

Abstract
A wide range of central nervous system (CNS) disorders, particularly those related to sleep, are associated with the abnormal function of orexin (OX) receptors. Several orexin receptor antagonists have been reported in recent years, but currently there are no imaging tools to probe the density and function of orexin receptors in vivo. To date there are no published data on the pharmacokinetics (PK) and accumulation of some lead orexin receptor antagonists. Evaluation of CNS pharmacokinetics in the pursuit of positron emission tomography (PET) radiotracer development could be used to elucidate the association of orexin receptors with diseases and to facilitate the drug discovery and development. To this end, we designed and evaluated carbon-11 labeled compounds based on diazepane orexin receptor antagonists previously described. One of the synthesized compounds, [11C]CW4 showed high brain uptake in rats and further evaluated in non-human primate (NHP) using PET-MR imaging. PET scans performed in a baboon showed appropriate early brain uptake for consideration as a radiotracer. However, [11C]CW4 exhibited fast kinetics and high nonspecific binding, as determined after co-administration of [11C]CW4 and unlabeled CW4. These properties indicate that [11C]CW4 has excellent brain penetrance and could be used as a lead compound for developing new CNS-penetrant PET imaging probes of orexin receptors.
Keywords: orexin, PET radiotracers, imaging, hypocretin, carbon-11
Introductions
Orexin A and orexin B (hypocretins), first discovered in 1998,[1] are neuropeptides essential for a number of hypothalamic functions. Two receptor subtypes, termed orexin-1 (OX1) and orexin-2 (OX2), have since been identified.[2] The orexins play an important role in regulation of the sleep-wake cycle, modulating feeding behavior, and energy homeostasis.[3] Distribution studies in rat brain using in situ hybridization and immunohistochemistry (IHC) have shown that OX1 receptors are most abundantly expressed in the locus coeruleus while OX2 receptors are expressed in regions controlling arousal, such as tuberomammillary nucleus, which is an important site for the regulation of sleep and wakefulness.[4]
Both selective and non-selective orexin receptor antagonists have been reported. SB-334867-A (1-(2-methylbenzoxazol- 6-yl)- 3-[1,5]naphthyridin- 4-yl urea) was the first non-peptide OX1 receptor antagonist reported and was used for in vivo studies of orexin-A physiological effects.[5] Cp-5 ((S)-1-(6,7-Dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-3,3-dimethyl-2-[(pyridin-4-ylmethyl)-amino]-butan-1-one) was the first non-peptide selective OX2 receptor antagonist.[6] Almorexantis a dual (OX1R/OX2R) orexin receptor antagonist, which was shown to significantly decrease wakefulness in rats, dogs and humans.[7] There are two antagonists current in clinical trials for the treatment of insomnia. The first one is Suvorexant (MK-4305), a dual orexin receptor antagonist in development by Merck & Co, and it has completed three Phase III trials.[8, 9] SB-649868, the other one in a Phase II clinical trial, is a dual orexin receptor antagonist in development by GlaxoSmithKline.[10-12]
A selective OX2R antagonist N-ethyl-2-[(6-methoxy-pyridin-3-yl)-(toluene-2-sulphonyl)-amino]-N-pyridin- 3-ylmethyl-acetamide (EMPA)was labeled with tritium (3H), which measured ex vivo and in vitro binding with rat brain sections.[13] However to date, the in vivo selectivity, distribution and involvement of individual receptors in the pathophysiology of orexin-mediated disorders are not available. The regional abundance of [3H]EMPA (94.3 Ci/mmol) specific binding was measured by quantitative autoradiography in the range of 40-140 fmol·mg−1 protein. A high density of OX2R was observed in the CA3 region of the hippocampus (140 fmol·mg−1 protein), cortical layer 6, tuberomammillary nucleus, induseumgriseum and nucleus accumbens. Development of an imaging tool that permits detection and quantification of orexin receptors in vivo will accelerate research in this domain to deepen our understanding the function of orexins in the regulation of sleeping, food uptake and drug addiction. PET imaging would be an excellent tool for this purpose because radiotracers developed and tested in animals can be translated rapidly for human imaging. The autoradiography results with [3H]EMPA further encouraged us to label orexin antagonists with radioisotopes (e.g., carbon-11 and fluorine-18). Currently, there are no PET tracers available for imaging orexin receptors. Radiosynthesis of [11C]BBAC and [11C]BBPC have been reported recently[14], but PET scans performed in a rhesus monkey did not show tracer retention or appropriate brain uptake (less than 0.0001%ID/cc in the brain).
Herein, our goals were to develop CNS-penetrant PET imaging probes of orexin 2 receptors and evaluate the uptake and distribution of these probes in the rodent and the non-human primate brain; additionally, these information can help us to understand if these radiolabeled compounds can be used as CNS drugs or only for the peripheral organs To accomplish this, we incorporated the positron emitting isotope carbon-11 (t1/2=20.4 min) into labeling precursors using [11C]CH3I. Herein we describe the design and synthesis of [11C]CW3, [11C]CW4 and [11C]CW6, which are N,N-disubstituted-1,4-diazepanesfor OX2R PET imaging. Results from our imaging studies indicate that [11C]CW4 has good BBB penetration in rats and non-human primates and it would be a lead compound for further radiotracer development.
Results and discussions
A new class of dual orexin receptor antagonists based on a 1,4-diazepane central scaffold were identified and some of them are reported as potent, brain penetrating dual orexin receptor antagonists able to block orexin signaling both in vitro and in vivo.[15]However, there is no evidence to show which the regions has maximal biological effects with these compounds. Hence, three N,N-disubstituted-1,4-diazepanes were chosen for our study to investigate the potentials to be used as a PET tracer and to investigate the PK and distributions in the brain. According to the literature,[15] CNS penetration could be predicted based on their physical chemical properties. All three compounds showed preferred physicochemical properties for CNS penetration (molecular weight, calculated or measured log P and Topological polar surface area (tPSA)) as well as favorable Ki values for OX2R (see Table 1). Preliminary analysis of physicochemical properties indicates that CW3, CW4 and CW6 could be used as imaging agents for OX2R;[16] albeit the affinities may be slight too low given the Bmax of OX2 (measured in rats) up to 140 fmol/mg.[10] To be a PET imaging probe for OX2 receptor, the binding potential (BP, which equals the product of receptor density (Bmax) and affinity (1/Kd or 1/Ki)) is usually at least 10 in the regions with high receptor density to obtain images for quantification.[17] The three compounds we chose to pursue have BP values (we assumed that fmol/mg equals nM) in the range of 3-30, allowing us to test the potential of these compounds use as quantitative PET imaging agents for the OX2 receptor.
Table 1.
Physiochemical properties and reported activities of orexin antagonists evaluated in this study.
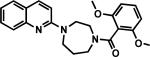
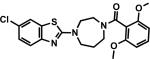
Synthesis of nonradioactive standards CW3, CW4, CW6and their O-desmethyl precursors were achieved in good yield as shown in Scheme 1.[16] Briefly, 1, 4-diazepane was coupled with 2-chloroquinoline or 2, 6-dichlorobenzo[d]thiazole to form the monosubstituted-1, 4-diazepane intermediates. The intermediates were then reacted with 2, 6-dimethoxybenzoic acid or 2-methoxybenzoic acid to yield CW3, CW4 and CW6 as reference standards for labeling. Radiolabeling precursors 4, 8 and 10were synthesized through similar coupling reactions with 2-hydroxy-6-methoxybenzoic acid or 2-hydroxybenzoic acid.
Scheme 1.

