

Abstract
Infantile neuronal ceroid lipofuscinosis (INCL) is a devastating childhood neurodegenerative lysosomal storage disease (LSD) that has no effective treatment. It is caused by inactivating mutations in the palmitoyl-protein thioesterase-1 (PPT1) gene. PPT1-deficiency impairs the cleavage of thioester linkage in palmitoylated proteins (constituents of ceroid), preventing degradation by lysosomal hydrolases. Consequently, accumulation of lysosomal ceroid leads to INCL. Thioester linkage is cleaved by nucleophilic attack. Hydroxylamine, a potent nucleophilic cellular metabolite, may have therapeutic potential for INCL but its toxicity precludes clinical application. Here we report that a hydroxylamine-derivative, N-(tert-Butyl) hydroxylamine (NtBuHA), is non-toxic, cleaves thioester linkage in palmitoylated proteins and mediates lysosomal ceroid depletion in cultured cells from INCL patients. Importantly, in Ppt1−/− mice, which mimic INCL, NtBuHA crossed the blood-brain-barrier, depleted lysosomal ceroid, suppressed neuronal apoptosis, slowed neurological deterioration and extended lifespan. Our findings provide the proof of concept that thioesterase-mimetic and antioxidant small molecules like NtBuHA are potential drug-targets for thioesterase deficiency diseases like INCL.
INTRODUCTION
Although individually lysosomal storage diseases (LSDs) are rare, cumulatively they affect 1 in 5–7,500 live births1, 2. Neurodegeneration is a devastating manifestation in the majority of the more than 50 LSDs. Neuronal ceroid-lipofuscinoses (NCLs), commonly known as Batten disease3, constitute a group of the most common (1 in 12,500 births), genetically distinct, neurodegenerative storage disorders, characterized by progressive psychomotor deterioration with onset around early childhood to adolescence. Mutations in eight different genes underlie various types of NCLs and several more NCL genes have been predicted to exist4. Autofluorescent cytoplasmic inclusion granules in neurons and in other cell types are associated with selective destruction of neurons in the brain and in the retina5. The NCLs as a group are broadly divided into two categories: one in which the gene mutations underlie a defect in a transmembrane protein, and the other in which activities of soluble lysosomal enzymes are impaired6. Interestingly, the deficiency of two soluble lysosomal enzymes, cathepsin D and palmitoyl-protein thioesterase-1 (PPT1) underlies two of the most lethal NCLs: congenital NCL and infantile NCL (INCL), respectively.
More than a decade ago, the purification and characterization of PPT1 from bovine brain significantly advanced our understanding of protein de-acylation7. PPT1 catalyzes the hydrolysis of the thioester linkage that attaches a 16-carbon fatty acid (predominantly palmitate) to cysteine residues in polypeptides. Subsequent molecular cloning and expression of bovine and rat PPT1 (ref. 8) facilitated further characterization and the demonstration that PPT1 is a lysosomal enzyme9, 10. Previous linkage analysis already localized the candidate locus for INCL on chromosome 1p32 (ref. 11), a region of chromosome 1 in which the PPT1 gene co-localized. Finally, the identification of inactivating mutations in the PPT1 gene (ref. 12) in INCL patients clearly established the molecular basis of this autosomal recessive neurodegenerative LSD. Children afflicted with INCL are normal at birth, but by 11–18 months of age they develop psychomotor retardation and by two years of age they are completely blind due to retinal degeneration. By age four, these children manifest virtually no brain activity and they remain in a vegetative state for several more years before eventual death13. These grim facts underscore an urgent need for the development of rational and effective therapeutic strategies for this devastating childhood neurodegenerative LSD.
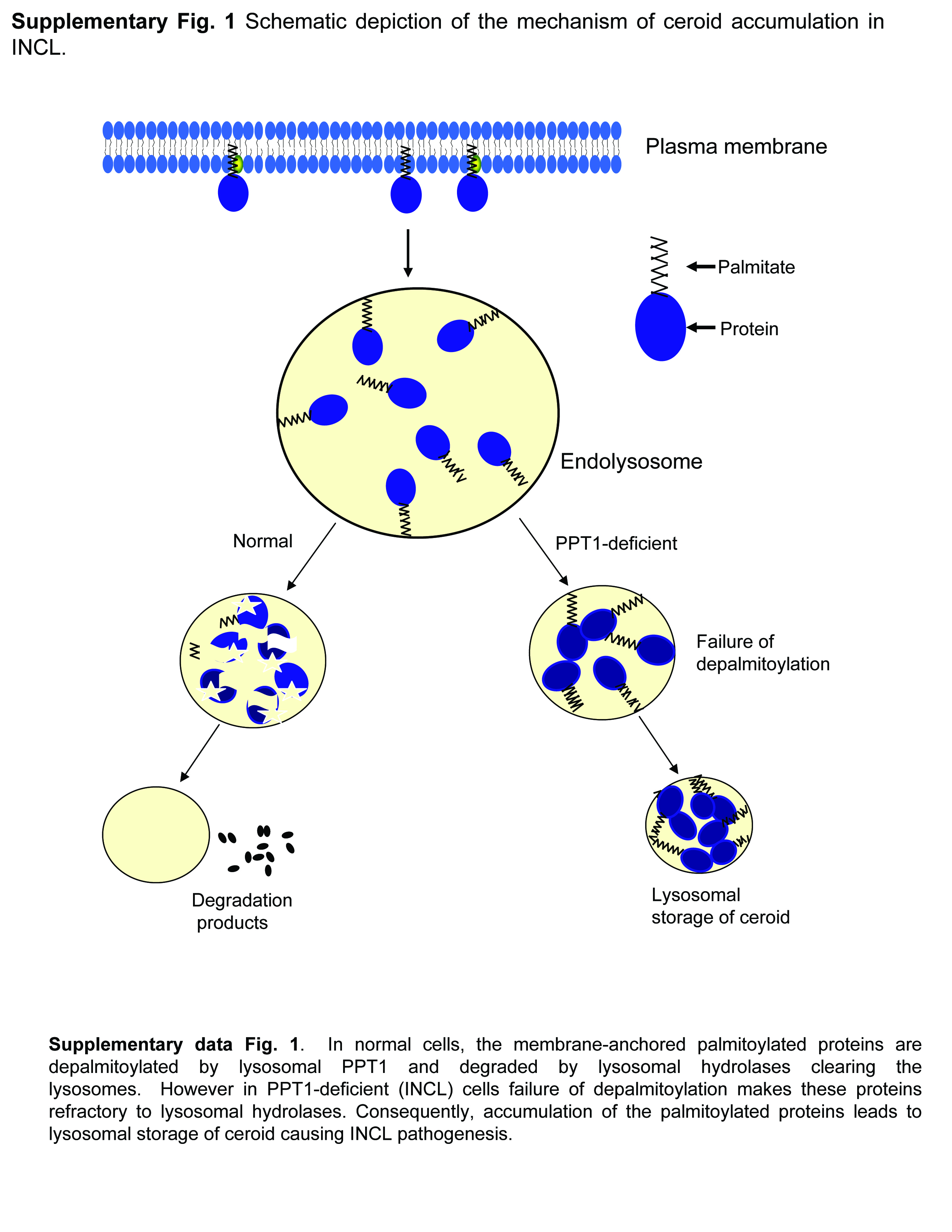
Palmitoylation (S-acylation) has emerged as an important and the only reversible posttranslational lipid-modification of proteins14, 15. Thus, dynamic palmitoylation (palmitoylation-depalmitoylation) provides an important regulatory mechanism for the function of many proteins16, 17. While palmitoylation plays many critical roles in protein function, depalmitoylation is equally important for these proteins to recycle or undergo degradation by lysosomal hydrolases. PPT1-deficiency impairs degradation of palmitoylated proteins (constituents of ceroid) by lysosomal hydrolases and consequently, abnormal ceroid accumulation in lysosomes is suggested to cause INCL pathogenesis18 (Supplementary Fig. 1). Because the thioester linkage is labile and nucleophilic attack cleaves this linkage19, we hypothesized that nucleophilic small molecules may have therapeutic potential for INCL. Hydroxylamine20, a potent nucleophilic metabolite present in all plants and animals21, cleaves thioester linkage with high potency and specificity14, 15. Thus, functionally hydroxylamine mimics all thioesterases, including PPT1. However, hydroxylamine at a high concentration stimulates the production of methemoglobin, which unlike hemoglobin, does not transport oxygen to the tissues. This toxicity precludes its clinical application.
We rationalized that derivatives of hydroxylamine may be non-toxic while retaining its ability to cleave thioester linkage. In this study, we first screened a panel of 12 hydroxylamine-derivatives and identified N-(tert-Butyl) hydroxylamine (NtBuHA), which is non-toxic, cleaved thioester linkage in palmitoylated proteins, mediated depletion of lysosomal ceroid deposits and suppressed apoptosis in both cultured cells from INCL patients and in the brain of Ppt1−/− mice, which recapitulate virtually all clinical and pathological features of INCL. Moreover, we found that NtBuHA is neuroprotective and modestly, albeit significantly extends lifespan in these mice without elevating methemoglobin levels. Our findings provide the proof of principle that non-toxic nucleophilic small molecules with antioxidant property such as NtBuHA are potential drug-targets for diseases like INCL caused by thioesterase-deficiency.
RESULTS
Screening of hydroxylamine derivatives that mimic thioesterases
Thioester linkage in palmitoyl~CoA as well as in palmitoylated proteins (constituents of ceroid)18 is cleaved by hydroxylamine with high specificity14,15,22. To determine if hydroxylamine-derivatives retain this property, we first screened a panel of 12 hydroxylamine-derivatives (Suplementary Table 1) for their ability to cleave thioester linkage in [14C]palmitoyl~CoA. We used [14C]palmitoyl~CoA because it is a model substrate of thioesterases including PPT1 and mediates the release of free [14C]palmitic acid (Fig. 1a). As a positive control, we used the parent compound, hydroxylamine. Our results showed that like hydroxylamine, both water-soluble- (Fig. 1b, upper panel) and DMSO-soluble (Fig. 1c, upper panel) hydroxylamine-derivatives cleaved thioester linkage in [14C]-palmitoyl~CoA releasing free [14C]-palmitate, albeit at varying degrees of efficiency. The densitometric quantitation of the released free [14C]palmitate bands in TLC (Fig. 1b, c, lower panels) confirmed these results.
Figure 1.

Hydroxylamine derivatives cleave thioester linkage in [14C]palmitoyl~CoA.
