

Abstract
Sudden infant death syndrome (SIDS) is defined as the sudden and unexpected death of an infant less than 12 months of age that is related to a sleep period and remains unexplained after a complete autopsy, death scene investigation, and review of the clinical history. The cause of SIDS is unknown, but a major subset of SIDS is proposed to result from abnormalities in serotonin (5-HT) and related neurotransmitters in regions of the lower brainstem that result in failure of protective homeostatic responses to life-threatening challenges during sleep. Multiple studies have implicated gene variants that affect different elements of 5-HT neurotransmission in the pathogenesis of these abnormalities in SIDS. In this review I discuss the data from these studies together with some new data correlating genotype with brainstem 5-HT neurochemistry in the same SIDS cases and conclude that these gene variants are unlikely to play a major role in the pathogenesis of the medullary 5-HT abnormalities observed in SIDS.
1. Introduction
The identification of gene variants underlying the pathogenesis of the sudden infant death syndrome (SIDS) is of major significance in SIDS research. Identification of such variants could potentially form the basis of genetic screening to identify living infants at risk at or soon after birth. Such an advance would allow targeted parental counseling to minimize exposure to environmental risk factors and potentially form the basis of therapeutic intervention strategies. Evidence suggests that a major subset of SIDS cases results from abnormalities in serotonin (5-HT) and related neurotransmitters in regions of the lower brainstem that result in failure of protective homeostatic responses to life-threatening challenges during sleep. Multiple studies have identified associations between variants in genes related to 5-HT signaling in the central nervous system and SIDS suggesting that they contribute to the brainstem 5-HT abnormalities observed and, thus, represent risk factors for SIDS. Here, I review and examine the evidence for the potential role of these gene variants in the pathogenesis of SIDS by comparing data from the studies of others to those of our own studies in the San Diego SIDS Dataset. In addition, I also present new (previously unpublished) data correlating medullary 5-HT neurochemical markers with genotype in the same SIDS cases in order to determine if these polymorphisms actually alter brainstem 5-HT neurochemistry, and, thus, contribute to SIDS pathogenesis in the hypothesized manner.
2. The Sudden Infant Death Syndrome (SIDS)
2.1 Definition and Incidence
The sudden infant death syndrome (SIDS) is defined as the sudden death of an infant <12 months of age that is associated with a sleep period and remains unexplained after a complete autopsy and death scene investigation (Krous et al. 2004). Typically a seemingly healthy infant (i.e., without a clinical prodrome) dies following a sleep period, with death presumably occurring either during sleep or during one of the many transitions to waking that occur during sleep (Krous et al. 2004; Kinney et al. 2009). The incidence of SIDS has fallen by at least 50% in the last 20 years largely due to the identification of the increased risk of SIDS associated with the prone sleep position (Beal 1986; Engelberts et al. 1990; Fleming et al. 1990; Ponsonby et al. 1993; Willinger et al. 1994; Dwyer et al. 1995; Moon et al. 2007) and the subsequent implementation of risk-reduction campaigns promoting safe sleeping practices (Mitchell et al. 1994; Willinger et al. 1994; Beal et al. 2000). Nevertheless, there has been a plateau in the SIDS rate over the last decade, and it remains the leading cause of postneonatal infant mortality and the third leading cause of infant mortality in the United States with an incidence of 0.57/1,000 live births (Moon et al. 2007; Hauck et al.). In effect, there are at least 6 infant deaths due to SIDS each day in the United States.
2.2 The Triple Risk Model of SIDS
The pathogenesis of SIDS is unknown, but it has become clear that it has a complex etiology involving multiple and simultaneous factors (Takashima et al. 1990; Hoffman et al. 1992; Arnestad et al. 2001; Trachtenberg et al. 2012). Risk factors associated with the sleep environment include prone or face down sleeping (Fleming et al. 1990; Fleming et al. 1996; Blair et al. 2006), bed sharing (Blair et al. 1999; Hauck et al. 2003; Tappin et al. 2004; Tappin et al. 2005; Pelayo et al. 2006; Mitchell 2007; O’Mara 2007; Vennemann et al. 2009) and over-bundling (Fleming et al. 1990; Fleming et al. 1996). Additional risk factors are associated with the mother and pregnancy, including prematurity, low birth weight, prenatal and postnatal cigarette smoke exposure (Schoendorf et al. 1992; Haglund 1993; Blair et al. 1996; Fleming et al. 1996; MacDorman et al. 1997; Wisborg et al. 2000; Anderson et al. 2005; Mitchell et al. 2006) and maternal alcohol ingestion during pregnancy (Scragg et al. 1993; Alm et al. 1999; Iyasu et al. 2002; Kinney et al. 2003; Klug et al. 2003; Duncan et al. 2008; O’Leary et al. 2013). The “Triple Risk Model” of SIDS (Filiano et al. 1994) has proven useful in thinking about how these multiple factors interact to cause SIDS (Fig. 1). According to this model, SIDS occurs when three factors simultaneously impinge upon the infant: 1) an underlying vulnerability in the infant; 2) a critical developmental period, i.e., the first year of life; and 3) an exogenous stressor, e.g., prone sleep. According to this model, normal infants do not die of SIDS, but rather, only infants with an underlying disease process. The model also explains why only a few of the many infants placed prone to sleep die of SIDS (Kinney et al. 2011) and reinforces the important concept that environmental stressors, such as prone sleep, do not cause SIDS.
Figure 1. Triple Risk Model for SIDS Demonstrating Genetic-Environmental Interactions.

The Triple Risk Model for SIDS proposes that death occurs when three factors simultaneously impinge upon the infant: 1) an underlying vulnerability in the infant; 2) a critical developmental period, i.e., the first year of life; and 3) an exogenous stressor, e.g., prone sleep. According to this model, normal infants do not die of SIDS, but rather, only infants with an underlying disease process. Gene variants are thought to contribute to SIDS risk either by directly causing or contributing to the failure of homoestatic mechanisms or by rendering the infant less resilient to environmental stressors.
2.3 The Role of Genetic Factors in the Pathogenesis of SIDS
While SIDS is not inherited in Mendelian fashion (Irgens et al. 1984; Peterson et al. 1986; Irgens et al. 1988) evidence suggests that genetic factors are involved. The rate of SIDS in twins is twice that in singletons and the rate in identical twins is 2.5 times that in dizygotic twins with birth weight >3000g, indicating that the increased mortality in identical twins is not related to low birth weight (Pharoah et al. 2007). Similarly, the risk of a second SIDS death within a family is estimated to be 1-5 times that in the general population, although the influence of environmental factors in these studies cannot be completely ruled out (Oyen et al. 1996; Getahun et al. 2004; Hunt 2004; Pharoah et al. 2007). There are also marked racial disparities in SIDS, with rates 2-7 times the national averages observed in indigenous populations in the United States (Kitsantas et al.; Kinney et al. 2009; Hauck et al. 2011), New Zealand (Moon et al. 2007) and Australia (Moon et al. 2007), as well as in the Cape Colored population in South Africa (Kibel et al. 2005). While these populations are frequently socioeconomically disadvantaged this does not completely account for the disparity in SIDS rates (Kattwinkel et al. 2005). Indeed, the SIDS rates in Hispanics in the United States, who are also recognized as a socially disadvantaged population, are lower than the national average (Kattwinkel et al. 2005). In addition, there is an increased rate of SIDS in male compared to female (3:2) infants (Froggatt et al. 1968; Beal 1972; Stewart 1975; Arneil et al. 1985; Millar et al. 1993). While this observation may be due, at least in part, to normally occurring differences in hormone levels, it also suggests that there may be an X-chromosome linked genetic susceptibility to SIDS. Finally, and perhaps most significantly, prior to the identification of the genetic mutations responsible for medium-chain acyl-CoA dehydrogenase (MCAD) fatty acid oxidation disorders (Bennett et al. 1994; Opdal et al. 2004) and cardiac channelopathies responsible for long QT syndrome (LQTS) (Schwartz et al. 1998; Ackerman et al. 2001; Arnestad et al. 2007), infant deaths resulting from these entities were considered to be SIDS. These observations support the idea that rare functionally significant gene mutations underlie the pathogenesis of different subsets of SIDS cases.
3. Medullary Homoestatic Network Dysfunction as a Key Underlying Abnormality in SIDS
Research consensus implicates dysfunction of homeostatic mechanisms that result in failure of protective responses to life-threatening stressors during sleep as the “underlying vulnerability” that precipitates the death of the infant (Shannon et al. 1982b; Shannon et al. 1982a; Hunt et al. 1987; Harper 2001; Fifer et al. 2002; Kinney et al. 2005; Kinney et al. 2009; Harper et al. 2010; Waters 2010; Kinney et al. 2011). Indeed, evidence implicates abnormalities in several pathways related to homeostasis, including in cardio-respiratory function (Kelly et al. 1979; Kelly et al. 1986; Antila et al. 1990; Pincus et al. 1993; Franco et al. 1998; Ledwidge et al. 1998; Rantonen et al. 1998; Schwartz et al. 1998; Franco et al. 1999; Edner et al. 2000; Ramanathan et al. 2001; Kinney et al. 2005), neurodevelopment (Ambler et al. 1981; Kinney et al. 1991; Filiano et al. 1992; O’Kusky et al. 1994; O’Kusky et al. 1995; Waters et al. 1999; Biondo et al. 2003; Weese-Mayer et al. 2004), immune function (Vege et al. 1995; Moscovis et al. 2004; Prandota 2004; Moscovis et al. 2006), and metabolic/energy pathways (Arens et al. 1993; Prandota 2004). However, among the most consistent abnormalities reported in SIDS cases are neurotransmitter and related abnormalities including in 5-HT (Panigrahy et al. 2000; Ozawa et al. 2002a; Ozawa et al. 2002b; Kinney et al. 2003; Paterson et al. 2006b; Machaalani et al. 2009; Duncan et al. 2010), γ-amino-butyric acid (GABA) (Broadbelt et al. 2011), and the family of 14-3-3 signal transduction proteins (Broadbelt et al. 2012) in the medulla oblongata of SIDS cases. Based upon these observations it is proposed that an important subset of SIDS cases results from abnormalities in a multi-neurotransmitter modulated medullary homeostatic network. These abnormalities are proposed to result in defective modulation and co-ordination of homeostatic function when the infant is in a compromised sleep environment (e.g., face-down or face-covered), initiating a chain of events ultimately leading to sleep-related sudden death.
4. Gene Variants Affecting Serotonin Neurotransmission Implicated in SIDS
Multiple studies have attempted to identify genetic variants associated with SIDS (see Table 1). The majority of these studies have been “candidate gene” studies focusing on single nucleotide polymorphisms (SNP) or small insertion/deletion polymorphisms that alter either protein structure or function or expression level of genes related to SIDS susceptibility pathways. Given the abnormalities in markers of 5-HT neurotransmission reported in SIDS cases significant research effort has focused upon gene variants that alter 5-HT neurotransmission (Fig. 2). The evidence for a role of these gene variants in the pathogenesis of SIDS is discussed in detail below.
Table 1.
