile nodule of substantial size. Furthermore, the sarcomatous cells show prominent cytologic atypia, robust mitotic activity, an architecturally complex growth pattern, and usually show distinct heterologous differentiation.
In summary, we studied the development of SC in a large series of GCTs that arose exclusively in the testitis. Rhabdomyosarcoma is by far the most common SC in testicular GCTs, and other histologic types are also observed. SC may occur in the primary testicular GCT or metastasis. Our results suggest that patients with SC confined to the primary testicular GCT may not have a higher risk of mortality than those at a comparable stage without SC. However, patients with SC in the metastasis have an increased risk of mortality, and they may benefit from sarcoma-orientated drugs.
References
- 1.Ahmed T, Bosl GJ, Hajdu SI. Teratoma with malignant transformation in germ cell tumors in men. Cancer. 1985;56:860–863. doi: 10.1002/1097-0142(19850815)56:4<860::aid-cncr2820560426>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Carver BS, Serio AM, Bajorin D, et al. Improved clinical outcome in recent years for men with metastatic nonseminomatous germ cell tumors. J Clin Oncol. 2007;25:5603–5608. doi: 10.1200/JCO.2007.13.6283. [DOI] [PubMed] [Google Scholar]
- 3.Comiter CV, Kibel AS, Richie JP, et al. Prognostic features of teratomas with malignant transformation: a clinicopathological study of 21 cases. J Urol. 1998;159:859–863. [PubMed] [Google Scholar]
- 4.Donadio AC, Motzer RJ, Bajorin DF, et al. Chemotherapy for teratoma with malignant transformation. J Clin Oncol. 2003;21:4285–4291. doi: 10.1200/JCO.2003.01.019. [DOI] [PubMed] [Google Scholar]
- 5.El Mesbahi O, Terrier-Lacombe MJ, Rebischung C, et al. Chemotherapy in patients with teratoma with malignant transformation. Eur Urol. 2007;51:1306–1311. doi: 10.1016/j.eururo.2006.10.021. Discussion 1311–1312. [DOI] [PubMed] [Google Scholar]
- 6.Eble JN, Sauter G, Epstein JI, et al. World Health Organization Classification of Tumours. Lyon: IARC Press; 2004. Pathology and genetics of tumours of the urinary system and male genital organs; pp. 218–249. [Google Scholar]
- 7.Greene LF, Page DL, Fleming ID, et al. AJCC Cancer Staging Manual. Sixth. New York, NY: Springer-Verlag; 2002. [Google Scholar]
- 8.Hartmann JT, Nichols CR, Droz JP, Horwich A, Gerl A, Fossa SD, Beyer J, Pont J, Kanz L, Einhorn L, Bokemeyer C. Prognostic variables for response and outcome in patients with extragonadal germ-cell tumors. Ann Oncol. 2002;13:1017–28. doi: 10.1093/annonc/mdf176. [DOI] [PubMed] [Google Scholar]
- 9.International Germ Cell Cancer Collaborative Group International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. J Clin Oncol. 1997;15:594–603. doi: 10.1200/JCO.1997.15.2.594. [DOI] [PubMed] [Google Scholar]
- 10.Iwasa A, Oda Y, Kaneki E, Ohishi Y, et al. Squamous cell carcinoma arising in mature cystic teratoma of the ovary: an immunohistochemical analysis of its tumorigenesis. Histopathology. 2007;51:98–104. doi: 10.1111/j.1365-2559.2007.02727.x. [DOI] [PubMed] [Google Scholar]
- 11.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 12.Jones TD, Wang M, Sung MT, et al. Clonal origin of metastatic testicular teratomas. Clin Cancer Res. 2006;12:5377–5383. doi: 10.1158/1078-0432.CCR-06-0444. [DOI] [PubMed] [Google Scholar]
- 13.Korski K, Breborowicz D, Filas V, et al. A case of primary testicular germ cell tumor with rhabdomyosarcoma metastases as an example of applying the FISH method to diagnostic pathology. APMIS. 2007;115:1296–301. doi: 10.1111/j.1600-0643.2007.00582.x. [DOI] [PubMed] [Google Scholar]
- 14.Malagón HD, Valdez AM, Moran CA, et al. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol. 2007;31:1356–1362. doi: 10.1097/PAS.0b013e318033c7c4. [DOI] [PubMed] [Google Scholar]
- 15.Michael H, Lucia J, Foster RS, et al. The pathology of late recurrence of testicular germ cell tumors. Am J Surg Pathol. 2000;24:257–273. doi: 10.1097/00000478-200002000-00012. [DOI] [PubMed] [Google Scholar]
- 16.Moran CA, Suster S, Koss MN. Primary germ cell tumors of the mediastinum: III. Yolk sac tumor, embryonal carcinoma, choriocarcinoma, and combined nonteratomatous germ cell tumors of the mediastinum--a clinicopathologic and immunohistochemical study of 64 cases. Cancer. 1997;80:699–707. [PubMed] [Google Scholar]
- 17.Mostofi FK. Proceedings: Testicular tumors. Epidemiologic, etiologic, and pathologic features. Cancer. 1973;32:1186–1201. doi: 10.1002/1097-0142(197311)32:5<1186::aid-cncr2820320527>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Motzer RJ, Amsterdam A, Prieto V, et al. Teratoma with malignant transformation: diverse malignant histologies arising in men with germ cell tumors. J Urol. 1998;159:133–138. doi: 10.1016/s0022-5347(01)64035-7. [DOI] [PubMed] [Google Scholar]
- 19.Preissing S, Smith HT, Huntington HW. Rhabdomyosarcoma arising in pineal teratoma. Cancer. 1979;44:281–284. doi: 10.1002/1097-0142(197907)44:1<281::aid-cncr2820440148>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 20.Richie JP, Steele GS. Neoplasms of the testis. In: Wein AJ, Kavoussi LR, Novick AC, editors. Campbell-Walsh Urology. 9th. Philadelphia, PA: Saunders Elsevier; 2007. pp. 893–925. [Google Scholar]
- 21.Rim SY, Kim SM, Choi HS. Malignant transformation of ovarian mature cystic teratoma. Int J Gynecol Cancer. 2006;16:140–144. doi: 10.1111/j.1525-1438.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez E, Mathew S, Reuter V, et al. Cytogenetic analysis of 124 prospectively ascertained male germ cell tumors. Cancer Res. 1992;52:2285–22891. [PubMed] [Google Scholar]
- 23.True LD, Otis CN, Delprado W, et al. Spermatocytic seminoma of testis with sarcomatous transformation. A report of five cases. Am J Surg Pathol. 1988;12:75–82. doi: 10.1097/00000478-198802000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Ulbright TM, Loehrer PJ, Roth LM, et al. The development of non-germ cell malignancies within germ cell tumors. A clinicopathologic study of 11 cases. Cancer. 1984;54:1824–1833. doi: 10.1002/1097-0142(19841101)54:9<1824::aid-cncr2820540910>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 25.Ulbright TM, Amin MB, Young RH. Atlas of Tumor Pathology. fascicle 25. Washington, D.C.: Armed Forces Institute of Pathology; 1999. Tumors of the Testis, Adnexa, Spermatic Cord, and Scrotum; pp. 147–164. (third). [Google Scholar]