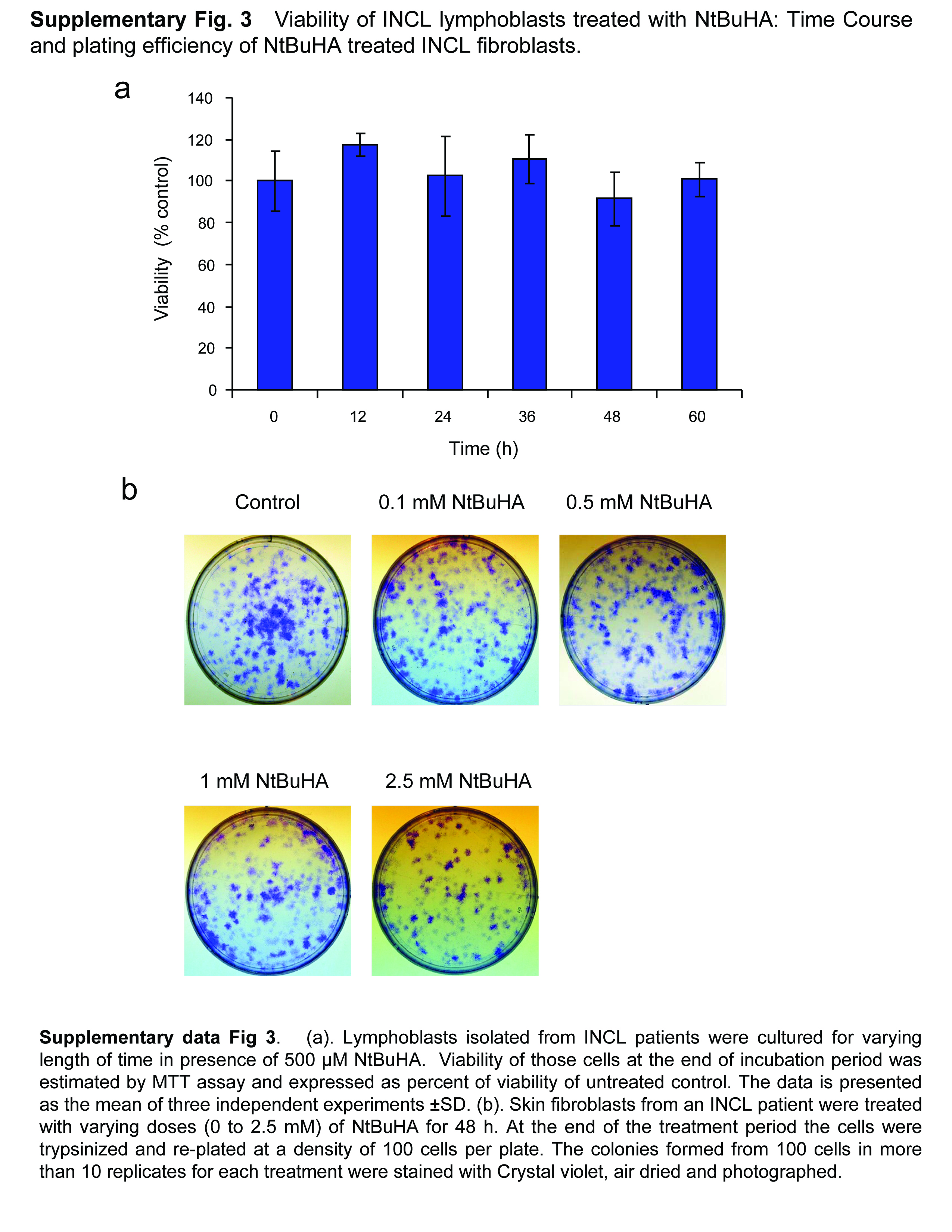
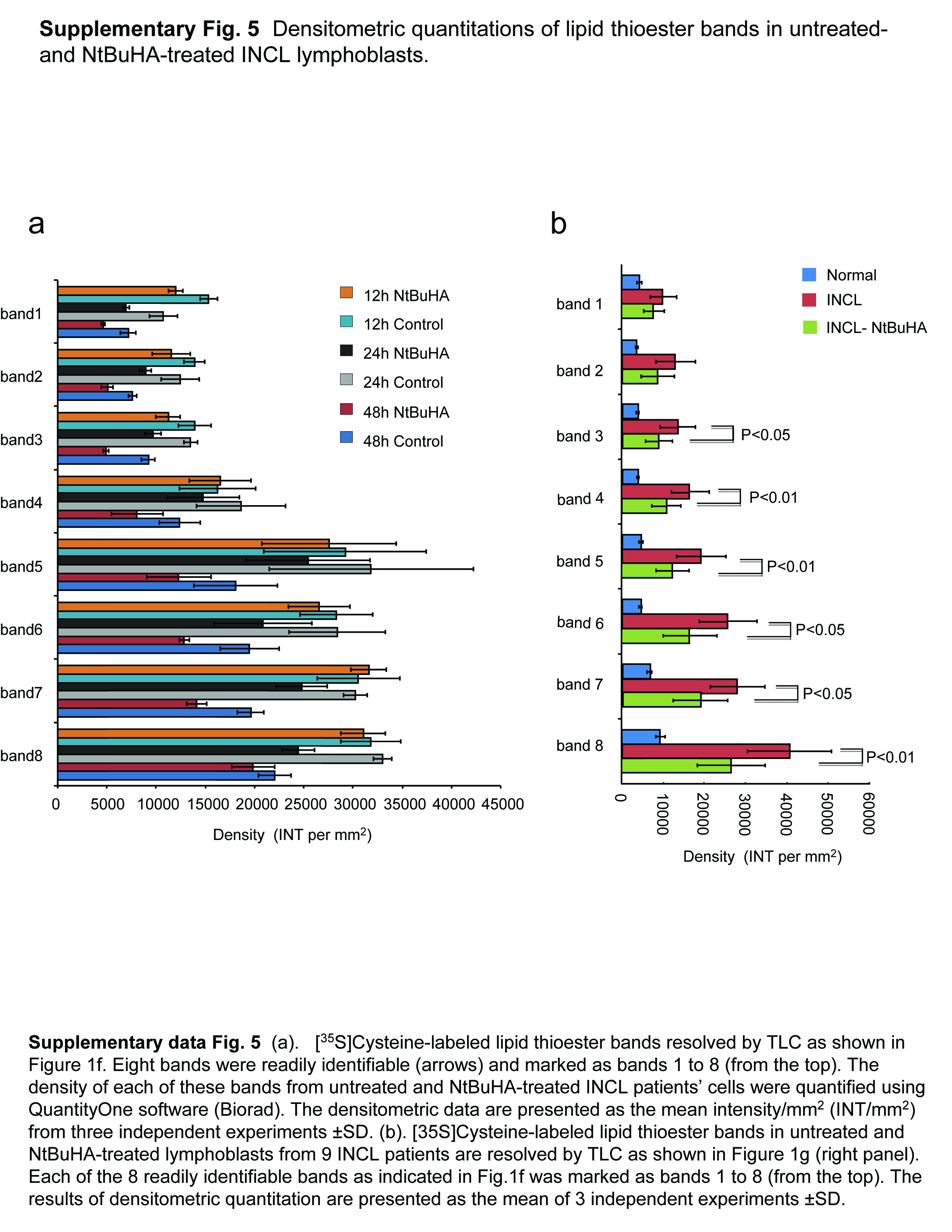
(Panel a) Method of screening of hydroxylamine-derivatives for the cleavage of thioester linkage in [14C]palmitoyl~CoA. Palmitoyl~CoA is a model substrate of PPT1 in which [14C]palmitic acid (asterisk) is attached to CoA via thioester linkage (arrow). PPT1 or nucleophilic compound cleaving the thioester linkage would release radioactive palmitate from [14C]-palmitoyl~CoA. (Panel b) Water-soluble and (Panel c) DMSO-soluble hydroxylamine-derivatives were screened by incubating [14C]palmitoyl~CoA with each of the 12 hydroxylamine-derivatives (Supplementary Table 1) for 1 h at room temperature. Varying degrees of [14C]palmitate were released by both water-soluble (Panel b, lanes 3–9) and DMSO-soluble (Panel c, lanes 3–7) hydroxylamine-derivatives resolved by TLC. (Panel b: lanes: (1): [14C] palmitic acid standard; (2) [14C]-palmitoyl~CoA standard; (3) N-(tert-Butyl)hydroxylamine; (4): N-Benzylhydroxylamine; (5): N-Methylhydroxylamine; (6): N-Cyclohexylhydroxylamine; (7): N,N-Dimethylhydroxylamine; (8): N,N-Diethylhydroxylamine (9): N,O-Bis(trimethylsilyl) hydroxylamine; (10): hydroxylamine (control); (11): recombinant human PPT1 (control); (12): [14C]palmitate standard. (Panel c), lanes: (1): [14C] palmitic acid standard; (2) [14C]-palmitoyl~CoA standard; (3) N-Benzyloxycarbonyl hydroxylamine; (4), N,N-Dibenzylhydroxylamine; (5): N-Benzoyl-N-phenyl hydroxylamine; (6): N-tert-Butyl-O-[1-[4-(chloromethyl) phenyl]ethyl]- N-(2-methyl-1-phenylpropyl) hydroxylamine; (7): N,O-Di-Boc-hydroxylamine; (8): hydroxylamine (control); (9): recombinant human PPT1 (control); (10): [14C]palmitate standard. The arrow indicates the released free [14C] palmitate band. The densitometric quantitation of released free [14C]palmitate bands is represented graphically. (Panel d) Effect of varying concentrations of NtBuHA treatment for 48 h on the viability of cultured lymphoblasts from an INCL patient was evaluated by MTT assay23. (Panel e). Plating efficiency of the NtBuHA-treated INCL fibroblasts compared with that of the normal control cells. Note that compared with the cells treated with up to 1 mM NtBuHA, those treated with 2.5mM NtBuHA manifested slightly lower level of plating efficiency. (Panel f). Time-course of NtBuHA-mediated depletion of [35S]labeled lipid thioester compounds from INCL lymphoblasts. Cultured lymphoblasts from an INCL patient were treated with 500 µM NtBuHA for varying lengths of time (12–48h) after being labeled with [35S]-cysteine. Fresh NtBuHA was added every 12 h. The [35S]-labeled lipid thioesters were extracted and resolved by TLC. Note a marked decrease in intensity of the radioactive bands at 48h. The arrows indicate the positions of each of the 8 identifiable lipid thioester bands (1–8: counting from the top) and the quantitations of these bands are presented graphically (Supplementary Fig. 5a). (Panel g) Depletion of [35S]Cysteine-labeled lipid thioesters in lymphoblasts from INCL patients, which were either untreated or treated with NtBuHA for 48 h. The culture medium was replaced every 12h with fresh NtBuHA containing medium. Because all the normal controls and 9 INCL patients’ cells could not be accommodated in one TLC plate the samples are resolved using two TLC plates. The smaller TLC (left): INCL, INCL patient’s lymphoblast; N1–N3, 3 normal control lymphoblasts; Larger TLC (right) N4, normal lymphoblasts (control), which in addition to N1–N3 (smaller TLC) show virtually no dense thioester bands. However, lymphoblasts from 9 INCL patients (Supplementary Table 2) show clearly identifiable lipid thioester bands. The [35S]labeled lipid thioesters were resolved by TLC as previously reported18. Lanes marked ‘–’ or ‘+’ indicate untreated and NtBuHA-treated samples, respectively. Note the reduction in density of the radioactive lipid thioester bands in NtBuHA-treated samples. Quantitative analyses of each of the 8 identifiable lipid thioester bands marked in panel f were performed and the results are shown in Supplementary Fig.5b.
To determine whether the hydroxylamine-derivatives are non-toxic, we treated cultured fibroblasts from INCL patients with each of the 10 out of 12 derivatives and evaluated their viability. We excluded N, N-dibenzylhydroxylamine and N-tert-Butyl-O-[1-[4-(chloromethyl) phenyl] ethyl]-N-(2-methyl-1-phenylpropyl) hydroxylamine as these derivatives were insoluble in tissue culture medium. The results showed that all 10 compounds tested, except for N-Benzoyl-N-phenyl hydroxylamine, were non-toxic up to a concentration of 1 mM (Supplementary Fig. 2).
NtBuHA is non-toxic to cultured cells from INCL patients
To evaluate whether NtBuHA is toxic to the cells, we cultured lymphoblasts from INCL patients in the absence and presence of varying concentrations of NtBuHA (0 to 5mM) and determined the viability of the cells by MTT method23. It should be noted that while NtBuHA at 1 mM concentration is non-toxic to cultured INCL lymphoblasts, viability of the cells gradually declined beyond this dose (Fig. 1d). However, when these cells were cultured in medium containing 500 µM NtBuHA for varying lengths of time (12–60 h), it did not alter the cell viability (Supplementary Fig. 3a).
To further confirm these results, we tested the plating efficiency of the cells after treatment with NtBuHA at varying concentrations ranging from 0.1mM to 2.5mM. The results showed that the plating efficiency of the cells remained unaffected after treatment with NtBuHA at concentrations up to 1mM although the plating efficiency slightly declined when the NtBuHA concentration was raised to 2.5 mM (Fig. 1e and Supplementary Fig. 3b). Taken together, these results clearly indicated that NtBuHA, at wide range of concentrations, is non-toxic to cultured cells from INCL patients.