Gene variants analyzed in SIDS
| Gene | Variant | Studies |
|---|---|---|
| FEV | IVS2-191_190insA | Rand et al., 2009; Broadbelt et al., 2009 |
| TPH2 | G1463A exon 6 (rs1200074175) | Marzano et al., 2008 |
| 5-HTT (SLC6A4) | 5-HTTLPR |
Narita et al., 2001; Weese-Mayer et al., 2003a
b; Marzano et al., 2008; Opdal et al., 2008; Haas et al., 2009; Paterson et al., 2010 |
| Intron 2 |
Weese-Mayer et al., 2003a
b; Marzano et al., 2008; Opdal et al., 2008; Haas et al., 2009 |
|
| 3′ UTR SNP | Maher et al., 2006 | |
| MAO-A | MAO-A promoter VNTR | Filzoni et al., 2008; Klintschar et al., 2012; Courts et al., 2013 |
| Interleukin 1 | Multiple | Ferrante et al., 2010 |
| Interleukin 6 | −174G/C |
Moscovis et al., 2006; Dashash et al., 2006; Opdal et al., 2007; Ferrante et al., 2012 |
| Interleukin 10 | −592A/C | Summers et al., 2000; Opdal et al., 2003 Moscovis et al.,2004; Korachi et al., 2004 Opdal et al., 2004 |
| TNFα | Multiple | Ferrante et al., 2008 |
| VEGF | −1154G/A | Dashash et al., 2006 |
| Phox2A | 287C/A | Weese-Mayer et al., 2004 |
| RET | Multiple | Weese-Mayer et al., 2004 |
| ECE1 | 1060A/G | Weese-Mayer et al., 2004 |
| TLX3 | 196C/T 152G/A |
Weese-Mayer et al., 2004 |
| EN1 | 719C/T 986C/A |
Weese-Mayer et al., 2004 |
| C4 CB |
Partial gene Deletions |
Opdal et al.,1999 |
| HVR-I | Multiple | Opdal et al.,1998,1999,2002 |
| KCNQ1, KCNH2, SCN5A | Multiple |
Ackerman et al., 2001; Arnestad et al., 2007; Otagiri et al., 2008; Millat et al., 2009. |
Figure 2. Model of 5-HT neurotransmission.

Schematic of 5-HT synthesis, release, and metabolism in a pre-synaptic neuron showing 5-HT pathway genes analyzed in SIDS. 1) FEV, Fifth Ewing Variant gene-transcription factor critical in differentiation of the 5-HT neuronal phenotype. Promotes the expression of TPH2, 5-HTT and 5-HT receptor genes in 5-HT neurons. 2) TPH2, tryptophan hydroxylase-2 gene-rate limiting enzyme in biosynthesis of 5-HT, catalyses the formation of 5-hydoxytryptophan from tryptophan (Tryp), which is then converted to 5-HT by aromatic acid decarboxylase (not shown). 3) 5-HT1A and 5-HT2A receptor genes-mediate the downstream effects of 5-HT. 4) 5-HTT, serotonin transporter gene-transports 5-HT released into the synapse back into the neuron for re-packing in vesicles for future release or degradation by MAO-A. 5) MAO-A, mononamine oxidase A-mitochondrial enzyme primarily responsible for the degradation of 5-HT into 5-hydoxy-indole-acetaldehyde (not shown) that is then converted into 5-hydroxy-indole-acetic acid (5-HIAA) by aldehyde dehydrogenase (not shown).
4.1 Fifth Ewing Variant (FEV)
The pheochromocytoma 12 (PC12) E26 transformation specific (Ets) factor (PET-1) plays a critical role in the differentiation and maintenance of the 5-HT neuron phenotype in rodents and chickens during neural development (Hendricks et al. 1999; Pfaar et al. 2002; Cheng et al. 2003; Hendricks et al. 2003; Scott et al. 2005). The Fifth Ewing Variant (FEV) is the human homologue of the PET-1, sharing 96% of its amino acid sequence, consistent with the idea that is plays a similar function (Pfaar et al. 2002; Maurer et al. 2004). A FEV gene variant that results in altered expression or function of the transcription factor could, therefore, potentially result in abnormal development and function of 5-HT neurons and the medullary 5-HT abnormalities observed in SIDS. In a study of 96 African American and Caucasian SIDS cases and 96 ethnically matched controls Rand et al (2007) identified what appeared to be a rare insertion mutation in the FEV gene (IVS2-191_190insA) exclusively in African American SIDS cases (Rand et al. 2007). This observation suggested that the mutation contributes to medullary 5-HT abnormalities selectively in African American SIDS infants, and potentially plays a role in the increased incidence of SIDS in this population. In our laboratory, we sought to replicate these observations by studying 78 SIDS cases from the San Diego SIDS Dataset, with an additional 296 control DNA samples of Caucasian, African American, and Mexican ethnicity (HD100CAU, HD100AA, and HD100MEX) from Coriel Cell Repositories. In contrast to Rand et al., however, we observed the insertion to be present in all ethnicities and to be a common mutation in African American controls-31% (33/108). Notably, we observed the insertion to be present in significantly higher frequency in Hispanic SIDS cases-9% (3/34) compared to ethnicity matched controls (Broadbelt et al. 2009). These observations indicate that the polymorphism appears to be a common, likely non-pathogenic, variant in the African-American population, but may be associated with increased SIDS risk in Hispanic populations.
4.2 Tryptophan Hydroxylase 2 (TPH2)
Tryptophan hydroxylase is the rate-limiting enzyme for the biosynthesis of 5-HT (Walther et al. 2003). Two isoforms of TPH exist: TPH1, which is responsible for 5-HT synthesis in peripheral tissues, and TPH2, which is expressed in the brain (Walther et al. 2003). Analysis of TPH2 gene is relevant to SIDS research as a functional variation in the gene that results in altered expression or function of the enzyme could result in a reduction in 5-HT synthesis, and potentially cause or contribute to the reduced levels of tissue 5-HT and TPH2 itself observed in the medulla of SIDS cases (Duncan et al. 2010). To date, however, only one study investigating genetic variation in the TPH2 gene in SIDS has been published. Marzano et al., (2008) analyzed a cohort of 20 Italian SIDS cases for a rare SNP (rs1200074175) in the TPH2 gene that affects the catalytic site of the enzyme and results in reduced 5-HT synthesis (Zhang et al. 2004; McKinney et al. 2008). The mutation has a frequency of approximately 0.7% in the general population, so it is unsurprising that authors did not observed the SNP in any of their SIDS cases (Nonnis Marzano et al. 2008). This mutation has not been studied in any other SIDS populations but given its rarity it is unlikely to play a role in the pathogenesis of a significant number of SIDS cases.
4.3 5-HT1A and 5-HT2A receptors
The 5-HT1A and 5-HT2A receptors represent the two main functional classes of receptors mediating the downstream affects of 5-HT neurotransmission. The 5-HT1A receptor is predominantly recognized as a somato-dendritic autoreceptor (although there are significant populations of 5-HT1A heteroreceptors) that inhibits neuron function (Barnes et al. 1999). In contrast, the 5-HT2A receptor is considered to be an excitatory post-synaptic (to 5-HT neurons) heteroreceptor that stimulates neuron function (Barnes et al. 1999). Both of these receptors are densely distributed in medullary regions mediating homeostatic function, (Paterson et al. 2004; Paterson et al. 2006b; Paterson et al. 2009; Paterson et al. 2010) and play an important role in neural development as well as 5-HT mediated regulation of autonomic and respiratory function (Gillis et al. 1989; King et al. 1991; Lalley 1994; Lalley et al. 1994; Bach et al. 1996; Berner et al. 1999; Mitchell et al. 2001; Pena et al. 2002; Raul 2003; Baker-Herman et al. 2004; Ramirez et al. 2004; Tryba et al. 2006; Comet et al. 2007; Dergacheva et al. 2007; Shen et al. 2007; Villalon et al. 2007). Significant reductions in 5-HT1A receptor binding (Paterson et al. 2006b; Duncan et al. 2010) and 5-HT2A receptor immunostaining (Ozawa et al. 2002a; Ozawa et al. 2002b) have been observed in the medulla of SIDS cases. Analysis of functional genetic variants that alter the expression or function of these receptors is, therefore, relevant to genetic research in SIDS.
In two separate studies, analyzing the same SIDS cohort as in the FEV study, Morley et al., (2008) and Rand et al., (2009), respectively, sequenced the 5-HT1A (HTR1A) and 5-HT2A (HTR2A) genes in order to identify functional variants that potentially contributed to the pathogenesis of SIDS. Both studies identified SNPs, including some novel variants, which resulted in protein-coding changes in the 5-HT1A and 5-HT2A receptors (Morley et al. 2008; Rand et al. 2009). Morley et al., (2008) observed a greater frequency of variation in the HTR1A gene in male SIDS cases compared to female SIDS cases, raising the possibility that increased variation in general in the 5-HT1A receptor gene may play a role in the increased incidence of SIDS in male versus female infants. However, neither study observed a significant association between any gene variant and SIDS.
4.4 The 5-HT Transporter Promoter Polymorphism (5-HTTLPR)
The 5-HT transporter (5-HTT) plays a key role in 5-HT neurotransmission via its control of synaptic 5-HT levels (Blakely et al. 2005); thus, genetic variations that alter 5-HTT expression or function may affect synaptic availability of 5-HT and contribute to the deficit in 5-HT levels observed in the medulla in SIDS cases. The 5-HTT gene is the most commonly studied 5-HT pathway gene in SIDS, with 8 studies to date analyzing different functional genetic variants (Fig.3). The gene variants studied each alter gene expression and include two insertion/deletion polymorphisms, one in the promoter region (5-HTT promoter polymorphism or 5-HTTLPR) (Heils et al. 1995; Heils et al. 1997; Greenberg et al. 1999) and the other in the second intron (Ogilvie et al. 1996), and a SNP (rs1042173) in a polyadenylation site in the 3′ region of the UTR (Battersby et al. 1999; Seneviratne et al. 2009) (Fig. 3). While the 3′ UTR SNP was not observed to be associated with SIDS (Maher et al. 2006), significant associations with both the 5-HTTLPR and Intron 2 polymorphisms have been reported in multiple studies, and I now consider these latter two polymorphisms in more detail.
Figure 3. Serotonin gene variants studied in SIDS.

Schematic of the 5-HTT gene (SLC6A4) and the location of the 5-HTTLPR, Intron 2 and 3′ UTR polymorphisms and their putative effects on gene transcription.
The 5-HTT promoter polymorphism consists of a 22-23 base-pair insertion-deletion in the 5′ promoter region of the gene producing either a short “S” allele that has 14 copies or a long “L” allele that has 16 copies of the insertion. The L allele is associated with increased gene transcription and protein expression compared to the S allele (Heils et al. 1995; Heils et al. 1997; Greenberg et al. 1999) (Fig.3). Similarly, the LL genotype results in greater gene transcription and protein expression than the SS genotype. However, gene and protein expression levels in the SL genotype are comparable with those observed with the SS genotype (Lesch et al. 1996; Heils et al. 1997; Hranilovic et al. 2004). These observations indicate that the S allele exerts dominance over the L allele in regulation of gene expression (Lesch et al. 1996; Heils et al. 1997; Hranilovic et al. 2004) and that there is no “allele dosage” effect on gene transcription and protein expression. In 2001, Narita et al., reported that the LL genotype and the L allele were present in significantly higher frequency in SIDS cases compared to controls in a Japanese cohort (Narita et al. 2001). This observation led to the hypothesis that infants with the LL genotype have increased 5-HT uptake, and thus, are at greater risk of SIDS, particularly if there is an existing medullary 5-HT deficiency. Subsequently, multiple studies attempted to confirm the observations of Narita et al., analyzing different SIDS populations. Weese-Mayer et al., (2003a) replicated the association between the LL genotype and L allele with SIDS in a combined Caucasian and African American SIDS cohort. However, when the ethnic subgroups were analyzed individually, no significant associations between genotype or allele were observed in African Americans and an association with the L allele only was observed in Caucasians (Weese-Mayer et al. 2003a; Weese-Mayer et al. 2003b). Nonnis-Marzano et al., (2008) also reported a significant association between the LL genotype and L allele with SIDS in an Italian population (Nonnis Marzano et al. 2008), albeit with a very limited sample size of 20 SIDS cases. In another study, Opdal et al., (2008) observed a borderline significant association (p=0.05) of the L allele with SIDS in a Norwegian Caucasian cohort. In contrast, in a Swiss Caucasian population Hass et al., (2009) observed no association between genotype or allele frequency and SIDS (Haas et al. 2009). Similarly, we did not observed any association between genotype or allele and SIDS in our predominantly Caucasian (SIDS n=94, Controls n=100) San Diego Dataset containing 179 SIDS cases and 139 controls, either in the total cohort or when stratifying by ethnicity (Paterson et al. 2010).