Synthesis of orexin antagonists and their labeling precursors. Reagents and conditions: (a) Na2CO3, DMSO, 120 °C, 10 h; (b) 2-hydroxy-6-methoxybenzoic acid or 2,6-dimethoxybenzoic acid, EDC·HCl, DCM, rt, 1 h; two steps yields: 24% for 4, 30% for CW3, 27% for 8, 33% for CW6; (c) 2-hydroxybenzoic acid or 2-methoxybenzoic acid, EDC·HCl, DCM, rt, 1 h; two steps yield: 34% for 10, 40% for CW4.
The synthesis of the target carbon-11 labeled compounds were accomplished using O-desmethyl precursors in DMSO with [11C]methyl iodine ([11C]CH3I) as outlined in Scheme 2. [11C]CH3I was trapped in a TRACERlab FX-M synthesizer reactor (General Electric) preloaded with a solution of precursor (4, 8 or 10) (1.0 mg) and Cs2CO3 (6.0 mg) in dry DMSO (300 μL). The solution was stirred at 50 °C for 3 min and water (1.2 mL) was added. The reaction mixture was purified by reverse phase semi-preparative HPLC and the desired fraction was collected. The final product was reformulated by loading onto a solid-phase exchange (SPE) C-18 cartridge rinsing with H2O (5 mL), eluting with EtOH (1 mL), and diluting with saline (0.9%, 9 mL) to formulate the product as isotonic solution. The chemical and radiochemical purity of the final product was tested by analytical HPLC. The identity of the product was confirmed by analytical HPLC with additional co-injection of reference standard. The average time required for the synthesis from end of cyclotron bombardment to end of synthesis was 40 min (CW3), 35 min (CW4) and 35 min (CW6). The radiochemical yield was 7.5% ([11C]CW3), 16-21% ([11C]CW4) and 15.9% ([11C]CW6) (decay-uncorrected to trapped [11C]CH3I). The specific activity was 0.6 Ci/μmol ([11C]CW3), 1.5 Ci/μmol ([11C]CW4) and 1.8 Ci/μmol ([11C]CW6) (end of synthesis, EOS).Chemical and radiochemical purities were ≥ 95 % for all radiotracers.
Scheme 2.