NtBuHA cleaves thioester linkage in [14C]Palmitoyl ~CoA
We chose to study NtBuHA in further detail because a previous report indicated that in normal mice and rats this compound manifested potent mitochondrial anti-oxidant activity and prolonged longevity24. These properties of NtBuHA are especially relevant because high levels of ER- and oxidative-stress have been reported to cause apoptosis in cultured cells from INCL patients as well as in brain neurons of Ppt1−/− mice25 contributing to neurodegeneration26–28 and shortened lifespan25. Consistent with these findings, high levels of apoptosis have been reported in brain tissues of INCL patients29. Thus, we first sought to determine whether NtBuHA-mediated cleavage of thioester linkage is dose- and time-dependent. Accordingly, we incubated 0.1µCi of [14C]-palmitoyl~CoA with varying concentrations of NtBuHA (0 to 500 µM) and measured the levels of free [14C]-palmitate. We found that NtBuHA efficiently cleaved thioester linkage and released free [14C]-palmitate in a dose-dependent manner (Supplementary Fig. 4a).
We then incubated 0.1µCi of [14C]-palmitoyl~CoA with 500µM NtBuHA, which yielded the highest amount of free [14C]-palmitate release and stopped the reaction at varying incubation times. The results showed that NtBuHA-mediated the cleavage of thioester linkage in [14C]-palmitoyl~CoA and the release of free [14C]-palmitate occurred in a time-dependent manner (Supplementary Fig. 4b). Taken together, these results clearly demonstrated that NtBuHA efficiently cleaves thioester linkage in a dose- and time-dependent manner.
NtBuHA cleaves thioester linkage in palmitoylated proteins
To determine whether NtBuHA cleaves thioester linkage in palmitoylated proteins, we used cultured lymphoblasts from INCL patients labeled with [35S]cysteine as previously reported22. The results showed that compared with the cells grown in medium without NtBuHA, those grown in NtBuHA-containing medium for 24 and 48 hours yielded appreciably less intense [35S]cysteine-labeled lipid thioester bands (Fig. 1f and Supplementary Fig 5a). To further confirm the above results, we performed identical experiments using cultured lymphoblasts from 9 different INCL patients who carried varying inactivating mutations in the PPT1 gene (Supplementary Table 2). The results showed that compared with the density of the [35S]cysteine-labeled lipid thioester bands from untreated INCL cells those of the NtBuHA-treated counterparts yielded significantly less dense bands (Fig. 1g and Supplementary Fig.5b).
The loss of intensity of [35S]cysteine-labeled lipid thioester bands in NtBuHA-treated cells also indicates the cleavage of the thioester linkage in S-acylated proteins like its parent compound, hydroxylamine. As expected, the normal control cells (n=4) exhibited extremely low level of accumulation of [35S]cysteine-labeled lipid thioesters unlike the more dense bands characteristically found in INCL cells (Fig. 1g). Collectively, these results confirmed that NtBuHA cleaves thioester linkage in palmitoylated proteins in cultured cells from INCL patients in a dose- and time-dependent manner and mediates depletion of lipid thioesters in these cells.
NtBuHA depletes granular osmiophilic deposits in INCL cells
A physical correlate of ceroid deposition in the cells of INCL patients is the presence of GRODs (granular osmiophilic deposits)13. We sought to determine whether NtBuHA mediates the depletion of GRODs in INCL cells, which are detectable by transmission electron microscopy (TEM). Accordingly, we analyzed the levels of GRODs by TEM of cultured INCL lymphoblasts that were either untreated or treated with NtBuHA for three weeks before TEM analysis was performed. The results showed that compared with the untreated controls (Fig. 2a, left panel), NtBuHA-treated cells contained either no GRODs (Fig. 2a, middle panel (1)) or a substantially reduced number of GRODs (Fig. 2a, right panel (2). Quantitative analysis confirmed a significantly decreased (t (77) =7.61, P<0.001, permutation t-test) number of GRODs in NtBuHA-treated cells (Fig. 2b). The morphometric analysis of the GRODs showed that the size of the GRODs, when present in NtBuHA-treated cells, is also significantly smaller (t (66) =4.68, P<0.001 permutation t-test) than those of the untreated cells (Fig. 2c).
Figure 2.

NtBuHA mediates the depletion of ceroid and GRODs in cultured INCL cells.
(Panel a) Electron micrographs of cultured lymphoblasts from INCL patients showing the intracellular accumulation of GRODs (inset) and effect of NtBuHA treatment on the accumulation of GRODs. The left panel (with magnified inset below) represents untreated control. The middle and right panels and corresponding magnified insets represent two representative cells treated with NtBuHA (250 µM) for 3 weeks. Note that in most NtBuHA-treated cells GRODs are completely depleted [Panel a (1)] while an infrequent cell contained a few GRODs [Panel a (2); Scale bar: 500nm]. Box plot analysis of GRODs (Panel b) counted from the TEM in untreated (n = 23) and NtBuHA-treated (n = 56) cells. Untreated- versus NtBuHA-treated group (t (77) =7.61, P<0.001, permutation t-test). Morphometric analysis of the GRODs in untreated- (n=47) and NtBuHA-treated (n=21) cells (Panel c) (t (66) =4.68, P<0.001 permutation t-test) when the uuntreated-INCL cells were compared with. NtBuHA-treated cells. Electron micrographs of cerebral cortices of 6-month old WT (Panel d, left panel) and those of untreated- (Panel d, middle panel) and NtBuHA-treated Ppt1−/− mice (Panel d, right panel). Note that compared with WT mouse cortices, which show no GRODs (left panel), those of the untreated Ppt1−/− mice showed a very high level of GRODs (middle panel), whereas those treated with NtBuHA showed significantly reduced number (t (16) =2.89, P=0.012, permutation t-test) of GRODs (right panel; Scale bar: 2µm). (Panel e) Quantitation of GRODs/cell in the brain sections from WT, untreated- and NtBuHA-treated Ppt1−/− mice is presented graphically. Representative electron micrographs from 3–6 animals from each group were analyzed. (Panel f) Autofluorescence visualized by dark field microscopic analysis of cortical tissue sections of 6-month old WT mice (left panel) and untreated- (middle panel) as well as NtBuHA-treated (right panel) Ppt1−/− mice. Densitometric analyses of autofluorescence in the brain tissues of untreated- and NtBuHA-treated Ppt1−/− mice (Panel g) show significant decline in autofluorescence in treated mice; (t(28)=11.78, P<0.001, permutation t-test). Scale bar=50µm.
We also conducted similar experiments using cultured fibroblasts from INCL patients. The results showed that compared with the untreated INCL fibroblasts, which contained numerous highly dense GRODs (Supplementary Fig. 6a), the NtBuHA-treated cells contained significantly lower number of GRODs (Supplementary Figs. 6b, c). These results demonstrated that NtBuHA is effective in mediating the depletion of GRODs in both cultured lymphoblasts and fibroblasts from INCL patients.
Normal serum methemoglobin in NtBuHA-treated Ppt1−/− mice
Because hydroxylamine stimulates methemoglobin production, it was important to determine whether the hydroxylamine-derivative (i.e. NtBuHA) caused elevation of serum methemoglobin levels in the NtBuHA-treated mice. Accordingly, we measured the blood methemoglobin levels in WT, untreated- and NtBuHA-treated Ppt1−/− mice. The results showed that treatment of Ppt1−/− mice with NtBuHA for up to 3 months did not elevate the methemoglobin levels (Table 1). We concluded that this derivative of hydroxylamine, at the dosages used in our experiments, is unlikely to have toxicity from the standpoint of increased methemoglobin levels.
Table 1.
Serum methemoglobin levels in WT, untreated- and NtBuHA-treated Ppt1−/− mice
| Animals | Treatment | Methemoglobin (%) ± SD | No. of Animals |
|---|---|---|---|
| WT | Untreated | 1.60 ± 0.69 | 8 |
| Ppt1−/− | Untreated | 1.50 ± 0.66 | 8 |
| Ppt1−/− | NtBuHA | 1.59 ± 0.58 | 8 |
NtBuHA crosses the blood-brain barrier
Next, we sought to determine whether NtBuHA crossed the blood-brain barrier. Accordingly, we provided WT and Ppt1−/− mice with NtBuHA in their drinking water and determined the levels of NtBuHA in perfused brain tissues by multiple reaction-monitoring mass spectrometry as previously reported30. Our results clearly showed that NtBuHA given to 3- and 6-month old Ppt1−/−, and WT mice was readily detectable in the brain (Table 2). These results demonstrated that orally administered NtBuHA is readily absorbed from the gastrointestinal tract and crossed the blood-brain barrier.
Table 2.
NtBuHA levels in brain tissues of mice receiving NtBuHA in their drinking water
| Genotype of Mice | Age | NtBuHA in Brain Tissues (ng/mg brain tissue)±SD |
|---|---|---|
| WT | 3 months | 4.44± 0.50 (n=3) |
| Ppt1−/− | 3 months | 4.87 ±0.25 (n=3) |
| WT | 6 months | 4.73 ±0.48 (n=3) |
| Ppt1−/− | 6 months | 5.65 ±0.51 (n=3) |
NtBuHA-treatment depletes GRODs in Ppt1−/− mouse brain
To determine whether the beneficial effects of NtBuHA observed in cellulo are reproducible in vivo, we treated 3-month old Ppt1−/− mice (n=6) with NtBuHA continuously for three months. We tested these mice at 6 months of age because at this age they begin to manifest signs of neurological impairment as well as show characteristic pathological features of INCL (refs. 26–28, 31). Untreated age- and sex-matched Ppt1−/− mice (n=6) served as controls. At the end of the treatment period, the mice were sacrificed and the cortical tissues were examined by TEM. The results showed that while the WT mice had no GRODs (Fig. 2d, left panel) high levels of GRODs were readily detectable in cortical tissues of untreated Ppt1−/− mice (Fig. 2d, middle panel). Notably, the NtBuHA-treated Ppt1−/− mice showed markedly reduced number of GRODs (Fig. 2d, right panel). Quantitation of the GRODs in the brain cells of untreated and NtBuHA-treated Ppt1−/− mice showed significant reduction (t (16) =2.89, P=0.012, permutation t-test) in the number of the GRODs (Fig. 2e). These results demonstrated that NtBuHA-treatment mediates depletion of GRODs in the brain of Ppt1−/− mice.
NtBuHA reduces autofluorescence in Ppt1−/− mouse brain
Accumulation of intracellular autofluorescent lipopigment material in the brain and in other tissues is a characteristic pathological finding in NCLs including INCL (ref. 13), and in Ppt1−/− mice25, 26. Accordingly, we analyzed the brain tissues of NtBuHA-treated Ppt1−/− mice for autofluorescence and compared the results with those of untreated-Ppt1−/− and WT mice. Our results showed that while the brain tissues from WT mice had no autofluorescence (Fig. 2f, left panel), those of the untreated Ppt1−/− mice manifested intense autofluorescence (Fig. 2f, middle panel). Importantly, NtBuHA-treated Ppt1−/− mice had significantly reduced (t(28)=11.78, P<0.001, permutation t-test) autofluorescence (Fig. 2f, right panel). The quantitative analysis showed that compared with the intensity of autofluorescence in the brain tissues of untreated-Ppt1−/− mice, that of the NtBuHA-treated Ppt1−/− mice was significantly lower (Fig. 2g). Together these results suggested that NtBuHA mediated depletion of intracellular ceroid, decreased the number of GRODs per cell and the intensity of autofluorescence.
NtBuHA ameliorates ER- and oxidative-stress
Previously, we reported that ER- and oxidative-stress contribute to neuropathology in INCL (refs. 26–28). Thus, we sought to determine whether NtBuHA-treatment of Ppt1−/− mice ameliorated these stresses. Accordingly, we first performed Western blot analysis of proteins in brain lysates from untreated- and NtBuHA-treated Ppt1−/− mice and determined the levels of ER-stress markers, Grp-78, Grp-94 and ATF6. The results showed that compared with the levels of all three ER-stress markers in untreated mice those in the brain tissues of NtBuHA-treated Ppt1−/− mice were markedly reduced (Supplementary Fig. 7).
We then performed Western blot analysis of brain lysates from NtBuHA-treated and untreated Ppt1−/− mice to determine the levels of oxidative stress-markers, superoxide dismutase-2 (SOD2) and catalase. The levels of these two enzymes increase in response to elevated oxidative-stress to maintain cellular homeostasis and a decrease in the levels of SOD2 and catalase occur if a treatment is effective in reducing oxidative-stress. Our results showed that compared with untreated Ppt1−/− mice, NtBuHA-treatment significantly reduced the levels of both SOD2 and catalase (Supplementary Fig. 7). Taken together, these results showed that NtBuHA-treatment is effective in reducing both ER- and oxidative-stress in the brain of Ppt1−/− mice.