As is evident from the description of the studies above, the vast majority of SIDS cases analyzed for the 5-HTTLPR have been Caucasian. Taken together, observations from these studies do not support a role for the 5-HTTLPR as significant risk factor for SIDS in this ethnicity. Two out of the three studies reporting positive associations with the LL genotype and/or the L allele with SIDS were significantly underpowered by sample size (i.e., Weese-Mayer et al., n=44 SIDS; Nonnis-Marzano et al., n=20 SIDS), and reported allele frequencies in their Control populations studies that were the reverse of those expected for this ethnicity, i.e., exhibiting a higher frequency of the S allele over the L allele (Fig. 4). Thus, the significant associations observed in these two studies may well be a product of population bias. Thus, out of the studies of Caucasian populations with reasonable statistical power to detect differences in genotype and allele frequency distribution, only one, Opdal et al., (2008) reported a borderline association of SIDS with the L allele. Moreover, given that increased 5-HTT expression and 5-HT uptake is observed only with the LL genotype and not with the L allele, this observation does not support the idea that the 5-HTTLPR is a risk factor for SIDS. Therefore, when considered together, the data from the studies described above provide, at best, only weak evidence to support a link between the 5-HTTLPR and SIDS in Caucasians. The role of this polymorphism in conferring SIDS risk in other ethnicities remains to be fully elucidated, but it seems unlikely that it plays a major pathogenic role in any population.
Figure 4. 5-HTTLPR allele frequencies in Caucasians.

Graphs comparing the S and L allele frequencies in Caucasian control populations in different studies. Note that the S allele is the major allele (i.e., allele present in highest frequency) in the populations analyzed by Weese-Mayer et al., (2003a) and Nonnis-Marzano et al., (2008), the reverse of the other study populations and the expected allele frequency for this polymorphism in Caucasians. This suggests that sample bias may influence the observations in these studies. Names under each data group refer to the first author of the study from which the data is extracted with the exceptions of “San Diego” and “Coriell” that refer to the San Diego SIDS Dataset and the HD100CAU human variation DNA panel from the Coriell Institute Cell Repository (Camden, NJ) from Paterson et al., (2010).
4.5 The 5-HTT Intron 2 Polymorphism
This polymorphism consists of a variable number tandem repeat (VNTR) of 9, 10 or 12 copies of a 17bp element in the second intron the 5-HTT gene (Ogilvie et al. 1996) (Fig. 2). Similar to the L allele of the 5-HTTLPR, cells transfected with 12 copies of the repeat element display increased gene expression compared to cells with 10 copies (Fiskerstrand et al. 1999; MacKenzie et al. 1999). In 2003, Weese-Mayer et al., analyzed the same dataset of 96 SIDS cases and ethnicity matched controls for this poymorphism that they had previously analyzed for the 5-HTTLPR. They identified a significant correlation between the 1212 genotype and the 12 allele in African American but not Caucasian SIDS infants (Weese-Mayer et al. 2003b). Hass et al., (2009), Opdal et al., (2008) and Nonnis-Marzano et al., (2008) did not observe any association between genotype or allele frequency and SIDS when analyzing Caucasian only cohorts. In our San Diego SIDS Dataset we genotyped 147 SIDS cases and 122 controls, including n=18 SIDS and n=7 Controls of African American origin but did not observe any significant association between genotype or allele with SIDS in this ethnicity (Table 2). This observation may be due to the small number of African American cases available for analysis. However, genotype and allele frequency distributions in the African American SIDS cases were consistent with those in n=100 African American Coriell Controls (Table 2), supporting the idea that there is no association with this polymorphism and SIDS in African Americans. Interestingly, we observed a significantly higher frequency (p<0.002) of the 1010 genotype (14.29%) and a lower frequency of the 1212 genotype (4.1%) in SIDS cases compared to controls when we analyzed the total cohort (Table 2). However, these observations appear to be driven by our Caucasian control population as they exhibited a higher frequency of the 1212 genotype and 12 allele compared to Caucasian Coriell controls, which are accepted as representative of the Caucasian population (Table 2) (Fig 5). Therefore, we think it is unlikely that there is a true association between the 1010 genotype and SIDS. In summary, to date studies of the intron 2 polymorphism do not provide any data to support a role for the polymorphism in the pathogenesis of SIDS in Caucasian populations. However, while recognizing that the observations of Weese-Mayer et al., require confirmation in larger datasets, it remains possible that the intron 2 polymorphism is associated with increased SIDS risk in African American, and potentially other non-Caucasian, populations.
Table 2.
Genotype Distribution and Allele Frequencies for the Intron 2 Polymorphism in the San Diego Database and Coriell Controls
| 1010 | 1012 | 1212 | χ2 test p-value |
10 | 12 | χ2 test p-value |
||
|---|---|---|---|---|---|---|---|---|
| All Cases |
SIDS
n=147 |
21 (14.3%) |
61 (41.5%) |
65 (44.2%) |
0.002* | 103 (35.0%) |
191 (65.0%) |
<0.001** |
|
Controls
n=122 |
5 (4.1%) |
41 (33.6%) |
76 (62.3%) |
51 (20.9%) |
193 (79.1%) |
|||
|
| ||||||||
| Caucasians |
SIDS
n=122 |
12 (16.4%) |
29 (39.7 %) |
32 (43.8%) |
0.017* | 53 (36.3%) |
93 (63.7%) |
0.002* |
|
Controls
n=88 |
5 (5.7%) |
27 (30.7%) |
56 (63.6%) |
27 (21.0 %) |
139 (79.0 %) |
|||
|
Coriell
n=95 |
11 (11.6%) |
48 (51.6%) |
36 (37.9%) |
70 (37.0%) |
120 (63.0%) |
|||
|
| ||||||||
|
African
American |
SIDS
n=18 |
2 (11.1%) |
8 (44.4%) |
8 (44.4%) |
0.12 | 12 (33.3%) |
24 (66.7%) |
0.39 |
|
Controls
n=7 |
0 (0%) |
1 (14.3%) |
6 (85.7%) |
1 (7.1%) |
13 (92.9%) |
|||
|
Coriell n=100 |
7 (7.0%) |
49 (49.0%) |
44 (44.0%) |
63 (31.5%) |
137 (68.5%) |
|||
|
| ||||||||
| Hispanic |
SIDS
n=33 |
6 (15.0%) |
15 (37.5%) |
19 (47.5%) |
0.58 | 27 (33.7%) |
53 (33.7%) |
0.27 |
|
Controls
n=18 |
0 (0%) |
9 (50.0%) |
0 (50.0%) |
9 (25.0%) |
27 (75.0%) |
|||
|
Coriell
n=100 |
3 (3.0%) |
45 (45.0%) |
52 (52.0%) |
51 (25.5%) |
149 (75.5%) |
|||
Table shows number of cases/controls and (%) for each genotype and allele. The frequency of the 1010 genotype and the 10 allele was significantly higher in SIDS cases compared to controls in the total cohort and in Caucasian SIDS cases compared to Caucasian controls.
P<0.05 χ2 test for comparisons between SIDS cases and controls.
Figure 5. Intron 2 allele frequencies in San Diego and Coriell Caucasians.
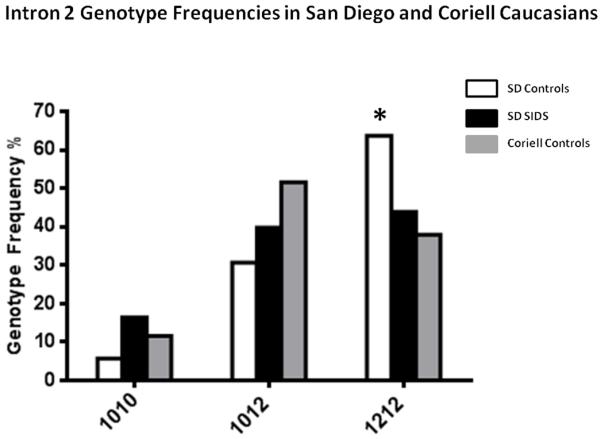
Graph comparing the intron 2 genotype frequencies in Caucasian San Diego SIDS cases, Controls and Coriell controls. The frequency of the 1212 genotype is significantly higher in the San Diego Control population compared to the SIDS cases and Coriel Controls. χ2 =12.23 df=2 p=0.002 for San Diego Controls vs. Coriell and χ2 =8.17 df=2 p=0.017 for San Diego Controls vs. San Diego SIDS.
4.6 Monoamine Oxidase A (MAOA)
Mono-Amine Oxidase (MAO) is the mitochondrial enzyme primarily responsible for the intracellular degradation of 5-HT (Shih et al. 1999b; Shih et al. 1999a). Two isoforms of MAO enzyme exist, A and B. MAOA has greater affinity for 5-HT and plays the major role in 5-HT degradation (Shih et al. 1999b; Shih et al. 1999a) (Fig. 2). Elevated or reduced expression of MAO resulting from genetic variation associated with this polymorphism may potentially alter the level of 5-HT available for release, via increased or reduced enzymatic degradation of 5-HT, and, therefore, alter 5-HT function in SIDS. The MAOA gene is also of interest to SIDS research because it is located on the X-chromosome. As males have only one X-chromosome they exhibit hemizygosity (i.e., have only one copy of the gene and, thus, a single allele) for any X-chromosome located gene, and thus, functional allelic variants in these genes are likely to have a greater impact on the biological process in males compared to females. It is possible, therefore, that functional genetic variants in the MAOA gene may underlie the excess of male-to-female SIDS deaths.
A VNTR polymorphism in the promoter region of the MAOA gene that alters expression has been analyzed in the three independent SIDS populations. The polymorphism consists of a 30bp repeat sequence present in 2, 3, 3.5, 4, or 5 repeats, with 3.5 and 4 copies (“high” alleles) of the repeat associated with increased gene transcription compared to 2 and 3 copies (“low” alleles) (Sabol et al. 1998; Deckert et al. 1999). A significant association between the “high” allelic variants and SIDS was reported by FIlzoni et al., (2009), albeit in only 20 SIDS cases (Filonzi et al. 2009). Nevertheless, this observation was essentially replicated by Courts et al., (2013), who reported a significantly higher frequency of “high” allelic variants in their total cohort (n=142) and in female (n=52), but not male (n=90), SIDS cases compared to controls (n=280) (Courts et al. 2013). In contrast, however, Klintschar et al., (2012) reported a significant association between the “low” 2 and 3 allelic variants and male SIDS cases, but not in female SIDS cases (Klintschar et al. 2012). In our San Diego SIDS Dataset, we analyzed the genotype and allele frequency of the MAOA promoter VNTR in n=140 SIDS cases (n=85 male and n=55 female) and 92 controls (n=52 males and n=40 females). Consistent with the observations of Klintschar et al., (2012), we observed an increased frequency of the “low” (2 and 3) alleles in Caucasian male SIDS cases compared to controls (χ2 =3.98, df=1, p=0.046) (Table 3). However, there was no significant difference in allele frequency between the Caucasian male SIDS cases and Coriel Caucasian male controls (Table 3). In addition, the frequency of the “high” alleles in our Caucasian male controls (86%) was higher than in Coriel Caucasian male controls (67%) (Table 3). Therefore, as with our observations of the intron 2 polymorphism, this data suggests that the observed differences in allele frequency between SIDS and controls in the San Diego Dataset are being driven by admixture in the control population and likely to do not represent a true association with SIDS.
Table 3.
Allele Frequencies for the MAO-A VNTR Polymorphism in the San Diego Database and Caucasian Coriell Controls
| Male | Female | ||||||
|---|---|---|---|---|---|---|---|
| All Cases | Allele |
SIDS
n=84 |
Control
n=53 |
Exact Test |
SIDS
n=55 |
Control
n=39 |
Exact Test |
| 2 | 2 (2.4%) | 1 (1.9%) | p=0.78 | 0 (0%) | 0 (0%) | p=0.86 | |
| 3 | 23 (27.4%) | 12 (22.6%) | 30 (27.3%) | 18 (23.1%) | |||
| 4 | 57 (67.9%) | 40 (75.5%) | 77 (70.0%) | 58 (74.4%) | |||
| 5 | 2 (2.4%) | 0 (0%) | 3 (0.7%) | 2 (2.5%) | |||
| χ2 test | χ2 test | ||||||
| 2&3 | 25 (29.8%) | 13 (24.5%) | p=0.51 | 30 (27.3%) | 18 (23.1%) | p=0.52 | |
| 4&5 | 59 (70.2%) | 40 (75.5%) | 80 (72.7%) | 60 (76.9%) | |||
|
| |||||||
| Caucasians |
n=43 |
n=36 |
Exact Test |
n=23 |
n=30 |
Exact Test | |
| 2 | 2 (4.7%) | 0 (0%) | p=0.09 | 0 (0%) | 0 (0%) | p=0.36 | |
| 3 | 12 (27.9%) | 5 (13.9%) | 13 (28.3%) | 12 (20.0%) | |||
| 4 | 28 (65.1%) | 31 (86.1%) | 32 (69.6%) | 46 (76.7%) | |||
| 5 | 1 (2.3%) | 0 (0%) | 1 (2.1%) | 2 (3.3%) | |||
| χ2 test | χ2 test | ||||||
| 2&3 | 14 (32.6%) | 5 (13.9%) | p=0.046* | 13 (28.3%) | 12 (20.0%) | p=0.32 | |
| 4&5 | 29 (67.4%) | 31 (86.1%) | 33 (71.7%) | 48 (80.0%) | |||
|
| |||||||
|
Caucasian
Coriell |
n=50 |
Exact Test† |
n=46 |
χ2 test | |||
| 2 | 0 (0%) | p=0.34 | 0 (0%) | p=0.34 | |||
| 3 | 16 (32.0%) | 25 (25.5%) | |||||
| 4 | 34 (68.0%) | 73 (74.5%) | |||||
| 5 | 0 (0%) | 0 (0%) | |||||
| 2&3 | 16 (32.0%) | p=0.34 | 25 (25.5%) | p=0.34 | |||
| 4&5 | 34 (68.0%) | 73 (74.5%) | |||||
Table shows number of cases/controls and (%) for each allele. The frequency of the combined 3&4 alleles is significantly higher in Caucasian male controls compared to Caucasian SIDS cases in the San Diego Dataset. However, no significant difference in combined allele frequency was observed between Caucasian SIDS cases and Caucasian Coriell controls.
P-values for comparison with Caucasian SIDS cases.
P<0.05 χ2 test.
The observations from the studies with reasonable statistical power to detect differences in genotype and allele frequency appear, therefore, to be entirely contradictory. That is, one study identifying increased SIDS risk with increased MAOA expression in females (Courts et al.), another with reduced MAOA expression in males (Klinstschar et al.,) and in our study of the San Diego SIDS Dataset we essentially identified no association at all. Further studies on larger datasets, including on different ethnicities, will be necessary to determine whether the MAOA polymorphism is associated with increased SIDS risk.
5. Correlation of 5-HT Signaling Pathway Gene Variants with Brainstem 5-HT Markers in SIDS Cases
The 5-HTTLPR, Intron 2 and MAOA polymorphisms are hypothesized to increase the risk of SIDS by altering 5-HT neurotransmission thereby putatively contributing to the medullary 5-HT defects observed in SIDS cases. In a subset of SIDS cases in the San Diego SIDS Dataset we have quantitative data on markers of medullary 5-HT function including, 5-HTT binding density, 5-HT levels, TPH2 levels and 5-HT1A receptor binding density (Paterson et al. 2006a; Duncan et al. 2010). By correlating genotype information with this data we are in the unique position of being able to test the above hypothesis directly in SIDS cases. To date, in our San Diego SIDS Dataset, we have 55 SIDS cases and 15 Controls for which we have brainstem neurochemical data that we can correlate with genotype information on the 5-HTTLPR, Intron 2 and MAO-A polymorphisms. The small number of Controls available in our database is, thus, impractical to allow accurate correlation of neurochemistry with genotype. Therefore, we individually correlated 5-HTTLPR, Intron 2 and MAOA genotypes with previously determined neurochemistry data in SIDS cases only. For these analyses, we tested the hypothesis that the LL genotype of the 5-HTTLPR, 1212 genotype of the Intron 2, and the 4 allele (or 44 genotype) of the MAOA polymorphisms are associated with: 1) higher 5-HTT binding density, 2) lower 5-HT1A receptor binding density, and 3) lower TPH2 and 5-HT tissue levels in the medulla compared to SIDS cases with other genotypes.
In contrast to our hypothesis we observed 5-HTT binding density to be significantly lower (23-34%) in the intermediate reticular zone (p=0.04), paragigantocellularis lateralis (p=0.02), and gigantocellularis (p=0.008) in SIDS cases with SL genotype of the 5-HTTLPR (Fig.7). Similarly contradictory in nature, we observed 5-HTT binding to be significantly higher in the nucleus of the solitary tract (p=0.04) in SIDS cases with the 1010 genotype of the intron 2 polymorphism (Fig 8A). The 1010 genotype was also associated with marginally significantly higher 5-HT1A receptor binding density in the gigantocellularis nucleus (p=0.05) (Fig. 8B). We did not observe any significant associations between genotype and 5-HT levels or TPH2 levels with genotype for either of the 5-HTT gene polymorphisms. For the MAOA polymorphism, we observed a significantly higher density of 5-HT1A receptor binding sites in the raphe obscurus (RO) of male SIDS cases with the 3 allele (Fig. 9A) and a significantly higher level of 5-HT in the RO in female SIDS cases with the 33 genotype (Fig. 9B).
Figure 7. Medullary 5-HTT binding density correlated with 5-HTTLPR genotype in SIDS cases.

Medullary 5-HTT binding density is significantly lower in SIDS cases with the SL genotype in the gigantocellularis (GC) (*p<0.001), paragigantocellularlis lateralis (PGCL) (*p=0.02), and intermediate reticular (IRN) (*p=0.04) nucleus by genotype (ANCOVA).
Figure 8. Medullary 5-HTT binding and 5-HT1A receptor binding density correlated with Intron 2 genotype in SIDS cases.

A. Medullary 5-HTT binding density in SIDS cases in the intermediate reticular nucleus (IRN) and nucleus of the solitary tract (NTS); binding density is significantly lower in SIDS cases with the 1010 genotype in the NTS (*p=0.05, ANCOVA) and shows a trend to be lower in the IRZ (p=0.06 ANCOVA). B. Medullary 5-HT1A receptor binding density in SIDS cases in the gigantocellularis (GC) nucleus; receptor binding density is significantly lower in SIDS cases with the 1010 genotype (*p=0.05, ANCOVA).
Figure 9. 5-HT1A receptor binding density and 5-HT tissue level correlated with MAO-A genotype in SIDS cases.

A. Medullary 5-HT1A binding density in the raphe obscurus (ROB) is significantly lower in male SIDS cases with the 3 allele compared to SIDS cases with the 4 allele nuclei. *p=0.02, ANCOVA. B. Medullary 5-HT tissue level in the raphe obscurus (ROB) is significantly lower in female SIDS cases with the 33 genotype compared to SIDS cases with the 34 or 44 genotypes. *p=0.01, ANCOVA.
Interpretation of the above observations is limited by the relatively small sample sizes (i.e., as little as n=3 for some MAOA analyses) as well as by the lack control data. Without appropriate genotype-correlation data from controls, it is not possible to determine if the significant correlations observed in SIDS are pathogenic or if they simply result from a normal genotype association, e.g., that 5-HTT binding density is also reduced in controls with the SL genotype. Alternatively, it is possible that the hypothesized genotype-marker correlation exists in controls but not in SIDS. Nevertheless, based on these limited data, we propose that the observations above do not support a role for the 5-HTTLPR, Intron 2 and MAOA polymorphisms in the pathogenesis of the medullary 5-HT abnormalities in SIDS in the hypothesized way, i.e., by contributing directly to the “5-HT deficit”, via elevated 5-HTT or MAO-A expression. Specifically, the LL or 1212 genotypes of the 5-HTTLPR and intron 2 polymorphisms were neither associated with increased 5-HTT binding density nor reduced 5-HT levels in the medullary 5-HT system. Similarly, neither the “high” expressing 4 allele nor the “low” expressing 3 allele of the MAOA polymorphism were associated with reduced medullary 5-HT levels in SIDS cases. Indeed, the observations were consistent with the “low” expressing 3 allele conferring a protective effect against SIDS in that the 3 allele and 33 genotype were associated with increased 5-HT1A receptor binding density and increased 5-HT levels in the RO. Taken together, these observations suggest that it is unlikely that any SIDS risk that is conferred by these polymorphisms involves a simple mechanism whereby they alter 5-HTT or MAOA expression or function and lead to reduced synaptic 5-HT.
6. Conclusions
The observations of the genetic studies described above do not provide compelling evidence that any of the 5-HT pathway genes studied to date play a major role in the pathogenesis of SIDS at least in Caucasian populations. If any of the gene variants studied was a strong predictor of SIDS it is expected that a consistent association of the risk-related genotype with SIDS would be observed across the studies even with the modest sample sizes analyzed. It is possible that these polymorphisms may be more strongly associated with SIDS in non-Caucasians, e.g., African Americans, as only a handful of studies have been performed on these populations and the observations of these studies have yet to be replicated or refuted. However, the risk-related genotype-neurochemistry correlation studies performed by us in our San Diego SIDS Database, while limited by sample size, do not provide any evidence to support that the idea that these gene variants alter makers of 5-HT neurotransmission in the expected way. Thus, it is unclear, how, if at all, they would contribute to the medullary 5-HT abnormalities observed in SIDS, regardless of ethnicity. Overall, it appears unlikely that any of the 5-HT pathway gene polymorphisms studied to date impart any significant SIDS risk. Furthermore, any role that they do play in the pathogenesis of SIDS is likely to be part of a complicated interaction of multiple other factors (physiological, environmental and genetic) that is currently unclear. Ultimately, in the opinion of this author, analysis of these polymorphisms does not provide a meaningful index of SIDS risk.