In cultured INCL cells NtBuHA suppresses apoptosis
Next, we sought to determine whether NtBuHA-treatment protects cultured INCL lymphoblasts from apoptosis, which has been reported to be one of the major contributors of neuronal death in INCL patients29. Because PPT1 is ubiquitously expressed in all mammalian tissues and organs32, 33, we used cultured cells from INCL patients in our in vitro experiments. Moreover, these cultured cells manifest virtually identical pathological changes (i.e. accumulation of GRODs and increased apoptosis) that are also found in postmortem brain tissues from INCL patients26–28. Accordingly, we pretreated cultured lymphoblasts from INCL patients with 1 mM NtBuHA for 12 h and then treated them with hydrogen peroxide (500 µM) in presence or absence of NtBuHA (1mM) for 3 h. As control, we treated INCL lymphoblasts without NtBuHA-pretreatment with the same concentration of hydrogen peroxide for 3 h.
Apoptosis was detected by annexin V-staining and quantified by FACS analysis. The results showed that H2O2treatment elevated the levels of apoptosis in cells that were not pretreated with NtBuHA (Figs. 3a, b). However, in NtBuHA-pretreated lymphoblasts H2O2 failed to induce apoptosis above the basal level (Figs. 3a, b). Similarly, cells which were first pretreated with NtBuHA and then incubated with H2O2 in presence of NtBuHA (co-treated) also showed no alteration in apoptosis above the basal level in these cells (Fig. 3a, b). Together, these results suggested that NtBuHA protects cultured INCL lymphoblasts from oxidative stress-mediated apoptosis.
Figure 3.

NtBuHA prevents H2O2-induced apoptosis in INCL lymphoblasts. (Panel a) Flow cytometric analysis of apoptotic cells using apoptosis-marker, annexin V and necrotic cells using necrotic cell marker PI: Untreated control INCL lymphoblasts (Upper left panel); NtBuHA-pretreated (Upper middle panel); and NtBuHA pre- as well as co-treated (Upper right panel). INCL lymphoblasts were either untreated and exposed to oxidative-stress by H2O2 (untreated control, Lower left panel) or the cells were first pretreated with NtBuHA for 12h and then incubated with H2O2 in the absence (Lower middle panel) or presence of NtBuHA (Lower right panel) for 3h. (Panel b) Graphical presentation of level of apoptotic cells in untreated and NtBuHA-treated INCL cells. Apoptotic cells represent annexinV positive cells detected by FACS (Summation of cell number in Q2 + Q3).
Reduced apoptosis in NtBuHA-treated Ppt1−/− mouse brain
To determine whether NtBuHA-treatment protects against neuronal apoptosis in vivo, we performed TUNEL assay using cortical tissue sections from 6-month old WT and untreated- as well as NtBuHA-treated Ppt1−/− mice. The results showed that while in the brains of WT mice virtually no TUNEL-positive apoptotic cells were detectable (Fig. 4a), those of the untreated Ppt1−/− mice contained numerous apoptotic cells (Fig. 4b). In contrast, the brain tissues of the NtBuHA-treated Ppt1−/− mice showed significantly lower levels (t(19) = 15.74, P<0.001, permutation t-test) of TUNEL-positive cells (Figs. 4c, d).
Figure 4.

NtBuHA suppresses apoptosis in Ppt1−/− mouse brain.
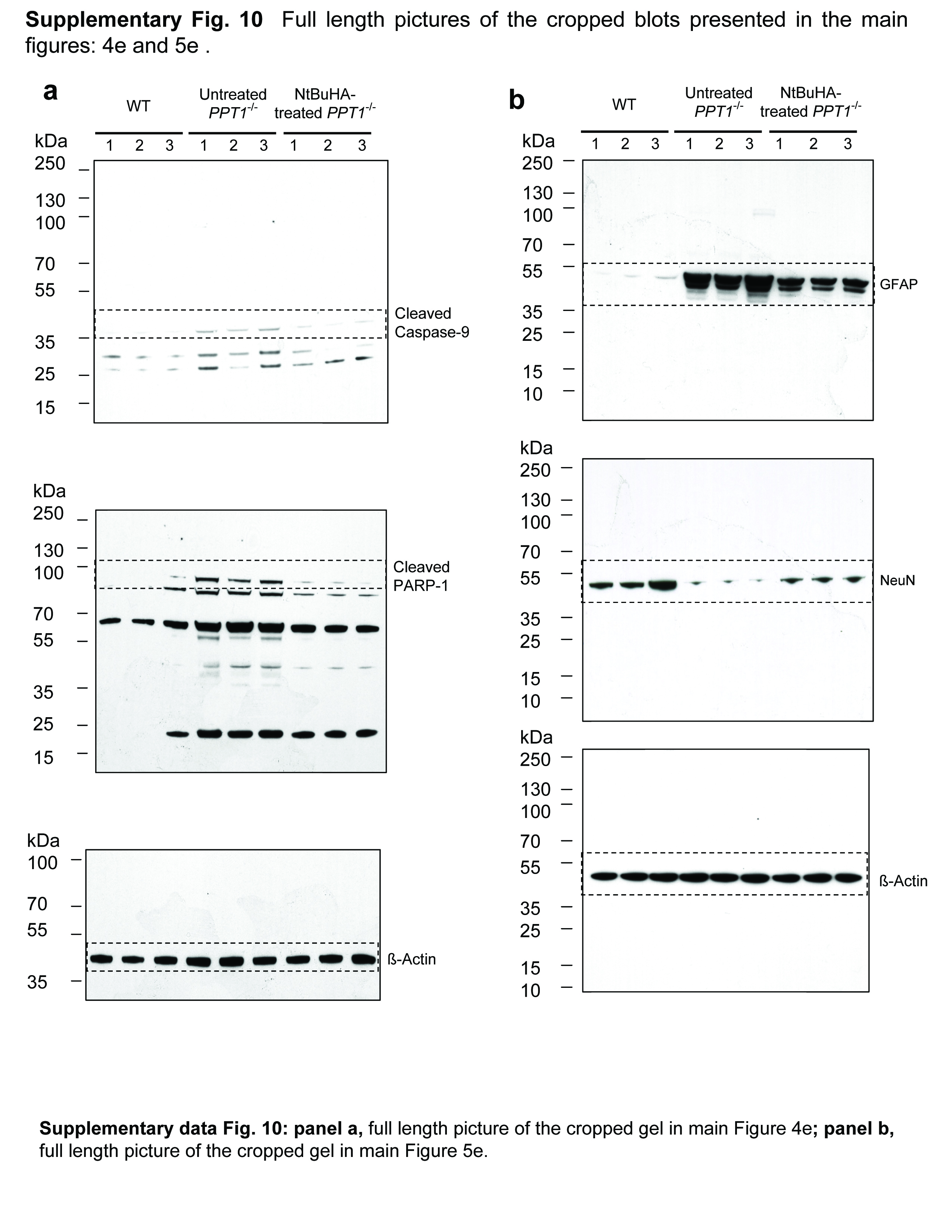
Apoptotic cells were detected by TUNEL assay in brain sections (Panel a) from 6-month old WT (n=9); (Panel b) untreated- (n=9) and (Panel c) NtBuHA-treated Ppt1−/− mice (n=12). TUNEL-positive apoptotic cells appear reddish brown. (Panel d) Graphic representation of the levels of apoptotic cells in the brain of WT, untreated- as well as NtBuHA-treated Ppt1−/− mice. (t(16)=27.63, P<0.001, permutation t-test) for WT vs. untreated-Ppt1−/−; (t(19) = 15.74, P<0.001, permutation t-test) for untreated-Ppt1−/− vs. NtBuHA-treated Ppt1−/−. (Panel e) Western blot analysis of cortical tissue homogenates prepared from 6-month old WT, untreated- as well as NtBuHA-treated Ppt1−/− mice for cleaved caspase-9 and cleaved PARP-1. Full-length blots of the cropped gel images used in 4e are provided in Sypplementary Fig. 10a. Densitometric quantitation of cleaved caspase-9 (Panel f) and cleaved PARP-1 (Panel g) from three independent experiments are presented. For panel f, WT vs. untreated-Ppt1−/− (t(10)=11.65, P=0.002, permutation t-test); Untreated-Ppt1−/− vs. NtBuHA-treated Ppt1−/− (t(10) = 8.23, P=0.002, permutation t-test); For panel g, WT vs. untreated Ppt1−/− (t(9) = 5.08, P=0.004, permutation t-test); Untreated Ppt1−/− vs. NtBuHA-treated Ppt1−/−(t(10) =4.73, P=0.015, permutation t-test).
To further confirm the results of TUNEL assay, we also performed Western blot analysis of brain tissue lysates for two biochemical correlates of oxidative-stress mediated mitochondrial pathway of apoptosis: cleaved caspase-9 and cleaved PARP-1. We found that compared with the brain tissues of WT and those of the untreated-Ppt1−/− mice had significantly higher levels of both cleaved caspase-9 and cleaved PARP-1 (Figs. 4e–g). These results suggested that NtBuHA protects the brain neurons from apoptosis in an INCL mouse model.
NtBuHA retards progression of brain atrophy in Ppt1−/− mice
Elevated levels of apoptosis in the brain of Ppt1−/− mice26–28 and in INCL patients29 are suggested to cause brain atrophy. Thus, we first determined the brain size and weight of untreated-, as well as NtBuHA-treated Ppt1−/− mice and compared the results with those of WT mice. We found that compared with WT mice (Figs. 5a, b) the size and weight of the brains of untreated-Ppt1−/− mice were markedly decreased (Figs. 5a, b). In contrast, both of these parameters were markedly higher in NtBuHA-treated Ppt1−/− mice compared with those of the untreated Ppt1−/− mice (Figs. 5a, b).
Figure 5.

The effects of NtBuHA on brain size, weight, neuron density, GFAP and NeuN expression.
(Panel a) The representative brain size of a WT (left panel), untreated-Ppt1−/−(middle panel) and NtBuHA-treated Ppt1−/− mouse (right panel). (Panel b) The brain weights of WT (n=12), untreated Ppt1−/− (n=5) and NtBuHA-treated Ppt1−/− (n=12) mice were determined. WT vs. untreated-Ppt1−/− (t(15) = 13.3, P<0.001, permutation t-test); Untreated-Ppt1−/− vs. NtBuHA-treated Ppt1−/− group (t(15) = 5.1, P<0.001, permutation t-test). (Panel c) The neuron densities in the cerebral cortex of representative WT (left panel), untreated- (middle panel) and NtBuHA-treated Ppt1−/− mouse (right panel) were determined after staining the cortical tissue sections using antibodies against a neuron specific marker protein, NeuN. Sections from at least 4 mice in each category (WT, untreated Ppt1−/− and NtBuHA-treated Ppt1−/−) were analyzed. (Panel d) Graphic representation of the levels of neurons in WT, untreated Ppt1−/− and NtBuHA-treated Ppt1−/− mice. WT vs. untreated-Ppt1−/−(t(8) = 21.8, P=0.008, permutation t-test); Untreated-Ppt1−/− vs. NtBuHA-treated Ppt1−/− group (t(9) = 8.17, P=0.002, permutation t-test). (Panel e) Western blot analysis of brain lysates from WT, untreated-Ppt1−/− and NtBuHA-treated Ppt1−/− mice for GFAP as well as NeuN and their densitometric quantitations (Panel f and Panel g). For Panels f, WT vs. untreated-Ppt1−/− (t(10) = 49.7, P=0.002, permutation t-test); Untreated-Ppt1−/− vs. NtBuHA-treated Ppt1−/− group (t(10) = 15.1, P=0.002, permutation t-test). For Panels g, WT vs. untreated-Ppt1−/− (t(10) = 8.7, P=0.002, permutation t-test); Untreated-Ppt1−/− vs. NtBuHA-treated Ppt1−/− group (t(10) = 8.76, P=0.002, permutation t-test). Full-length blots of the cropped gel images used in 5e are presented in Supplementary Fig. 10b. (Panel h) Immunohistochemistry of brain sections showing GFAP-positive cells and (Panel i) quantitation of GFAP-positive cells per mm2. All mice used in these experiments were 6 months old. WT vs. untreated-Ppt1−/− (t(6) = 36.35, P=0.03, permutation t-test); Untreated-Ppt1−/− vs NtBuHA-treated Ppt1−/− group (t(6) = 5.19, P=0.03, permutation t-test).