The likely heterogeneous nature of SIDS, i.e., putatively consisting of multiple distinct diseases with a final common phenotype rather than a single disease entity, indicates that it is likely to be polygenic in its pathogenesis. Within this paradigm the genetic component of SIDS may consist of either rare gene variants that have a significant deleterious impact upon a vital physiological function such that individually they are sufficient to result in the death of the infant, e.g., MCAD gene mutations; or multiple susceptibility gene variants that individually confer a small degree of SIDS risk, e.g. 5-HT pathway gene variants, that act synergistically with other variants and environmental factors to precipitate the death of the infant. On this basis, the search for rare or novel gene variants such as copy number variations (CNV) or rare point mutations are of utility in SIDS research. Array comparative genome hybridization (aCGH) has been successful in identifying such mutations in other sporadic diseases with complex etiology including autism, schizophrenia, Crohn’s disease, and susceptibility to HIV infection (Gonzalez et al. 2005; Fellermann et al. 2006; Lee et al. 2006; Scherer et al. 2007; Sebat et al. 2007; Morrow et al. 2008; Walsh et al. 2008). Indeed, by applying aCGH Toruner et al., (2009) identified CNVs in 3 out of 27 SIDS cases (11%), including large-scale duplications and deletions in the region of the genome where the major cluster of histone genes is located (Toruner et al. 2009). The precise pathway(s) through which the CNVs identified by Toruner et al., (2009) contributed to the death of the infants is unclear and remains to be determined. However, these observations illustrate the potential of CNV analysis to identify novel candidate genes and pathways that may contribute to the pathogenesis of SIDS. Alternatively, approaches to develop a “genetic profile” of SIDS susceptibility gene variants may be employed by applying next generation sequencing technologies. Whole exome and whole genome sequencing allow deep sequencing of gene coding regions across the entire genome and provide the opportunity to simultaneously genotype an individual for all previously identified gene variants associated with SIDS, as well as for novel gene variants in the same or previously un-recognized systems. As the cost of these technologies continues to fall in line with the increase in their capabilities, they will enable the characterization of a genetic susceptibility profile in SIDS cases, including how the gene variants in each of the SIDS susceptibility pathways overlap and combine to increase SIDS risk in an individual. This type of analysis would also create a database of genetic information for each infant (and individual SIDS populations) that could be analyzed for gene variants as new genetic information on SIDS became available. In this way, a comprehensive profile of gene variants contributing to SIDS risk could be developed. Ultimately, it seems likely that the genetic component to SIDS will include both rare causative mutations as well as a “catalogue” of susceptibility variants that predispose an infant to increased SIDS risk.
Highlights.
Discussion of the evidence for a role of 5-HT gene variants in the pathogenesis of SIDS
Unique correlation of 5-HT neurochemistry with genotype in the same SIDS cases
5-HT gene variants studied to date do not appear to play a major role in the pathogenesis of SIDS
SIDS is likely to be polygenic
Pathogenesis potentially involves rare point mutations and copy number variations
Figure 6. MAO-A gene promoter polymorphism.

Schematic of the MAO-A and the location of the promoter VNTR and its putative effects on gene transcription.
ACKNOWLEDGEMENTS
The author thanks the dedicated assistance of the medical examiners of the San Diego County Medical Examiner’s Office, San Diego, CA, without whom this work would not be possible. I would also like to thank the work of Dr Felicia Trachtenberg for providing consultation on the statistical analyses presented in the review, Dr Kyriacos Markianos for his insights and advice in the course of these studies, and Dr Hannah Kinney for her critical comments in manuscript preparation. This work was funded by the National Institute of Child Health and Development (R37-HD20991, PO1-HD036379 [HCK], and P30-HD18655 [Developmental Disabilities Research Center, Children’s Hospital Boston]), Scottish Cot Death Trust, First Candle/SIDS Alliance, CJ Murphy Foundation for Solving the Puzzle of SIDS, the Marley Jaye Cherella Memorial Fund and “Jon’s Run” and the Any Baby Can Center for Infant and Child Loss.
Sources of Support: National Institute of Child Health and Development (R01-HD20991, PO1-HD036379, and P30-HD18655 [Developmental Disabilities Research Center, Children’s Hospital Boston]), Scottish Cot Death Trust, First Candle/SIDS Alliance, CJ Foundation for SIDS, the Marley Jaye Chirella Memorial Fund, “Jon’s Run”, and the Any Baby Can Center for Infant and Child Loss.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, Towbin JA. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. Jama. 2001;286:2264–2269. doi: 10.1001/jama.286.18.2264. [DOI] [PubMed] [Google Scholar]
- Alm B, Wennergren G, Norvenius G, Skjaerven R, Oyen N, Helweg-Larsen K, Lagercrantz H, Irgens LM. Caffeine and alcohol as risk factors for sudden infant death syndrome. Nordic Epidemiological SIDS Study. Arch Dis Child. 1999;81:107–111. doi: 10.1136/adc.81.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambler MW, Neave C, Sturner WQ. Sudden and unexpected death in infancy and childhood: neuropathological findings. Am J Forensic Med Pathol. 1981;2:23–30. doi: 10.1097/00000433-198103000-00005. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Johnson DC, Batal HA. Sudden Infant Death Syndrome and prenatal maternal smoking: rising attributed risk in the Back to Sleep era. BMC Med. 2005;3:4. doi: 10.1186/1741-7015-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antila KJ, Valimaki IA, Makela M, Tuominen J, Wilson AJ, Southall DP. Heart rate variability in infants subsequently suffering sudden infant death syndrome (SIDS) Early Hum Dev. 1990;22:57–72. doi: 10.1016/0378-3782(90)90080-3. [DOI] [PubMed] [Google Scholar]
- Arens R, Gozal D, Jain K, Muscati S, Heuser ET, Williams JC, Keens TG, Ward SL. Prevalence of medium-chain acyl-coenzyme A dehydrogenase deficiency in the sudden infant death syndrome. Journal of Pediatrics. 1993;122:715–718. doi: 10.1016/s0022-3476(06)80010-7. [DOI] [PubMed] [Google Scholar]
- Arneil GC, Brooke H, Gibson AA, Harvie A, McIntosh H, Patrick WJ. National Post-Perinatal Infant Mortality and Cot Death Study, Scotland 1981-82. Lancet. 1985;1:740–743. doi: 10.1016/s0140-6736(85)91273-5. [DOI] [PubMed] [Google Scholar]
- Arnestad M, Andersen M, Vege A, Rognum TO. Changes in the epidemiological pattern of sudden infant death syndrome in southeast Norway, 1984-1998: implications for future prevention and research. Arch Dis Child. 2001;85:108–115. doi: 10.1136/adc.85.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL, Jr., Schwartz PJ. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Battersby S, Ogilvie AD, Blackwood DH, Shen S, Muqit MM, Muir WJ, Teague P, Goodwin GM, Harmar AJ. Presence of multiple functional polyadenylation signals and a single nucleotide polymorphism in the 3′ untranslated region of the human serotonin transporter gene. J Neurochem. 1999;72:1384–1388. doi: 10.1046/j.1471-4159.1999.721384.x. [DOI] [PubMed] [Google Scholar]
- Beal S. Sudden infant death syndrome. Med J Aust. 1972;2:1223–1229. [PubMed] [Google Scholar]
- Beal SM. Sudden infant death syndrome: epidemiological comparisons between South Australia and communities with a different incidence. Aust Paediatr J. 1986;22(Suppl 1):13–16. [PubMed] [Google Scholar]
- Beal SM, Baghurst P, Antoniou G. Sudden infant death syndrome (SIDS) in South Australia 1968-97. Part 2: the epidemiology of non-prone and non-covered SIDS infants. J Paediatr Child Health. 2000;36:548–551. doi: 10.1046/j.1440-1754.2000.00576.x. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Powell S. Metabolic disease and sudden, unexpected death in infancy. Hum Pathol. 1994;25:742–746. doi: 10.1016/0046-8177(94)90241-0. [DOI] [PubMed] [Google Scholar]
- Berner NJ, Grahn DA, Heller HC. 8-OH-DPAT-sensitive neurons in the nucleus raphe magnus modulate thermoregulatory output in rats. Brain Res. 1999;831:155–164. doi: 10.1016/s0006-8993(99)01426-2. [DOI] [PubMed] [Google Scholar]
- Biondo B, Lavezzi A, Tosi D, Turconi P, Matturri L. Delayed neuronal maturation of the medullary arcuate nucleus in sudden infant death syndrome. Acta Neuropathol (Berl) 2003;106:545–551. doi: 10.1007/s00401-003-0757-3. [DOI] [PubMed] [Google Scholar]
- Blair PS, Fleming PJ, Bensley D, Smith I, Bacon C, Taylor E, Berry J, Golding J, Tripp J. Smoking and the sudden infant death syndrome: results from 1993-5 case-control study for confidential inquiry into stillbirths and deaths in infancy. Confidential Enquiry into Stillbirths and Deaths Regional Coordinators and Researchers. Bmj. 1996;313:195–198. doi: 10.1136/bmj.313.7051.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair PS, Fleming PJ, Smith IJ, Platt MW, Young J, Nadin P, Berry PJ, Golding J, CESDI SUDI research group Babies sleeping with parents: case-control study of factors influencing the risk of the sudden infant death syndrome. Bmj. 1999;319:1457–1461. doi: 10.1136/bmj.319.7223.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair PS, Platt MW, Smith IJ, Fleming PJ. Sudden infant death syndrome and sleeping position in pre-term and low birth weight infants: an opportunity for targeted intervention. Arch Dis Child. 2006;91:101–106. doi: 10.1136/adc.2004.070391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely RD, Defelice LJ, Galli A. Biogenic amine neurotransmitter transporters: just when you thought you knew them. Physiology (Bethesda) 2005;20:225–231. doi: 10.1152/physiol.00013.2005. [DOI] [PubMed] [Google Scholar]
- Broadbelt KG, Barger MA, Paterson DS, Holm IA, Haas EA, Krous HF, Kinney HC, Markianos K, Beggs AH. Serotonin-related FEV gene variant in the sudden infant death syndrome is a common polymorphism in the African-American population. Pediatr Res. 2009;66:631–635. doi: 10.1203/PDR.0b013e3181bd5a31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbelt KG, Paterson DS, Belliveau RA, Trachtenberg FL, Haas EA, Stanley C, Krous HF, Kinney HC. Decreased GABAA receptor binding in the medullary serotonergic system in the sudden infant death syndrome. J Neuropathol Exp Neurol. 2011;70:799–810. doi: 10.1097/NEN.0b013e31822c09bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbelt KG, Rivera KD, Paterson DS, Duncan JR, Trachtenberg FL, Paulo JA, Stapels MD, Borenstein NS, Belliveau RA, Haas EA, Stanley C, Krous HF, Steen H, Kinney HC. Brainstem deficiency of the 14-3-3 regulator of serotonin synthesis: a proteomics analysis in the sudden infant death syndrome. Mol Cell Proteomics. 2012;11:M111–009530. doi: 10.1074/mcp.M111.009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Chen CL, Luo P, Tan M, Qiu M, Johnson R, Ma Q. Lmx1b, Pet-1, and Nkx2.2 coordinately specify serotonergic neurotransmitter phenotype. J Neurosci. 2003;23:9961–9967. doi: 10.1523/JNEUROSCI.23-31-09961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comet MA, Bernard JF, Hamon M, Laguzzi R, Sevoz-Couche C. Activation of nucleus tractus solitarius 5-HT2A but not other 5-HT2 receptor subtypes inhibits the sympathetic activity in rats. Eur J Neurosci. 2007;26:345–354. doi: 10.1111/j.1460-9568.2007.05673.x. [DOI] [PubMed] [Google Scholar]
- Courts C, Grabmuller M, Madea B. Monoamine Oxidase A Gene Polymorphism and the Pathogenesis of Sudden Infant Death Syndrome. J Pediatr. 2013 doi: 10.1016/j.jpeds.2012.12.072. [DOI] [PubMed] [Google Scholar]
- Deckert J, Catalano M, Syagailo YV, Bosi M, Okladnova O, Di Bella D, Nothen MM, Maffei P, Franke P, Fritze J, Maier W, Propping P, Beckmann H, Bellodi L, Lesch KP. Excess of high activity monoamine oxidase A gene promoter alleles in female patients with panic disorder. Hum Mol Genet. 1999;8:621–624. doi: 10.1093/hmg/8.4.621. [DOI] [PubMed] [Google Scholar]
- Dergacheva O, Griffioen KJ, Wang X, Kamendi H, Gorini C, Mendelowitz D. 5-HT(2) receptor subtypes mediate different long-term changes in GABAergic activity to parasympathetic cardiac vagal neurons in the nucleus ambiguus. Neuroscience. 2007;149:696–705. doi: 10.1016/j.neuroscience.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Paterson DS, Hoffman JM, Mokler DJ, Borenstein NS, Belliveau RA, Krous HF, Haas EA, Stanley C, Nattie EE, Trachtenberg FL, Kinney HC. Brainstem serotonergic deficiency in sudden infant death syndrome. Jama. 2010;303:430–437. doi: 10.1001/jama.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Randall LL, Belliveau RA, Trachtenberg FL, Randall B, Habbe D, Mandell F, Welty TK, Iyasu S, Kinney HC. The effect of maternal smoking and drinking during pregnancy upon (3)H-nicotine receptor brainstem binding in infants dying of the sudden infant death syndrome: initial observations in a high risk population. Brain Pathol. 2008;18:21–31. doi: 10.1111/j.1750-3639.2007.00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer T, Ponsonby AL, Blizzard L, Newman NM, Cochrane JA. The contribution of changes in the prevalence of prone sleeping position to the decline in sudden infant death syndrome in Tasmania [see comments] Jama. 1995;273:783–789. [PubMed] [Google Scholar]
- Edner A, Katz-Salamon M, Lagercrantz H, Ericson M, Milerad J. Heart rate variability in infants with apparent life-threatening events. Acta Paediatr. 2000;89:1326–1329. doi: 10.1080/080352500300002516. [DOI] [PubMed] [Google Scholar]
- Engelberts AC, de Jonge GA. Choice of sleeping position for infants: possible association with cot death. Arch Dis Child. 1990;65:462–467. doi: 10.1136/adc.65.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, Bevins CL, Reinisch W, Teml A, Schwab M, Lichter P, Radlwimmer B, Stange EF. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifer WP, Myers MM. Sudden fetal and infant deaths: shared characteristics and distinctive features. Semin Perinatol. 2002;26:89–96. doi: 10.1053/sper.2002.29854. [DOI] [PubMed] [Google Scholar]
- Filiano JJ, Kinney HC. Arcuate nucleus hypoplasia in the sudden infant death syndrome. Journal of Neuropathology & Experimental Neurology. 1992;51:394–403. doi: 10.1097/00005072-199207000-00002. [DOI] [PubMed] [Google Scholar]
- Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biology of the Neonate. 1994;65:194–197. doi: 10.1159/000244052. [DOI] [PubMed] [Google Scholar]
- Filonzi L, Magnani C, Lavezzi AM, Rindi G, Parmigiani S, Bevilacqua G, Matturri L, Marzano FN. Association of dopamine transporter and monoamine oxidase molecular polymorphisms with sudden infant death syndrome and stillbirth: new insights into the serotonin hypothesis. Neurogenetics. 2009;10:65–72. doi: 10.1007/s10048-008-0149-x. [DOI] [PubMed] [Google Scholar]
- Fiskerstrand CE, Lovejoy EA, Quinn JP. An intronic polymorphic domain often associated with susceptibility to affective disorders has allele dependent differential enhancer activity in embryonic stem cells. FEBS Lett. 1999;458:171–174. doi: 10.1016/s0014-5793(99)01150-3. [DOI] [PubMed] [Google Scholar]
- Fleming PJ, Blair PS, Bacon C, Bensley D, Smith I, Taylor E, Berry J, Golding J, Tripp J. Environment of infants during sleep and risk of the sudden infant death syndrome: results of 1993-5 case-control study for confidential inquiry into stillbirths and deaths in infancy. Confidential Enquiry into Stillbirths and Deaths Regional Coordinators and Researchers. Bmj. 1996;313:191–195. doi: 10.1136/bmj.313.7051.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming PJ, Gilbert R, Azaz Y, Berry PJ, Rudd PT, Stewart A, Hall E. Interaction between bedding and sleeping position in the sudden infant death syndrome: a population based case-control study [see comments] Bmj. 1990;301:85–89. doi: 10.1136/bmj.301.6743.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco P, Szliwowski H, Dramaix M, Kahn A. Polysomnographic study of the autonomic nervous system in potential victims of sudden infant death syndrome. Clin Auton Res. 1998;8:243–249. doi: 10.1007/BF02277969. [DOI] [PubMed] [Google Scholar]
- Franco P, Szliwowski H, Dramaix M, Kahn A. Decreased autonomic responses to obstructive sleep events in future victims of sudden infant death syndrome. Pediatr Res. 1999;46:33–39. doi: 10.1203/00006450-199907000-00006. [DOI] [PubMed] [Google Scholar]
- Froggatt P, Lynas MA, Marshall TK. Sudden death in babies: epidemiology. Am J Cardiol. 1968;22:457–468. doi: 10.1016/0002-9149(68)90152-5. [DOI] [PubMed] [Google Scholar]
- Getahun D, Demissie K, Lu SE, Rhoads GG. Sudden infant death syndrome among twin births: United States, 1995-1998. J Perinatol. 2004;24:544–551. doi: 10.1038/sj.jp.7211140. [DOI] [PubMed] [Google Scholar]
- Gillis RA, Hill KJ, Kirby JS, Quest JA, Hamosh P, Norman WP, Kellar KJ. Effect of activation of central nervous system serotonin 1A receptors on cardiorespiratory function. Journal of Pharmacology & Experimental Therapeutics. 1989;248:851–857. [PubMed] [Google Scholar]
- Gonzalez JM, Portillo MC, Saiz-Jimenez C. Multiple displacement amplification as a pre-polymerase chain reaction (pre-PCR) to process difficult to amplify samples and low copy number sequences from natural environments. Environ Microbiol. 2005;7:1024–1028. doi: 10.1111/j.1462-2920.2005.00779.x. [DOI] [PubMed] [Google Scholar]
- Greenberg BD, Tolliver TJ, Huang SJ, Li Q, Bengel D, Murphy DL. Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet. 1999;88:83–87. [PubMed] [Google Scholar]
- Haas C, Braun J, Bar W, Bartsch C. No association of serotonin transporter gene variation with sudden infant death syndrome (SIDS) in Caucasians. Leg Med (Tokyo) 2009;11(Suppl 1):S210–212. doi: 10.1016/j.legalmed.2009.01.051. [DOI] [PubMed] [Google Scholar]
- Haglund B. Cigarette smoking and sudden infant death syndrome: some salient points in the debate. Acta Paediatr Suppl. 1993;82(Suppl 389):37–39. doi: 10.1111/j.1651-2227.1993.tb12872.x. [DOI] [PubMed] [Google Scholar]
- Harper RM. Autonomic control during sleep and risk for sudden death in infancy. Arch Ital Biol. 2001;139:185–194. [PubMed] [Google Scholar]
- Harper RM, Kinney HC. Potential Mechanisms of Failure in the Sudden Infant Death Syndrome. Curr Pediatr Rev. 2010;6:39–47. doi: 10.2174/157339610791317214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck FR, Herman SM, Donovan M, Iyasu S, Merrick Moore C, Donoghue E, Kirschner RH, Willinger M. Sleep environment and the risk of sudden infant death syndrome in an urban population: the Chicago Infant Mortality Study. Pediatrics. 2003;111:1207–1214. [PubMed] [Google Scholar]
- Hauck FR, Tanabe KO, Moon RY. Racial and ethnic disparities in infant mortality. Semin Perinatol. 2011;35:209–220. doi: 10.1053/j.semperi.2011.02.018. [DOI] [PubMed] [Google Scholar]
- Heils A, Mossner R, Lesch KP. The human serotonin transporter gene polymorphism--basic research and clinical implications. J Neural Transm. 1997;104:1005–1014. doi: 10.1007/BF01273314. [DOI] [PubMed] [Google Scholar]
- Heils A, Teufel A, Petri S, Seemann M, Bengel D, Balling U, Riederer P, Lesch KP. Functional promoter and polyadenylation site mapping of the human serotonin (5-HT) transporter gene. J Neural Transm Gen Sect. 1995;102:247–254. doi: 10.1007/BF01281159. [DOI] [PubMed] [Google Scholar]
- Hendricks T, Francis N, Fyodorov D, Deneris ES. The ETS domain factor Pet-1 is an early and precise marker of central serotonin neurons and interacts with a conserved element in serotonergic genes. J Neurosci. 1999;19:10348–10356. doi: 10.1523/JNEUROSCI.19-23-10348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks TJ, Fyodorov DV, Wegman LJ, Lelutiu NB, Pehek EA, Yamamoto B, Silver J, Weeber EJ, Sweatt JD, Deneris ES. Pet-1 ETS gene plays a critical role in 5-HT neuron development and is required for normal anxiety-like and aggressive behavior. Neuron. 2003;37:233–247. doi: 10.1016/s0896-6273(02)01167-4. [DOI] [PubMed] [Google Scholar]
- Hoffman HJ, Hillman LS. Epidemiology of the sudden infant death syndrome: maternal, neonatal, and postneonatal risk factors. Clin Perinatol. 1992;19:717–737. [PubMed] [Google Scholar]
- Hranilovic D, Stefulj J, Schwab S, Borrmann-Hassenbach M, Albus M, Jernej B, Wildenauer D. Serotonin transporter promoter and intron 2 polymorphisms: relationship between allelic variants and gene expression. Biol Psychiatry. 2004;55:1090–1094. doi: 10.1016/j.biopsych.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Hunt CE. Genes and Sudden Infant Death Syndrome. Pediatr Res. 2004;56:321–322. doi: 10.1203/01.PDR.0000135999.55443.8B. [DOI] [PubMed] [Google Scholar]
- Hunt CE, Brouillette RT. Sudden infant death syndrome: 1987 perspective. J Pediatr. 1987;110:669–678. doi: 10.1016/s0022-3476(87)80001-x. [DOI] [PubMed] [Google Scholar]
- Irgens LM, Skjaerven R, Peterson DR. Prospective assessment of recurrence risk in sudden infant death syndrome siblings. J Pediatr. 1984;104:349–351. doi: 10.1016/s0022-3476(84)81093-8. [DOI] [PubMed] [Google Scholar]
- Irgens LM, Skjaerven R, Peterson DR. Sudden infant death syndrome and recurrence in subsequent siblings. J Pediatr. 1988;112:501–502. doi: 10.1016/s0022-3476(88)80352-4. [DOI] [PubMed] [Google Scholar]
- Iyasu S, Randall LL, Welty TK, Hsia J, Kinney HC, Mandell F, McClain M, Randall B, Habbe D, Wilson H, Willinger M. Risk factors for sudden infant death syndrome among northern plains Indians. Jama. 2002;288:2717–2723. doi: 10.1001/jama.288.21.2717. [DOI] [PubMed] [Google Scholar]
- Kattwinkel J, Hauck FR, Keenan ME, Malloy M, Moon RY. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics. 2005;116:1245–1255. doi: 10.1542/peds.2005-1499. [DOI] [PubMed] [Google Scholar]
- Kelly DH, Golub H, Carley D, Shannon DC. Pneumograms in infants who subsequently died of sudden infant death syndrome. J Pediatr. 1986;109:249–254. doi: 10.1016/s0022-3476(86)80380-8. [DOI] [PubMed] [Google Scholar]