To delineate if the brain size and weight correlated with neuron density, we performed immunohistochemical analyses of cortical tissue sections from WT, untreated- as well as NtBuHA-treated Ppt1−/− mice using antibodies against NeuN, a neuronal marker protein. Our results showed that compared with the brains of WT mice those of the untreated-Ppt1−/− mice had a significantly reduced number (t(8) = 21.8, P=0.008, permutation t-test) of NeuN-positive cells (Figs. 5c, d). In contrast, compared with the brains of untreated Ppt1−/− mice those of the NtBuHA-treated Ppt1−/− animals showed a modest but significantly higher (t(9) = 8.17, P=0.002, permutation t-test) number of NeuN-positive cells (Figs. 5c, d).
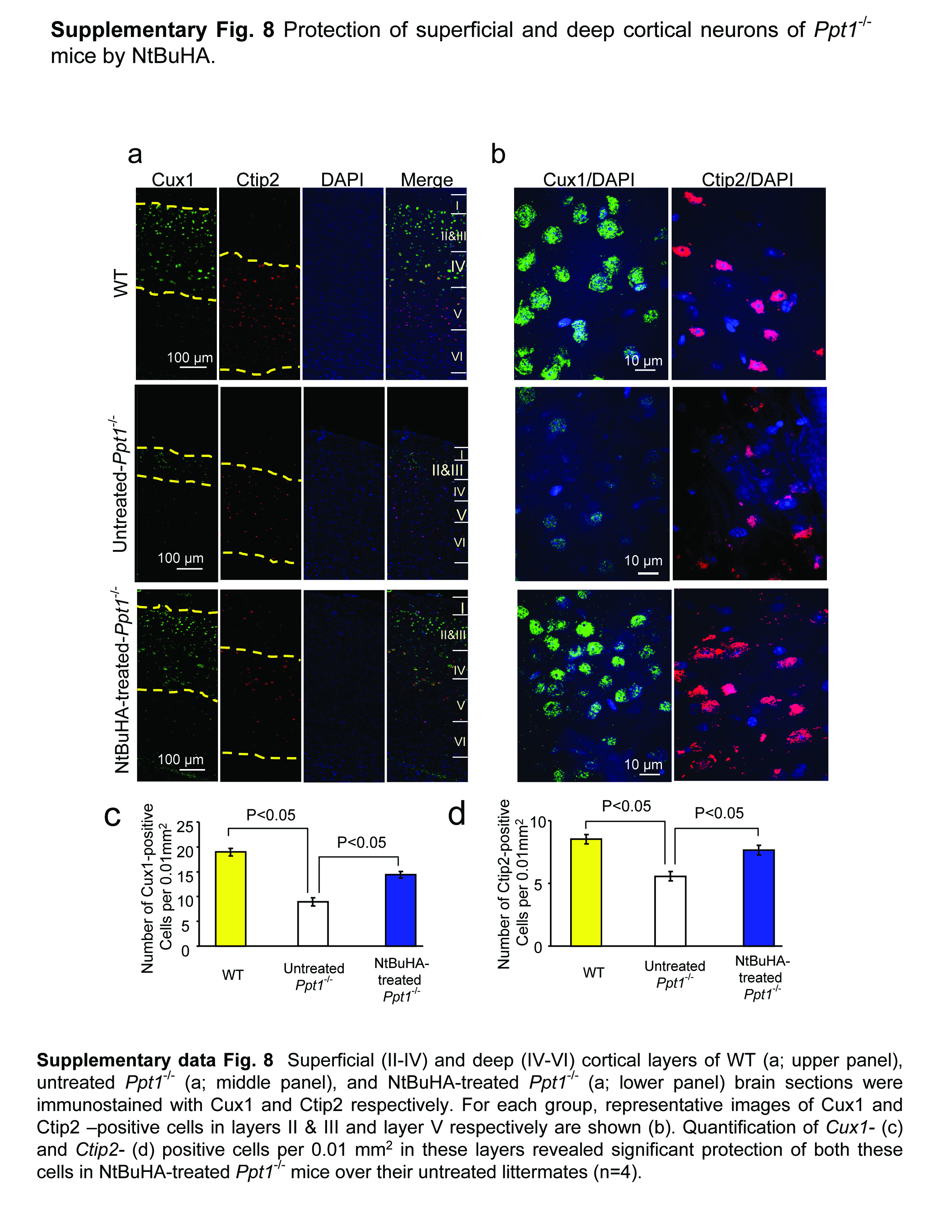
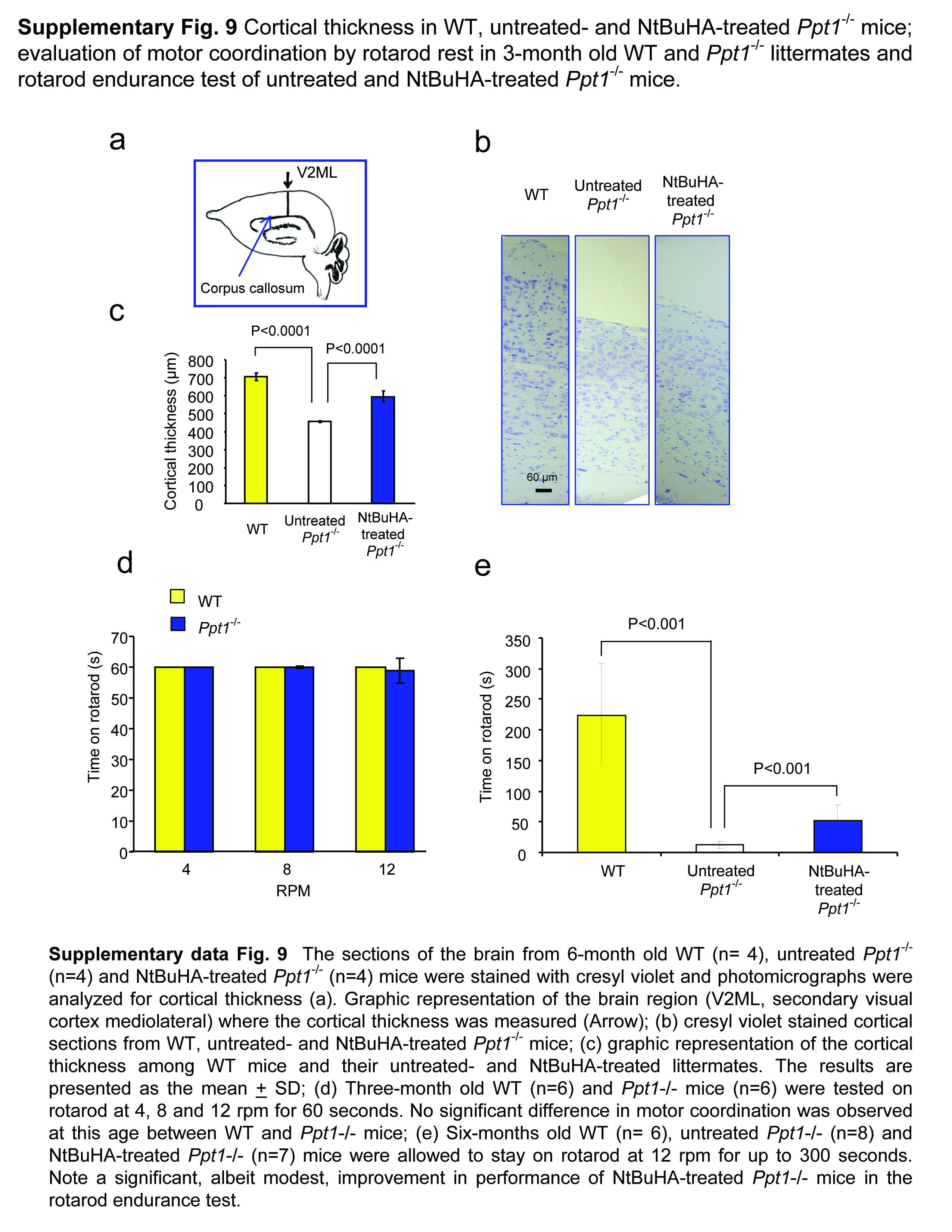
To evaluate the degree of protection provided by NtBuHA in neurons in various cortical layers, we immunostained the brain sections from WT, untreated Ppt1−/− and NtBuHA-treated Ppt1−/− mice using antibodies to Cux1 (cortical layers II-IV) and Ctip2 (cortical layers IV-VI). We then quantitated the Cux1- and Ctip2-positive neurons in these sections. The results showed that compared with the WT mice the numbers of Cux1- (Supplementary Figs. 8a–c) and Ctip2-positive (Supplementary Fig. 8a, b, d) neurons in the brains of Ppt1−/− mice were reduced to 53% and 35%, respectively. In contrast, the brains of the NtBuHA-treated Ppt1−/− mice showed a markedly increased number of both Cux1- and Ctip2-positive neurons in the mediolateral secondary visual cortex (V2ML) compared with those of the untreated Ppt1−/− mice (Supplementary Figs. 8a–d). Since one of the prominent neuropathological features of INCL is cortical atrophy, we also measured the cortical thickness in the V2ML region (Supplementary Fig. 9a) of cresyl violet stained brain sections from WT, untreated-Ppt1−/− and NtBuHA-treated Ppt1−/− mice (Supplementary Fig. 9b). Quantitative analysis showed that compared with WT, untreated Ppt1−/− mice had a marked decline in cortical thickness while this decline was less pronounced in the NtBuHA-treated Ppt1−/− mice (Supplementary Fig. 9c). Cumulatively, these results provided further supporting evidence that NtBuHA-treatment has neuroprotective effects in the brain of Ppt1−/− mice.
NtBuHA suppresses astroglia activation in Ppt1−/− mice
Next, we sought to determine whether NtBuHA-treatment of Ppt1−/− mice manifests any beneficial effect on astroglial activation, assessed by higher levels of expression of glial fibrillary acidic protein (GFAP) as it has been reported that increased GFAP expression precedes neuronal death in these mice34. Accordingly, we first performed Western blot analysis of cortical lysates from WT mice and those of the untreated- and NtBuHA-treated Ppt1−/− mice using GFAP and NeuN antibodies, respectively. The results showed that compared with the brain tissue lysates of WT mice, those of the untreated Ppt1−/− mice contained markedly higher levels of GFAP (Figs. 5e, f). However, the levels of NeuN were inversely proportional to those of the GFAP (Figs. 5e, g). Immunohistochemical analysis of cortical tissues corroborated the results of the Western blot analyses (Figs. 5h, i). Compared with the untreated-Ppt1−/− mice the NtBuHA-treated Ppt1−/− mice showed significantly decreased GFAP levels (t(6) = 5.19, P=0.03, permutation t-test) while those of NeuN were moderately elevated (Figs. 5e–i). These findings demonstrated that NtBuHA has beneficial effects in reducing the activation of astroglia and thus, may have played a role in preventing neurodegeneration in this mouse model of INCL.
NtBuHA preserves motor function in Ppt1−/− mice
The natural history of INCL shows that impaired motor coordination is one of the first signs of neurological deterioration in INCL patients13. Since NtBuHA functionally mimics PPT1, we tested WT, untreated- and NtBuHA-treated Ppt1−/− mice at 6- and 8-month of age for motor coordination by rotarod performance test35. We first performed rotarod test using 3-month old WT and Ppt1−/− mice because at this age they do not manifest signs of motor deficit. As expected, the results showed that there were virtually no difference in performance of the WT and Ppt1−/− mice at all 3 speeds (i.e. 4, 8 and 12 rpm for 60 seconds) (Supplementary Fig. 9d). We then performed the same test using 6-month old WT, untreated-Ppt1−/− and NtBuHA-treated Ppt1−/− mice. The results showed that untreated 6-month old Ppt1−/− mice manifested clear motor deficit while NtBuHA-treated ones had near-normal motor coordination (Figs. 6a–c and Supplementary Video). The rotarod test on eight-month old WT, untreated- and NtBuHA-treated Ppt1−/− mice yielded similar results (Fig. 6d).
Figure 6.

NtBuHA ameliorates neuropathology and extends lifespan in Ppt1−/− mice.
Motor coordination in WT, untreated- and NtBuHA-treated Ppt1−/− mice were evaluated using the rotarod test. Mice at 6 months of age were tested on rotarod at three different speeds: (Panel a) 4-rpm, (Panel b) 8-rpm and (Panel c) 12-rpm. Mice at 8 months of age were tested using only one speed of 4-rpm (Panel d) as the untreated mice could not withstand a higher speed at this age. At all three speeds, the direction of rotation of the rotarod was reversed every 15 s. Prior to the final test, the animals were trained for 3 d (1 min for each condition) and allowed to rest for 1 min between two successive trials. Each test was performed for 1 min. Latency (defined as the amount of time the animals stayed on the rotating rod) was recorded. For panel a, WT vs. untreated-Ppt1−/− (t(16) = 4.62, P<0.001, permutation t-test); Untreated-Ppt1−/− vs NtBuHA-treated Ppt1−/− group (t(14) = 4.0, P=0.006, permutation t-test). For panel b, WT vs untreated Ppt1−/− (t(15) = 6.4, P<0.001, permutation t-test); Untreated-Ppt1−/− vs NtBuHA-treated Ppt1−/− group (t(15) = 4.85, P<0.001, permutation t-test). For panel c, WT vs untreated-Ppt1−/− (t(14) = 13.4, P<0.001, permutation t-test); Untreated-Ppt1−/− vs NtBuHA-treated Ppt1−/− group (t(16) = 4.3, P=0.002, permutation t-test). For panel d, WT vs. untreated-Ppt1−/− (t(8) = 14.96, P=0.008, permutation t-test); Untreated-Ppt1−/− vs NtBuHA-treated Ppt1−/− group (t(7) = 9.74, P=0.008, permutation t-test). Exploratory behavior (open field test) of WT, untreated Ppt1−/− and NtBuHA-treated Ppt1−/− mice at 6 months of age (Panel e) and at 8 months of age (Panel f). The number of squares travelled by the animals over 3 minutes of time were counted. For 6-month old groups, WT vs untreated-Ppt1−/−(t(8) = 8.6, P=0.005, permutation t-test); Untreated-Ppt1−/− vs NtBuHA-treated Ppt1−/− group (t(13) = 3.2, P=0.01, permutation t-test). For 8-month old groups, WT vs. untreated-Ppt1−/− (t(10) = 37.0, P=0.002, permutation t-test); Untreated-Ppt1−/− vs NtBuHA-treated Ppt1−/− group (t(11) = 10.9, P=0.001, permutation t-test). (Panel g) Survival plots (Kaplan-Meyer analysis) of untreated and NtBuHA-treated Ppt1−/− mice. Untreated- (n=110) or NtBuHA treated- Ppt1−/− (n=80) animals were euthanized when they could not reach for food or water. Median Survival: 242d for untreated; 277d for treated, chi-square (161, df=1), P<0.001.
To determine whether NtBuHA-treatment of the Ppt1−/− mice improves their endurance, we repeated the rotarod test at the highest speed (12rpm) for 300 seconds instead of 60 seconds using WT, untreated- and NtBuHA-treated Ppt1−/− mice. The results (Supplementary Fig. 9e) showed that the endurance time (in seconds) of the WT mice was markedly higher than that of the untreated Ppt1−/− mice (WT:224±84 vs untreated-Ppt1−/−: 12±6). In contrast, compared with the endurance time of the untreated Ppt1−/− mice that of the NtBuHA-treated Ppt1−/− mice was modestly longer (untreated-Ppt1−/−:12±6 vs treated-Ppt1−/−: 53±24).
We also tested the Ppt1−/− mice for their exploratory behavior using open field test36. The results showed that compared with untreated 6-month old Ppt1−/− mice (Fig. 6e) and 8-month old Ppt1−/− mice (Fig. 6f) those treated with NtBuHA showed improved exploratory behavior (Figs. 6e, f). Taken together, these results suggested that NtBuHA-treatment of Ppt1−/− mice not only cleaved thioester linkage in palmitoylated proteins, mediated depletion of ceroid, reduced ER- and oxidative-stress and suppressed neuronal apoptosis but also considerably slowed the deterioration of motor coordination and exploratory behavior.
Extension of lifespan in Ppt1−/− mice treated with NtBuHA
To determine whether the observed beneficial effects of NtBuHA-treatment extended the lifespan, we divided 190 three-month old Ppt1−/− mice into two groups: one group (n=110) received no treatment (control) while the other group (n=80) was treated with NtBuHA. The status of all mice was monitored on a daily basis and when a mouse could not reach for food and water it was sacrificed. The results showed that compared with the lifespan of untreated Ppt1−/− mice that of the NtBuHA-treated animals had extended lifespan (Fig. 6g). The quantitative analysis confirmed that NtBuHA-treatment conferred a significant extension of median survival time (untreated: 242d vs treated: 277d; chi-square (161, df=1), P<0.001) and longer maximum lifespan (untreated:278d vs treated: 311d). These results strongly suggest that NtBuHA-treatment of Ppt1−/− mice not only slows down the progression of neuropathology but also modestly, albeit significantly, extends their lifespan.
DISCUSSION
In this study, we have demonstrated that thioesterase-mimetic, nucleophilic small molecules with antioxidant property like NtBuHA cleave thioester linkage in palmitoylated proteins (constituents of ceroid) with high specificity. We also show that this small molecule is non-toxic, mediates depletion of ceroid deposits and suppresses apoptosis in cultured cells from INCL patients. We further demonstrate that the results obtained in cellulo are reproduced in vivo when Ppt1−/− mice were treated with NtBuHA. Notably, in Ppt1−/− mice orally administered NtBuHA crossed the blood-brain-barrier (BBB), did not stimulate methemoglobin production, mediated ceroid-depletion, suppressed neuronal apoptosis, slowed neurological deterioration and caused a modest, albeit significant extension of lifespan.
In the central nervous system, a large number of proteins require dynamic palmitoylation for their function16, 37. Although posttranslational lipid-modifications of proteins such as palmitoylation are critical in many biological processes, removal of the lipid moieties (depalmitoylation) from these lipid-modified proteins is also critical for their recycling or degradation in lysosomes. Thus, genetic deficiency of a lysosomal thioesterase, PPT1, leads to a devastating neurodegenerative LSD, INCL. Because children afflicted with INCL are normal at birth and the disease manifestation does not occur before 11–18 months of age, it provides a window of opportunity to develop and implement therapeutic interventions that arrest disease pathology.
To date, INCL is the only well characterized neurodegenerative LSD, caused by the deficiency of a lysosomal thioesterase, PPT1. Mammalian thioesterases are both cytosolic (APT1 and APT2)38, 39 and lysosomal (PPT1 and PPT2)9, 10, which catalyze the cleavage of thioester linkage in S-acylated (S-palmitoylated) proteins. Hydroxylamine cleaves thioester linkage with high specificity14, 15 although it was not known whether its derivatives can cleave thioester linkage. Our results show that its derivatives like NtBuHA also cleave this linkage in S-acylated proteins and mediate ceroid depletion. However, although NtBuHA depalmitoylates S-acylated proteins, it is unlikely that it would be effective in depalmitoylating proteins in which the palmitate is linked to cysteine residues via amide linkage (N-palmitoylation).
In addition to its role in enhancing membrane-affinity, palmitoylation promotes protein-protein interaction14, 15. Interestingly, a recent report suggested that palmitoylation-induced aggregation of mutant cysteine-string protein-α (CSPα) and possibly other proteins that undergo palmitoylation may underlie the adult-onset NCL (ref. 40). Thus, thioesterase-mimetic small molecules such as NtBuHA may ameliorate the pathogenesis of diseases caused not only by thioesterase deficiency but also for diseases that may be caused by the accumulation of aggregate-prone palmitoylated proteins.
We previously reported that both in INCL and in the Ppt1−/− mice high levels of ER- and oxidative-stress leads to neuronal apoptosis26–28. It has been reported that NtBuHA has potent antioxidant property24. Our results show that NtBuHA not only mediates depletion of intracellular ceroid but also protects the PPT1-deficient cells from oxidative stress-mediated apoptosis due to its anti-oxidant property. This may suggest that the neuroprotective effects of NtBuHA may stem not only from its ability to cleave thioester linkage in S-acylated proteins but also from its anti-oxidant property. Since NtBuHA functionally mimics all thioesterases, we propose that nucleophilic small molecules with antioxidant property like NtBuHA may have therapeutic potential for INCL as well as other as yet unrecognized thioesterase-deficiency diseases.
Although currently several therapeutic approaches are being used for the LSDs with systemic (visceral) involvement41, the development of effective treatment strategies for LSDs affecting the central nervous system (CNS) remains challenging42. For INCL, several approaches are currently being tested in order to develop an effective treatment strategy for this uniformly fatal disease43–48. However, delivering macromolecules to the brain, as therapeutics for neurodegenerative diseases, remains challenging. Consequently, most of the potential therapies face this challenge, although novel strategies to overcome the obstacles are being tested49. In the present study, we took a mechanistic approach to identify non-toxic, small molecules with anti-oxidant and thioesterase-mimetic properties, which readily cross the BBB. The results of our present study show that NtBuHA fulfills at least some of these criteria required to be considered a potential drug-target for INCL.
Despite the positive beneficial effects of NtBuHA in Ppt1−/− mice, the animals ultimately succumbed to the disease. While at this time we do not have a clear understanding of the mechanism of death of the NtBuHA-treated animals, there are several possibilities that may account for this outcome. One of these possibilities is that the time of initiation of NtBuHA treatment may be critical. The other possibility is that NtBuHA dosage in our study may not have been adequate to maintain the full therapeutic level in the brain. Our ongoing studies are designed to address these and other possibilities. While to our knowledge, no off-target adverse effects of NtBuHA have been reported and in our present study, we uncovered no adverse effects of this small molecule, we are nonetheless mindful that like most drugs, NtBuHA may also have such adverse effects. Accordingly, our ongoing long-term investigations are designed to address this issue. Nevertheless, our findings provide the proof of concept that thioesterase-mimetic small molecules with antioxidant property like NtBuHA may offer a novel mechanism-based potential therapeutic option not only for INCL but also for as yet unrecognized diseases that may be caused by the deficiency of either cytosolic or lysosomal thioesterase.
ONLINE METHODS
Reagents
All hydroxylamine derivatives as well as all other reagents were purchased from Sigma. [14C]palmitoyl~CoA and [14C]-palmitic acid were from Perkin Elmer and [35S]-cysteine was purchased from MP Biomedicals.
Cell culture
Immortalized lymphoblasts from normal subjects and INCL patients were obtained from the laboratory of Late Dr. Krystina E. Wisniewski. PPT1-deficient fibroblasts were derived from skin biopsy samples from an INCL patient admitted to an ongoing clinical protocol (www. clinicaltrials.gov; NCT00028262; protocol #01-CH-0086), approved by the Institutional Review Board (IRB) of the NICHD, NIH. This patient was homozygous for one of the most lethal PPT1 gene mutations (R122W). A list of cell lines used and the PPT1-mutations they carried have been provided in Supplementary Table 2. Fibroblasts were cultured in DMEM supplemented with 10% heat inactivated fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin and streptomycin at 37° C in humidified atmosphere with 5% CO2. Lymphoblasts were cultured in RPMI supplemented with 16% FBS at 37° C in humidified atmosphere with 5% CO2.