- Kelly DH, Shannon DC. Periodic breathing in infants with near-miss sudden infant death syndrome. Pediatrics. 1979;63:355–360. [PubMed] [Google Scholar]
- Kibel MA, Molteno CD, De Decker R. Cot death controversies. S Afr Med J. 2005;95:853–857. [PubMed] [Google Scholar]
- King KA, McCall RB. The effects of 8-OH-DPAT on medullary 5-HT neurons and sympathetic activity in baroreceptor-denervated animals. European Journal of Pharmacology. 1991;200:357–360. doi: 10.1016/0014-2999(91)90596-i. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Broadbelt KG, Haynes RL, Rognum IJ, Paterson DS. The serotonergic anatomy of the developing human medulla oblongata: implications for pediatric disorders of homeostasis. J Chem Neuroanat. 2011;41:182–199. doi: 10.1016/j.jchemneu.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Brody BA, Finkelstein DM, Vawter GF, Mandell F, Gilles FH. Delayed central nervous system myelination in the sudden infant death syndrome. Journal of Neuropathology & Experimental Neurology. 1991;50:29–48. doi: 10.1097/00005072-199101000-00003. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Myers MM, Belliveau RA, Randall LL, Trachtenberg FL, Fingers ST, Youngman M, Habbe D, Fifer WP. Subtle autonomic and respiratory dysfunction in sudden infant death syndrome associated with serotonergic brainstem abnormalities: a case report. J Neuropathol Exp Neurol. 2005;64:689–694. doi: 10.1097/01.jnen.0000174334.27708.43. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Randall LL, Sleeper LA, Willinger M, Belliveau RA, Zec N, Rava LA, Dominici L, Iyasu S, Randall B, Habbe D, Wilson H, Mandell F, McClain M, Welty TK. Serotonergic Brainstem Abnormalities in Northern Plains Indians with the Sudden Infant Death Syndrome. Journal of Neuropathology and Experimental Neurology. 2003;62:1178–1191. doi: 10.1093/jnen/62.11.1178. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Thach BT. The sudden infant death syndrome. N Engl J Med. 2009;361:795–805. doi: 10.1056/NEJMra0803836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsantas P, Gaffney KF. Racial/ethnic disparities in infant mortality. J Perinat Med. 38:87–94. doi: 10.1515/jpm.2010.014. [DOI] [PubMed] [Google Scholar]
- Klintschar M, Heimbold C. Association between a functional polymorphism in the MAOA gene and sudden infant death syndrome. Pediatrics. 2012;129:e756–761. doi: 10.1542/peds.2011-1642. [DOI] [PubMed] [Google Scholar]
- Klug MG, Burd L, Kerbeshian J, Benz B, Martsolf JT. A comparison of the effects of parental risk markers on pre- and perinatal variables in multiple patient cohorts with fetal alcohol syndrome, autism, Tourette syndrome, and sudden infant death syndrome: an enviromic analysis. Neurotoxicol Teratol. 2003;25:707–717. doi: 10.1016/j.ntt.2003.07.018. [DOI] [PubMed] [Google Scholar]
- Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, Cutz E, Hanzlick R, Keens TG, Mitchell EA. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114:234–238. doi: 10.1542/peds.114.1.234. [DOI] [PubMed] [Google Scholar]
- Lalley PM. The excitability and rhythm of medullary respiratory neurons in the cat are altered by the serotonin receptor agonist 5-methoxy-N,N, dimethyltryptamine. Brain Res. 1994;648:87–98. doi: 10.1016/0006-8993(94)91909-7. [DOI] [PubMed] [Google Scholar]
- Lalley PM, Bischoff AM, Richter DW. 5-HT-1A receptor-mediated modulation of medullary expiratory neurones in the cat. J Physiol. 1994;476:117–130. [PMC free article] [PubMed] [Google Scholar]
- Ledwidge M, Fox G, Matthews T. Neurocardiogenic syncope: a model for SIDS. Arch Dis Child. 1998;78:481–483. doi: 10.1136/adc.78.5.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52:103–121. doi: 10.1016/j.neuron.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Muller CR, Hamer DH, Murphy DL. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- MacDorman MF, Cnattingius S, Hoffman HJ, Kramer MS, Haglund B. Sudden infant death syndrome and smoking in the United States and Sweden. Am J Epidemiol. 1997;146:249–257. doi: 10.1093/oxfordjournals.aje.a009260. [DOI] [PubMed] [Google Scholar]
- Machaalani R, Say M, Waters KA. Serotoninergic receptor 1A in the sudden infant death syndrome brainstem medulla and associations with clinical risk factors. Acta Neuropathol. 2009;117:257–265. doi: 10.1007/s00401-008-0468-x. [DOI] [PubMed] [Google Scholar]
- MacKenzie A, Quinn J. A serotonin transporter gene intron 2 polymorphic region, correlated with affective disorders, has allele-dependent differential enhancer-like properties in the mouse embryo. Proc Natl Acad Sci U S A. 1999;96:15251–15255. doi: 10.1073/pnas.96.26.15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher BS, Marazita ML, Rand C, Zhou L, Berry-Kravis EM, Weese-Mayer DE. 3′ UTR polymorphism of the serotonin transporter gene and sudden infant death syndrome: haplotype analysis. Am J Med Genet A. 2006;140:1453–1457. doi: 10.1002/ajmg.a.31261. [DOI] [PubMed] [Google Scholar]
- Maurer P, Rorive S, de Kerchove d’Exaerde A, Schiffmann SN, Salmon I, de Launoit Y. The Ets transcription factor Fev is specifically expressed in the human central serotonergic neurons. Neurosci Lett. 2004;357:215–218. doi: 10.1016/j.neulet.2003.12.086. [DOI] [PubMed] [Google Scholar]
- McKinney J, Johansson S, Halmoy A, Dramsdahl M, Winge I, Knappskog PM, Haavik J. A loss-of-function mutation in tryptophan hydroxylase 2 segregating with attention-deficit/hyperactivity disorder. Mol Psychiatry. 2008;13:365–367. doi: 10.1038/sj.mp.4002152. [DOI] [PubMed] [Google Scholar]
- Millar WJ, Hill GB. Prevalence of and risk factors for sudden infant death syndrome in Canada. Cmaj. 1993;149:629–635. [PMC free article] [PubMed] [Google Scholar]
- Mitchell EA. Sudden infant death syndrome: should bed sharing be discouraged? Arch Pediatr Adolesc Med. 2007;161:305–306. doi: 10.1001/archpedi.161.3.305. [DOI] [PubMed] [Google Scholar]
- Mitchell EA, Milerad J. Smoking and the sudden infant death syndrome. Rev Environ Health. 2006;21:81–103. doi: 10.1515/reveh.2006.21.2.81. [DOI] [PubMed] [Google Scholar]
- Mitchell EA, Scragg R. Observations on ethnic differences in SIDS mortality in New Zealand. Early Hum Dev. 1994;38:151–157. doi: 10.1016/0378-3782(94)90206-2. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr. Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Moon RY, Horne RS, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370:1578–1587. doi: 10.1016/S0140-6736(07)61662-6. [DOI] [PubMed] [Google Scholar]
- Morley ME, Rand CM, Berry-Kravis EM, Zhou L, Fan W, Weese-Mayer DE. Genetic variation in the HTR1A gene and sudden infant death syndrome. Am J Med Genet A. 2008;146:930–933. doi: 10.1002/ajmg.a.32112. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, Greenblatt R, Gleason D, Ertelt JA, Apse KA, Bodell A, Partlow JN, Barry B, Yao H, Markianos K, Ferland RJ, Greenberg ME, Walsh CA. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, Hall ST, Weir DM, Busuttil A, Blackwell CC. Interleukin-10 and sudden infant death syndrome. FEMS Immunol Med Microbiol. 2004;42:130–138. doi: 10.1016/j.femsim.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, Hall ST, Weir DM, Busuttil A, Blackwell CC. IL6 G-174C associated with sudden infant death syndrome in a Caucasian Australian cohort. Hum Immunol. 2006;67:819–825. doi: 10.1016/j.humimm.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Narita N, Narita M, Takashima S, Nakayama M, Nagai T, Okado N. Serotonin transporter gene variation is a risk factor for sudden infant death syndrome in the Japanese population. Pediatrics. 2001;107:690–692. doi: 10.1542/peds.107.4.690. [DOI] [PubMed] [Google Scholar]
- Nonnis Marzano F, Maldini M, Filonzi L, Lavezzi AM, Parmigiani S, Magnani C, Bevilacqua G, Matturri L. Genes regulating the serotonin metabolic pathway in the brain stem and their role in the etiopathogenesis of the sudden infant death syndrome. Genomics. 2008;91:485–491. doi: 10.1016/j.ygeno.2008.01.010. [DOI] [PubMed] [Google Scholar]
- O’Kusky JR, Kozuki DE, Norman MG. Sudden infant death syndrome: postnatal changes in the volumes of the pons, medulla and cervical spinal cord. Journal of Neuropathology & Experimental Neurology. 1995;54:570–580. [PubMed] [Google Scholar]
- O’Kusky JR, Norman MG. Sudden infant death syndrome: increased synaptic density in the central reticular nucleus of the medulla. Journal of Neuropathology & Experimental Neurology. 1994;53:263–271. [PubMed] [Google Scholar]
- O’Leary CM, Jacoby PJ, Bartu A, D’Antoine H, Bower C. Maternal Alcohol Use and Sudden Infant Death Syndrome and Infant Mortality Excluding SIDS. Pediatrics. 2013;131:e770–778. doi: 10.1542/peds.2012-1907. [DOI] [PubMed] [Google Scholar]
- O’Mara L. Review: bed sharing between parents and infants exposed to smoke may increase the risk of sudden infant death syndrome. Evid Based Nurs. 2007;10:119. doi: 10.1136/ebn.10.4.119. [DOI] [PubMed] [Google Scholar]
- Ogilvie AD, Battersby S, Bubb VJ, Fink G, Harmar AJ, Goodwim GM, Smith CA. Polymorphism in serotonin transporter gene associated with susceptibility to major depression. Lancet. 1996;347:731–733. doi: 10.1016/s0140-6736(96)90079-3. [DOI] [PubMed] [Google Scholar]
- Opdal SH, Rognum TO. The sudden infant death syndrome gene: does it exist? Pediatrics. 2004;114:e506–512. doi: 10.1542/peds.2004-0683. [DOI] [PubMed] [Google Scholar]
- Oyen N, Skjaerven R, Irgens LM. Population-based recurrence risk of sudden infant death syndrome compared with other infant and fetal deaths. Am J Epidemiol. 1996;144:300–305. doi: 10.1093/oxfordjournals.aje.a008925. [DOI] [PubMed] [Google Scholar]
- Ozawa Y, Okado N. Alteration of serotonergic receptors in the brain stems of human patients with respiratory disorders. Neuropediatrics. 2002a;33:142–149. doi: 10.1055/s-2002-33678. [DOI] [PubMed] [Google Scholar]
- Ozawa Y, Takashima S. Developmental neurotransmitter pathology in the brainstem of sudden infant death syndrome: a review and sleep position. Forensic Sci Int. 2002b;130(Suppl):S53–59. doi: 10.1016/s0379-0738(02)00139-1. [DOI] [PubMed] [Google Scholar]
- Panigrahy A, Filiano J, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, Foley E, White WF, Kinney HC. Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol. 2000;59:377–384. doi: 10.1093/jnen/59.5.377. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Belliveau RA, Trachtenberg F, Kinney HC. Differential development of 5-HT receptor and the serotonin transporter binding in the human infant medulla. J Comp Neurol. 2004;472:221–231. doi: 10.1002/cne.20105. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Darnall R. 5-HT2A receptors are concentrated in regions of the human infant medulla involved in respiratory and autonomic control. Auton Neurosci. 2009;147:48–55. doi: 10.1016/j.autneu.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson DS, Rivera KD, Broadbelt KG, Trachtenberg FL, Belliveau RA, Holm IA, Haas EA, Stanley C, Krous HF, Kinney HC, Markianos K. Lack of association of the serotonin transporter polymorphism with the sudden infant death syndrome in the San Diego Dataset. Pediatr Res. 2010;68:409–413. doi: 10.1203/PDR.0b013e3181f2edf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson DS, Thompson EG, Kinney HC. Serotonergic and glutamatergic neurons at the ventral medullary surface of the human infant: Observations relevant to central chemosensitivity in early human life. Auton Neurosci. 2006a;124:112–124. doi: 10.1016/j.autneu.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, Chadwick AE, Krous HF, Kinney HC. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. Jama. 2006b;296:2124–2132. doi: 10.1001/jama.296.17.2124. [DOI] [PubMed] [Google Scholar]