Animals
Ppt1−/− mice (a generous gift from Dr. S. L. Hofmann, University of Texas Southwestern Medical Center at Dallas, Dallas, Texas, USA) were generated by targeted disruption of the last exon in the Ppt1 gene in embryonic stem (ES) cells as previously reported25. These mice were subsequently backcrossed for 10 generations with wild type (WT) C57BL/6 mice in order to obtain congenic C57 background and a breeding pair was kindly given to us by Dr. Mark S. Sands to start our Ppt1−/− mouse colony at the NIH. Both male and female mice were used in this study. Mice were housed and maintained in a pathogen-free facility under a 12-hour light and 12-hour dark cycle. Animals were provided with food and water ad libitum. All procedures were carried out under an animal protocol approved by the Animal Care and Use Committee (ACUC) of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH. Three-month old Ppt1−/− mice were given NtBuHA in their drinking water (1 mM NtBuHA and 1mM NaCl) as previously reported24.
Screening for hydroxylamine derivatives that cleave thioester linkage
To determine whether hydroxylamine-derivatives, like its parent compound, hydroxylamine, cleave thioester linkage, we used the model thioester substrate of PPT1, [14C]palmitoyl~CoA. We screened 12 hydroxylamine derivatives (Supplementary Table 1) using hydroxylamine (1M) and recombinant human PPT1 (a generous gift from Dr. S. L. Hofmann, University of Texas Southwestern Medical Center at Dallas, Dallas, Texas, USA) as controls. Briefly, we incubated 0.1 µCi of [14C]palmitoyl~CoA with each of the 12 hydroxylamine-derivatives (1mM, pH 7.4) at room temperature for 1 h. [14C]-palmitate released was then extracted using a mixture of chloroform: methanol (1:1) and resolved by thin layer chromatography using a mixture of chloroform: methanol: water (65:25:4). As controls, [14C]palmitoyl~CoA was either treated with hydroxylamine or recombinant human PPT1.
MTT assay
Cell viability was determined according to a previously reported method23. Briefly, following treatment with hydroxylamine-derivatives INCL cells were incubated with MTT (Sigma) for 4 h at 37° C. Formazan crystals thus formed were dissolved in acidified isopropanol and absorption was measured at 570 nm. Viability was expressed as percent of untreated control.
Plating efficiency assay
Plating efficiency was evaluated as described previously50. Briefly after treatment with varying concentrations of NtBuHA (0–2.5mM), INCL fibroblast cells were harvested and plated at a density of 100 cells/50mm tissue culture dish. Ten days later cells were fixed with 2.5% glutaraldehye and stained with 2.5% crystal violet. Plates were air dried. Crystal violet positive cell colonies were then counted manually and plating efficiency was expressed as number of colonies per 100 cells plated.
Labeling Cells with [35S]-Cysteine
Lymphoblasts were labeled with [35S]-Cysteine as previously described18 with minor modifications. Briefly, labeled cells were treated with 500µM of NtBuHA at every 12 h for 48 h at 37° C. Cells were then washed x2 with ice-cold PBS and centrifuged at 2,250xg at 4 °C for 5 min. The pellet thus obtained was resuspended in PBS and lipid thioesters were extracted as described previously18.
Densitometric analyses of [35S] cysteine-labeled lipid thioester bands in TLCs
Intensities of the identifiable [35S] Cysteine labeled lipid thioester bands in autoradiographs of TLCs were quantified using QuantityOne software (Biorad). Band intensities from at least three different experiments were normalized and expressed as the intensity per mm2 (INT per mm2).
Transmission electron microscopy
Transmission electron microsocopy of INCL lymphoblasts and fibroblasts was performed as previously described26. Briefly, cells were first treated with 250 µM NtBuHA for 3 weeks. Medium was replaced with fresh 250 µM of NtBuHA at every 72 h. After treatment, cells were fixed with 2.5 % glutaraldehyde in sodium phosphate buffer, washed three times with Millonig’s phosphate buffer and stained with 2.5% uranyl acetate. Ultra-thin sections were then prepared using AO Reichert Ultracut ultramicrotome, stained with lead citrate and examined by using Zeiss EM10 CA. The number and size of the granular osmiophilic deposits (GRODs) were determined by using NIH Image J Software.
For brain TEM, cortical tissue sections were prepared from WT, untreated- and NtBuHA-treated Ppt1−/− mice. Cortical tissues (approximately 1mm3) were dissected from those animals, fixed in 2.5% glutaradlehyde in 0.1M sodium cacodylate buffer, pH 7.4 followed by three washing in 0.1 M sodium cacodylate buffer at room temperature. Tissues were then post-fixed with 1% osmium tetroxide, dehydrated by sequential treatment of 50%, 70%, 90% and 100% ethanol and treated with Spurr’s resin/ethanol using variable wattage Pelco BioWave Pro microwave oven (Ted Pella, Inc., Redding, CA.). Cortical tissues were embedded and polymerized in 100% resin for 18 hours at 70°C. Tissue sections (50nm thick) were prepared using Reichert-Jung Ultracut-E ultramicrotome and collected on LuxFilm grids (Ted Pella, Inc.) of 30nm film thickness. The grids were then post-stained with uranyl acetate and lead citrate and examined in a FEI Tecnai G2 transmission electron microscope operating at 80kV.
Detection of apoptosis
To evaluate whether NtBuHA protects against oxidative stress-induced apoptosis in INCL lymphoblasts we first treated the cells with 1mM NtBuHA for 12 h and then incubated with 500 µM H2O2 for 3 h in presence or absence of NtBuHA (1mM). Untreated INCL lymphoblasts (control) were also treated with H2O2 for 3 h. For the detection of apoptosis the cells were stained with annexin V using Apoptosis detection kit (Biovision) and analyzed by FACS (Guava EasyCyte Mini System, Millipore).
TUNEL assay
To determine the level of apoptosis in the brain sections of mice TUNEL assay was performed using TUNEL assay kit (Trevigen) as per manufacturer’s instruction.
Western blot analysis
Western blot analysis was performed as previously described26. Briefly 30 µg of protein were resolved in 4–12% Bis Tris gel (Invitrogen) and electro-transferred in PVDF membrane. The membranes were blocked with 5% non fat dried milk in TBS-T buffer for 1 h at room temperature and were then incubated overnight with primary antibodies at 4° C. After washing three times the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Santacruz Biotechnology) for 1 h at room temperature and developed using enhanced chemiluminescence detection reagents (Pierce). Primary antibodies used in this study: ATF6 (Imagenex), SOD2 & caspase-9 (Upstate), cleaved PARP-1 (BD Pharmingen), catalase, (Abcam), GFAP, Grp78/Grp 94 (Cell Signaling), NeuN (Chemicon) and β-Actin (US Biological). Densitometric quantitations were performed using QuantityOne software (BioRad).
Immunohistochemistry
The NtBuHA-treated and untreated mouse brain tissues were fixed in 3.7% paraformaldehyde, embedded in paraffin and processed for histological analyses. Briefly, after being treated with Xylene and then successively with different concentration of ethanol in water (100% to 0%) tissue sections were incubated overnight in sodium citrate buffer at 95° C. Sections were then blocked with 5% BSA in PBS, probed overnight with anti-GFAP (Cell Signaling) and anti-NeuN (Chemicon). Sections were then washed three times with 1 × PBS, incubated with anti-mouse biotinylated secondary antibody (1:500; Vector Laboratories) for 1 h at room temperature followed by washing for three times with PBS and incubation with ABC complex according to the protocol provided by the manufacturer (Vector Laboratories). Cells were counted using SPOT Advanced Plus software, version 4.7 (Diagnostic Instruments, Inc.).
Immunofluorescence of neurons in various cortical layers
Six months old WT, untreated Ppt1−/− and NtBuHA-treated Ppt1−/− animals were anesthetized and perfused with cold PBS followed by 4% paraformaldehyde. Brains were placed in the same fixative overnight at 4°C and were frozen in 30% sucrose solution in PBS. Frozen sections were permeabilized in 0.4% Triton-X100, blocked in 5% bovine serum albumin and incubated in primary antibodies overnight at 4°C. The primary antibodies used were Cux1 (Santacruz Biotechnology) and Ctip 2 (Abcam). Sections were washed and incubated with Alexafluor conjugated secondary antibodies (Invitrogen) at room temperature for 1 h. Nuclei were stained with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma) and fluorescence was visualized using an LSM-510 inverted confocal microscope (Zeiss). The captured images were processed using the LSM Image Browser, version 4.2 (Zeiss).
Cresyl violet staining
Five micron thick brain sagittal sections from 6 months old WT, untreated- or NtBuHA- treated Ppt1−/− mice were deparaffinized, hydrated and stained with 0.5% cresyl violet solution. At least 4 comparable sections from each brain were used for measurement of cortical thickness and a similar area (secondary visual cortex mediolateral) for all the sections was used.
Motor coordination performance test
Motor coordination of the untreated- and NtBuHA-treated Ppt1−/− mice was assessed using rotarod (UGO Basile, Italy) performance test35 at three different speeds (4, 8 and 12 rpm). In all these speeds the direction of rotation was reversed every 15 seconds. Animals were trained twice at all speed settings for 1 min each for 3 days. Animals were then given a rest for 1 min between two trials. rotarod experiments were performed for 60 seconds on day 4 and the amount of time a mouse was on the rotarod before falling from the rotating rod was recorded. Video recordings of the test were also made. These tests were performed by 2 independent investigators each of whom performed at least 3 separate experiments following identical protocol and the results were compared to determine replicability.
Rotarod endurance test
For testing endurance on rotarod we used 6-month old WT (n=6), untreated Ppt1−/− (n=8) and NtBuHA-treated Ppt1−/− (n=7) mice. The mice were trained on rotarod at 12rpm (300 seconds/day) for 3 consecutive days before performing the test. The direction of rotation was reversed every 15 seconds and the animals were allowed to stay on the rotating rod for up to 300 seconds. After 3 days of training, the mice were designated as group A (WT), group B (untreated-KO) and group C (NtBuHA-treated). The recordings were conducted on the 4th day by an operator who was unaware as to the identity of the groups. For all experiments, the training and the rotarod test were conducted in the afternoon between 3–5 PM.
Open field test
The exploratory behavior of untreated and NtBuHA-treated Ppt1−/− mice was evaluated by open field test as previously described36. These tests were performed by 2 independent investigators each of whom carried the identical protocol in at least 3 separate experiments and the results were compared to determine if they were replicable.
Detection of NtBuHA in brain tissues by mass spectrometry
Targeted detection of NtBuHA was performed using multiple reaction-monitoring mass spectrometry. For metabolite extraction, 3 volumes 50% chilled methanol containing internal standards was added to the tissue sections in the Magna-Lyser tubes containing ceramic beads (Roche, USA). The tissue samples were homogenized by spinning the tubes 3 times with 10–30 sec pulse in the Magna Lyser homogenizer (Roche, USA) at 7000rpm. The supernatant was transferred to a fresh tube and an equal volume of chilled 100% acetonitrile was added, vortexed and incubated on ice for 15 minutes. The tubes were centrifuged at 14,000rpm at 4°C for 15 minutes. The supernatant was transferred to a fresh tube and dried under vacuum and stored at −80°C until MS analysis. The samples were resolved on an Acquity UPLC BEH C18 1.7 µm, 2.1 × 100 mm column online with a triple quadruple mass spectrometer (Xevo-TQ, Waters Corporation, USA) operating in the multiple reactions monitoring (MRM) mode as previously described30.
Determination of lifespan in untreated and NtBuHA-treated animals
Both the untreated (n=110) and NtBuHA-treated (n=80) Ppt1−/− mice were used to evaluate longevity. When the mice were either dead or were unable to reach for food and water they were sacrificed according to the protocol. Kaplan–Meier analysis was performed to measure cumulative survival and log rank test was used to determine whether the median lifespan in NtBuHA treated animals was significantly different from that of untreated counterparts.
Statistical Analysis
All quantitative results including motor function and exploratory behavioral analyses were performed by investigators who were unaware of the genotype or the treatment of the respective mice. For in vivo experiments the ‘n’ numbers reported are biological replicates. To confirm the interassay reproducibility of the results, at least three independent experiments were performed. In some of the experiments, due to the relatively small sample sizes, two-sample permutation t-tests were conducted to compare the independent groups and p<0.05 was considered significant. All data are expressed as the mean of at least three independent experiments ± SD, and are also summarized in the legend of each figure with Box-Whisker plots, along with p-values for group comparisons. The results of lifespan studies were compared by Kaplan-Meier curves and log rank test.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank Dr. S. L. Hofmann for the generous gift of Ppt1−/− mice and for a sample of recombinant human PPT1 enzyme used as a positive control. Dr. Mark Sands provided a pair of C57 congenic Ppt1−/− mice used for establishing our colony at the NIH. We thank Drs. S.W. Levin, J.Y. Chou and I. Owens for critical review of the manuscript and helpful suggestions. We also thank Ms. A. Bouchelion and Mr. A. Chen for editorial assistance. We thank Mr. Louis Dye, Microscopy and Imaging Core facility, Eunice Kennedy-Shriver NICHD, for his expert assistance in performing the TEM of cultured lymphoblasts as well as the mouse brain tissues. We are grateful to Dr. H.-S. Jun for helping us in the FACS analysis. The mass spectrometric analyses for the detection of NtBuHA in brain tissues were performed in the laboratory of Dr. A. K. Cheema at Georgetown University School of Medicine. This research was supported in whole by the Intramural Research Program of the Eunice Kennedy Shriver NICHD, NIH.
Footnotes
AUTHOR CONTRIBUTIONS
C.S. and GC contributed in designing and performing the majority of the experiments. S.P. performed genotyping of the mice, densitometric analyses, motor function and behavior testing and analysis of the data. Z.Z. contributed in designing some of the experiments, performed data analysis and prepared the illustrations. A.L. provided statistical analyses of the data. A.B.M. conceived the project, designed some of the experiments and wrote major portions of the manuscript. All authors participated in writing, editing as well as in the preparation and approval of the manuscript before submission.
COMPETING FINANCIAL INTERESTS
The authors declare no financial interests. The NIH (DHHS) has filed an application for US patent protection of the results presented in this manuscript.
REFERENCES
- 1.Cox TM, Cachón-González MB. The cellular pathology of lysosomal diseases. J. Pathol. 2012;226:241–254. doi: 10.1002/path.3021. [DOI] [PubMed] [Google Scholar]
- 2.Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 2005;6:713–725. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- 3.Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim. Biophys. Acta. 2013;1832:1807–1826. doi: 10.1016/j.bbadis.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 4.Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum.Mutat. 2012;33:42–63. doi: 10.1002/humu.21624. [DOI] [PubMed] [Google Scholar]
- 5.Haltia M, Goebel HH. The neuronal ceroid lipofuscinoses: A historical introduction. Biochim Biophys Acta. 2013;1832:1795–1800. doi: 10.1016/j.bbadis.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Wong AM, Rahim AA, Waddington SN, Cooper JD. Current therapies for the soluble lysosomal forms of neuronal ceroid lipofuscinosis. Biochem. Soc. Trans. 2010;38:1484–1488. doi: 10.1042/BST0381484. [DOI] [PubMed] [Google Scholar]
- 7.Camp LA, Hofmann SL. Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J. Biol. Chem. 1993;268:22566–22574. [PubMed] [Google Scholar]
- 8.Camp LA, Verkruyse LA, Afendis SJ, Slaughter CA, Hofmann SL. Molecular cloning and expression of palmitoyl-protein thioesterase. J. Biol. Chem. 1994;269:23212–23219. [PubMed] [Google Scholar]
- 9.Verkruyse LA, Hofmann SL. Lysosomal targeting of palmitoyl protein thioesterase. J. Biol. Chem. 1996;271:15831–15836. doi: 10.1074/jbc.271.26.15831. [DOI] [PubMed] [Google Scholar]
- 10.Hellsten E, Vesa J, Olkkonen VM, Jalanko A, Peltonen L. Human palmitoyl protein thioesterase: evidence for lysosomal targeting of the enzyme and disturbed cellular routing in infantile neuronal ceroid lipofuscinosis. EMBO J. 1996;15:5240–5245. [PMC free article] [PubMed] [Google Scholar]
- 11.Jarvela I. Infantile form of neuronal ceroid lipofuscinosis (CLN1) maps to the short arm of chromosome 1. Genomics. 1991;9:170–173. doi: 10.1016/0888-7543(91)90235-7. [DOI] [PubMed] [Google Scholar]
- 12.Vesa J, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann SL, Peltonen L. The neuronal ceroid Lipofuscinosis. In: Scriver CR, Beud AL, Sly WS, Valle D, editors. The metabolic and Molecular Basis of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. pp. 3877–3894. [Google Scholar]
- 14.Smotrys JE, Linder ME. Palmitoylation of intracellular signaling proteins: regulation and function. Annu. Rev. Biochem. 2004;73:559–587. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- 15.Resh MD. Palmitoylation of ligands receptors and intracellular signaling molecules. Sci STKE. 2006;359:re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 16.Salaun C, Greaves J, Chamberlain LH. The intracellular dynamic of protein palmitoylation. J. Cell.Biol. 2010;191:1229–1238. doi: 10.1083/jcb.201008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwanaga T, Tsutsumi R, Noritake J, Fukata Y, Fukata M. Dynamic protein palmitoylation in cellular signaling. Prog. Lipid Res. 2009;48:117–127. doi: 10.1016/j.plipres.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Lu JY, Verkruyse LA, Hofmann SL. Lipid thioesters derived from acylated proteins accumulate in infantile neuronal ceroid lipofuscinosis: Correction of the defect in lymphoblasts by recombinant palmitoyl-protein thioesterase. Proc. Natl. Acad. Sci. USA. 1996;93:10046–10050. doi: 10.1073/pnas.93.19.10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jocelyn PC. Biochemistry of the SH groups. New York: Academic Press; 1972. pp. 63–93. [Google Scholar]
- 20.Gross P. Biologic activity of hydroxylamine: a review. Crit Rev Toxicol. 1985;14:87–99. doi: 10.3109/10408448509023765. [DOI] [PubMed] [Google Scholar]
- 21.Kirby AJ, et al. Hydroxylamine as an oxygen Nucleophile. Structure and reactivity of ammonia oxide. J. Am. Chem. Soc. 2006;128:12374–12375. doi: 10.1021/ja065147q. [DOI] [PubMed] [Google Scholar]
- 22.Drisdel RC, Alexander JK, Sayeed A, Green WN. Assays of protein palmitoylation. Methods. 2006;40:127–134. doi: 10.1016/j.ymeth.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Mossman T. Rapid colorimetric assay for cellular growth and survival. J. Immunol. Method. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 24.Atamna H, Robinson C, Ingersoli R, Elliott H, Aimes BN, et al. N-t-butyl hydroxylamine is an antioxidant that reverses age-related changes in mitochondria in vivo and in vitro. FASEB J. 2001;15:2196–2204. doi: 10.1096/fj.01-0134com. [DOI] [PubMed] [Google Scholar]
- 25.Gupta P, et al. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc. Natl. Acad. Sci. USA. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, et al. Palmitoyl-protein thioesterase-1 deficiency mediates the activation of the unfolded protein response and neuronal apoptosis in INCL. Hum Mol Genet. 2006;15:337–346. doi: 10.1093/hmg/ddi451. [DOI] [PubMed] [Google Scholar]
- 27.Kim SJ, Zhang Z, Lee YC, Mukherjee AB. Palmitoyl-protein thioesterase-1 deficiency leads to the activation of caspase-9 and contributes to rapid neurodegeneration in INCL. Hum. Mol. Genet. 2006;15:1580–1586. doi: 10.1093/hmg/ddl078. [DOI] [PubMed] [Google Scholar]
- 28.Wei H, et al. ER- and oxidative-stresses are common mediators of apoptosis in both neuro-degenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum.Mol.Genet. 2008;17:469–477. doi: 10.1093/hmg/ddm324. [DOI] [PubMed] [Google Scholar]
- 29.Riikonen R, Vanhanen SL, Tyynela J, Santavuori P, Turpeinen U. CSF insulin-like growth factor-1 in infantile neuronal ceroid lipofuscinosis. Neurology. 2000;54:1828–1832. doi: 10.1212/wnl.54.9.1828. [DOI] [PubMed] [Google Scholar]
- 30.Kaur P, et al. Metabolomic profiling for biomarker discovery in pancreatic cancer. Internatl. J. Mass Spectr. 2012;310:44–51. [Google Scholar]
- 31.Munasinghe J, Zhang Z, Kong E, Heffer A, Mukherjee AB. Evaluation of neurodegeneration in a mouse model of infantile Batten disease by magnetic resonance imaging and magnetic resonance spectroscopy. Neurodegener. Dis. 2012;9:159–169. doi: 10.1159/000334838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, et al. Palmitoyl-protein thioesterase gene expression in the developing mouse brain and retina: Implications for early loss of vision in infantile neuronal ceroid lipofuscinosis. Gene. 1999;231:203–211. doi: 10.1016/s0378-1119(99)00050-5. [DOI] [PubMed] [Google Scholar]
- 33.Isosomppi J, et al. Developmental expression of palmitoyl protein thioesterase in normal mice. Brain Res. Dev. Brain Res. 1999;118:1–11. doi: 10.1016/s0165-3806(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 34.Macauley SL, Pekny M, Sands MS. The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. J. Neurosci. 2011;31:15575–15585. doi: 10.1523/JNEUROSCI.3579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamm RJ, Pike BR, O’Dell DM, Lyeth V, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J. Neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- 36.Crawley JN. Exploratory behavior model of anxiety in mice. Neuroscience & Biobehavioral Reviews. 1985;9:37–44. doi: 10.1016/0149-7634(85)90030-2. [DOI] [PubMed] [Google Scholar]
- 37.el-Husseini Ael-D, Bredt DS. Protein palmitoylation: a regulator of neuronal development and function. Nat Rev Neurosci. 2002;3:791–802. doi: 10.1038/nrn940. [DOI] [PubMed] [Google Scholar]
- 38.Duncan JA, Gilman AG. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS) J. Biol. Chem. 1998;273:15830–15837. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 39.Tomatis VM, Trenchi A, Gomez GA, Daniotti JL. Acyl-protein thioesterase 2 catalyzes the decimation of peripheral membrane-associated GAP-43. PLoS One. 2010;5:e15045. doi: 10.1371/journal.pone.0015045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greaves J, et al. Palmitoylation-induced aggregation of cysteine-string protein mutants that cause neuronal ceroid lipofuscinosis. J. Biol. Chem. 2012 Aug 19; doi: 10.1074/jbc.M112.389098. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Platt FM, Lachmann RH. Treating lysosomal storage disorders: current practice and future prospects. Biochim Biophys Acta. 2009;1793:737–745. doi: 10.1016/j.bbamcr.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 42.Kohan R, et al. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr. Pharm.Biotechnol. 2011;12:867–883. doi: 10.2174/138920111795542633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu JY, Hu J, Hofmann SL. Human recombinant palmitoyl-protein thioesterase-1 (PPT1) for preclinical evaluation of enzyme replacement therapy for infantile neuronal ceroid lipofuscinosis. Mol.Genet. Metab. 2010;99:374–378. doi: 10.1016/j.ymgme.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu J, et al. Intravenous high –dose enzyme replacement therapy with recombinant palmitoyl-protein thioesterase reduces visceral lysosomal storage and modestly prolongs survival in a preclinical mouse model of infantile neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 2012;107:213–221. doi: 10.1016/j.ymgme.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffey MA, et al. CNS-directed AAV2-mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol. Ther. 2006;13:538–547. doi: 10.1016/j.ymthe.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 46.Chen YH, Chang M, Davidson BL. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat. Med. 2009;15:1215–1218. doi: 10.1038/nm.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lonnqvist T, et al. Hematopoietic stem cell transplantation in infantile neuronal ceroid lipofuscinosis. Neurology. 2001;57:1411–1416. doi: 10.1212/wnl.57.8.1411. [DOI] [PubMed] [Google Scholar]
- 48.Tamaki SJ, et al. Neuroprotection of host cells by human central nervous system stem cells in a mouse model of infantile neuronal ceroid lipofuscinosis. Cell Stem Cell. 2009;5:310–319. doi: 10.1016/j.stem.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 49.Srikanth M, Kessler JA. Nanotechnology-novel therapeutics for CNS disorders. Nat.Rev. Neurol. 2012;8:307–318. doi: 10.1038/nrneurol.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franken NA, Rodermond HM, Stap J, Haverman J, van Bree C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.