- Pelayo R, Owens J, Mindell J, Sheldon S. Bed sharing with unimpaired parents is not an important risk for sudden infant death syndrome: to the editor. Pediatrics. 2006;117:993–994. doi: 10.1542/peds.2005-2748. author reply 994-996. [DOI] [PubMed] [Google Scholar]
- Pena F, Ramirez JM. Endogenous activation of serotonin-2A receptors is required for respiratory rhythm generation in vitro. J Neurosci. 2002;22:11055–11064. doi: 10.1523/JNEUROSCI.22-24-11055.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DR, Sabotta EE, Daling JR. Infant mortality among subsequent siblings of infants who died of sudden infant death syndrome. J Pediatr. 1986;108:911–914. doi: 10.1016/s0022-3476(86)80926-x. [DOI] [PubMed] [Google Scholar]
- Pfaar H, von Holst A, Vogt Weisenhorn DM, Brodski C, Guimera J, Wurst W. mPet-1, a mouse ETS-domain transcription factor, is expressed in central serotonergic neurons. Dev Genes Evol. 2002;212:43–46. doi: 10.1007/s00427-001-0208-x. [DOI] [PubMed] [Google Scholar]
- Pharoah PO, Platt MJ. Sudden infant death syndrome in twins and singletons. Twin Res Hum Genet. 2007;10:644–648. doi: 10.1375/twin.10.4.644. [DOI] [PubMed] [Google Scholar]
- Pincus SM, Cummins TR, Haddad GG. Heart rate control in normal and aborted-SIDS infants. Am J Physiol. 1993;264:R638–646. doi: 10.1152/ajpregu.1993.264.3.R638. [DOI] [PubMed] [Google Scholar]
- Ponsonby AL, Dwyer T, Gibbons LE, Cochrane JA, Wang YG. Factors potentiating the risk of sudden infant death syndrome associated with the prone position [see comments] New England Journal of Medicine. 1993;329:377–382. doi: 10.1056/NEJM199308053290601. [DOI] [PubMed] [Google Scholar]
- Prandota J. Possible pathomechanisms of sudden infant death syndrome: key role of chronic hypoxia, infection/inflammation states, cytokine irregularities, and metabolic trauma in genetically predisposed infants. Am J Ther. 2004;11:517–546. doi: 10.1097/01.mjt.0000140648.30948.bd. [DOI] [PubMed] [Google Scholar]
- Ramanathan R, Corwin MJ, Hunt CE, Lister G, Tinsley LR, Baird T, Silvestri JM, Crowell DH, Hufford D, Martin RJ, Neuman MR, Weese-Mayer DE, Cupples LA, Peucker M, Willinger M, Keens TG. Cardiorespiratory events recorded on home monitors: Comparison of healthy infants with those at increased risk for SIDS. Jama. 2001;285:2199–2207. doi: 10.1001/jama.285.17.2199. [DOI] [PubMed] [Google Scholar]
- Ramirez JM, Tryba AK, Pena F. Pacemaker neurons and neuronal networks: an integrative view. Curr Opin Neurobiol. 2004;14:665–674. doi: 10.1016/j.conb.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Rand CM, Berry-Kravis EM, Fan W, Weese-Mayer DE. HTR2A variation and sudden infant death syndrome: a case-control analysis. Acta Paediatr. 2009;98:58–61. doi: 10.1111/j.1651-2227.2008.01018.x. [DOI] [PubMed] [Google Scholar]
- Rand CM, Berry-Kravis EM, Zhou L, Fan W, Weese-Mayer DE. Sudden infant death syndrome: rare mutation in the serotonin system FEV gene. Pediatr Res. 2007;62:180–182. doi: 10.1203/PDR.0b013e3180a725a0. [DOI] [PubMed] [Google Scholar]
- Rantonen T, Jalonen J, Gronlund J, Antila K, Southall D, Valimaki I. Increased amplitude modulation of continuous respiration precedes sudden infant death syndrome - detection by spectral estimation of respirogram. Early Hum Dev. 1998;53:53–63. doi: 10.1016/s0378-3782(98)00039-5. [DOI] [PubMed] [Google Scholar]
- Raul L. Serotonin2 receptors in the nucleus tractus solitarius: characterization and role in the baroreceptor reflex arc. Cell Mol Neurobiol. 2003;23:709–726. doi: 10.1023/A:1025096718559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet. 1998;103:273–279. doi: 10.1007/s004390050816. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Lee C, Birney E, Altshuler DM, Eichler EE, Carter NP, Hurles ME, Feuk L. Challenges and standards in integrating surveys of structural variation. Nat Genet. 2007;39:S7–15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoendorf KC, Kiely JL. Relationship of sudden infant death syndrome to maternal smoking during and after pregnancy. Pediatrics. 1992;90:905–908. [PubMed] [Google Scholar]
- Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, Grancini F, Marni ED, Perticone F, Rosti D, Salice P. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1709–1714. doi: 10.1056/NEJM199806113382401. [DOI] [PubMed] [Google Scholar]
- Scott MM, Krueger KC, Deneris ES. A differentially autoregulated Pet-1 enhancer region is a critical target of the transcriptional cascade that governs serotonin neuron development. J Neurosci. 2005;25:2628–2636. doi: 10.1523/JNEUROSCI.4979-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scragg R, Mitchell EA, Taylor BJ, Stewart AW, Ford RP, Thompson JM, Allen EM, Becroft DM, New Zealand Cot Death Study Group Bed sharing, smoking, and alcohol in the sudden infant death syndrome. Bmj. 1993;307:1312–1318. doi: 10.1136/bmj.307.6915.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seneviratne C, Huang W, Ait-Daoud N, Li MD, Johnson BA. Characterization of a functional polymorphism in the 3′ UTR of SLC6A4 and its association with drinking intensity. Alcohol Clin Exp Res. 2009;33:332–339. doi: 10.1111/j.1530-0277.2008.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon DC, Kelly DH. SIDS and near-sids (first of two parts) N Engl J Med. 1982a;306:959–965. doi: 10.1056/NEJM198204223061604. [DOI] [PubMed] [Google Scholar]
- Shannon DC, Kelly DH. SIDS and near-SIDS (second of two parts) N Engl J Med. 1982b;306:1022–1028. doi: 10.1056/NEJM198204293061704. [DOI] [PubMed] [Google Scholar]
- Shen FM, Wang J, Ni CR, Yu JG, Wang WZ, Su DF. Ketanserin-induced baroreflex enhancement in spontaneously hypertensive rats depends on central 5-HT(2A) receptors. Clin Exp Pharmacol Physiol. 2007;34:702–707. doi: 10.1111/j.1440-1681.2007.04626.x. [DOI] [PubMed] [Google Scholar]
- Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annu Rev Neurosci. 1999a;22:197–217. doi: 10.1146/annurev.neuro.22.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JC, Chen K, Ridd MJ. Role of MAO A and B in neurotransmitter metabolism and behavior. Pol J Pharmacol. 1999b;51:25–29. [PubMed] [Google Scholar]
- Stewart AM. Cot deaths. Nurs Mirror Midwives J. 1975;141:57–58. [PubMed] [Google Scholar]
- Takashima S, Mito T, Becker LE. Dendritic development of motor neurons in the cervical anterior horn and hypoglossal nucleus of normal infants and victims of sudden infant death syndrome. Neuropediatrics. 1990;21:24–26. doi: 10.1055/s-2008-1071452. [DOI] [PubMed] [Google Scholar]
- Tappin D, Brooke H, Ecob R. Bedsharing and sudden infant death syndrome (SIDS) in Scotland, UK. Lancet. 2004;363:994. doi: 10.1016/S0140-6736(04)15806-6. [DOI] [PubMed] [Google Scholar]
- Tappin D, Ecob R, Brooke H. Bedsharing, roomsharing, and sudden infant death syndrome in Scotland: a case-control study. J Pediatr. 2005;147:32–37. doi: 10.1016/j.jpeds.2005.01.035. [DOI] [PubMed] [Google Scholar]
- Toruner GA, Kurvathi R, Sugalski R, Shulman L, Twersky S, Pearson PG, Tozzi R, Schwalb MN, Wallerstein R. Copy number variations in three children with sudden infant death. Clin Genet. 2009;76:63–68. doi: 10.1111/j.1399-0004.2009.01161.x. [DOI] [PubMed] [Google Scholar]
- Trachtenberg FL, Haas EA, Kinney HC, Stanley C, Krous HF. Risk factor changes for sudden infant death syndrome after initiation of Back-to-Sleep campaign. Pediatrics. 2012;129:630–638. doi: 10.1542/peds.2011-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryba AK, Pena F, Ramirez JM. Gasping activity in vitro: a rhythm dependent on 5-HT2A receptors. J Neurosci. 2006;26:2623–2634. doi: 10.1523/JNEUROSCI.4186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vege A, Rognum TO, Scott H, Aasen AO, Saugstad OD. SIDS cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr. 1995;84:193–196. doi: 10.1111/j.1651-2227.1995.tb13608.x. [DOI] [PubMed] [Google Scholar]
- Vennemann MM, Bajanowski T, Brinkmann B, Jorch G, Sauerland C, Mitchell EA. Sleep environment risk factors for sudden infant death syndrome: the German sudden infant death syndrome study. Pediatrics. 2009;123:1162–1170. doi: 10.1542/peds.2008-0505. [DOI] [PubMed] [Google Scholar]
- Villalon CM, Centurion D. Cardiovascular responses produced by 5-hydroxytriptamine:a pharmacological update on the receptors/mechanisms involved and therapeutic implications. Naunyn Schmiedebergs Arch Pharmacol. 2007;376:45–63. doi: 10.1007/s00210-007-0179-1. [DOI] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Walther DJ, Peter JU, Bashammakh S, Hortnagl H, Voits M, Fink H, Bader M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299:76. doi: 10.1126/science.1078197. [DOI] [PubMed] [Google Scholar]
- Waters K. Serotonin in the sudden infant death syndrome. Drug News Perspect. 2010;23:537–548. doi: 10.1358/dnp.2010.23.9.1453626. [DOI] [PubMed] [Google Scholar]
- Waters KA, Meehan B, Huang JQ, Gravel RA, Michaud J, Cote A. Neuronal apoptosis in sudden infant death syndrome. Pediatr Res. 1999;45:166–172. doi: 10.1203/00006450-199902000-00002. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Maher BS, Silvestri JM, Curran ME, Marazita ML. Sudden infant death syndrome: association with a promoter polymorphism of the serotonin transporter gene. Am J Med Genet. 2003a;117A:268–274. doi: 10.1002/ajmg.a.20005. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Curran ME, Silvestri JM, Marazita ML. Sudden infant death syndrome: case-control frequency differences at genes pertinent to early autonomic nervous system embryologic development. Pediatr Res. 2004;56:391–395. doi: 10.1203/01.PDR.0000136285.91048.4A. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Zhou L, Berry-Kravis EM, Maher BS, Silvestri JM, Marazita ML. Association of the serotonin transporter gene with sudden infant death syndrome: a haplotype analysis. Am J Med Genet. 2003b;122A:238–245. doi: 10.1002/ajmg.a.20427. [DOI] [PubMed] [Google Scholar]
- Willinger M, Hoffman HJ, Hartford RB. Pediatrics. Vol. 93. National Institutes of Health; Bethesda, MD: 1994. Infant sleep position and risk for sudden infant death syndrome: report of meeting held January 13 and 14, 1994; pp. 814–819. [PubMed] [Google Scholar]
- Wisborg K, Kesmodel U, Henriksen TB, Olsen SF, Secher NJ. A prospective study of smoking during pregnancy and SIDS. Arch Dis Child. 2000;83:203–206. doi: 10.1136/adc.83.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science. 2004;305:217. doi: 10.1126/science.1097540. [DOI] [PubMed] [Google Scholar]