Radiosynthesis of [11C]CW3, [11C]CW4 and [11C]CW6. Yields are reported as non-decay corrected to trapped [11C]CH3I.
Using PET-CT in rodents, we determined that [11C]CW3 and [11C]CW6 exhibited very poor BBB penetration and low brain uptake over the scanning time (60 min) when the radiotracers (0.9-1.1mCi, i.v.) were administered, Figure 2. A concentration of less than 0.2% and 0.1% of the injected dose per cubic centimeter (%ID/cc) was distributed in the brain tissue for [11C]CW3 and [11C]CW6, respectively. The other carbon-11 labeled compound, [11C]CW4, showed higher brain uptake, up to 0.4% ID/cc (%ID/cc, is a measure of the concentration of a radiotracer in a defined region divided by the injected dose) distributed throughout brain tissue, and the injected dose per cubic centimeter was above 0.1% during the imaging session.
Figure 2.

Whole-brain time-activity curves generated from rodent PET imaging data for [11C]CW3, [11C]CW4 and [11C]CW6. (1.0 mCi/ rats, 50 minute scan with isoflurane)
To further investigate the pharmacokinetics and brain distribution of [11C]CW4, we used PET-MRI imaging in a Papio anubis baboon. [11C]CW4 brain-to-plasma ratios were found to increase after tracer administration; 1.4 after 10 min, 1.8 after 20 min, and 2.0, the maximum, after 30 min indicating binding and accumulation in brain tissue. Excellent brain penetrance of [11C]CW4 was further demonstrated by robust accumulation of radioactivity in PET images summed 30-80 min post injection (Figure 3), although little difference was observed in regional brain uptake of [11C]CW4. According to the literature, the hippocampus, thalamus and cortex are brain regions with high OX2R expression.[13] Consistent with these reported in vitro and ex vivo data, our PET imaging results showed subtle increases in radioactivity in regions including hippocampus, thalamus, frontal cortex and parietal cortex. However overall, radioactivity levels were nearly as high throughout the brain, including other regions such as pons, cerebellum, putamen and occipital cortex.
Figure 3.

PET-MRI Imaging (baboon brain). Summed PET images (30-80 min) superimposed with MEMPRAGE-MRI of the brain from the same baboon, following injection of [11C]CW4 (4.5 mCi/ baboon).
Radiolabeled compounds show different types of binding, specific binding (bind to the target receptor, saturable, reversible and can be inhibited by their unlabeled form.); nonspecific binding (adsorption to tissue, non-saturable). They are different from selective binding (radiolabeled compounds only bind to very limited types of receptors) and non-selective binding (radiolabeled compounds bind to several types of receptors). To test for specific binding with [11C]CW4, a second imaging study was conducted with the co-administration of [11C]CW4 and unlabeled CW4 (1 mg/kg, i.v.). We expected that the unlabeled CW4 would saturate the OX2R in the baboon brain to decrease the binding of [11C]CW4 to its target. However, no changes in the radioligand distribution or pharmacokinetics were observed in any brain region (Figure 4), suggesting that CW4 exhibits high non-specific binding.
Figure 4.

Time−activity curves for brain regions of interest in baseline [11C]CW4 (4.5 mCi/ baboon, 80 minute scan with isoflurane) study and co-administration study with 1 mg/kg CW4.The lack of change between the two scans indicates that accumulation is dominated by non-displaceable (non-specific) binding.
These studies demonstrate that [11C]CW4, a carbon-11 radiolabeled OX2R antagonist, has high brain permeability dominated by non-specific binding in rodents and non-human primates (NHPs) whereas the other carbon-11 labeled [11C]CW3 and [11C]CW6 showed limited brain penetration in rats. Non-specific uptake is common in radiotracer development even with molecules predicted to have proper physiochemical properties. While the density of OX2 receptors in rat brain in quite reasonable for PET (Bmax= 40-140 pmol·mg−1 protein) [13], and OX2-binding compound with greater binding affinity than CW4 is needed for further imaging. The development of higher affinity derivatives of CW4 may be a means to test whether specific binding is indeed limited by Bmax in the NHP brain. We hope this can be accomplished without compromising the excellent BBB penetration of CW4. Following imaging studies, all of the animals survived and showed no change in homecage behavior, which suggest the radiotracers and the ‘blocking doses’ have no apparent toxicity or any pharmacological effects. These results indicate that there is a potential to develop either selective (OX1R or OX2R) or non-selective (dual OX1R and OX2R) brain-penetrant orexin receptor imaging agents based on N, N-disubstituted-1, 4-diazepanes, which may facilitate the development of new orexin antagonists for treatment of neurological disorders. To this end, ongoing work is aimed at optimizing lead compound CW4 to improve the binding affinity and specificity. The results also indicate that CW3 and CW6 may not be useful as imaging probes in the brain due to low brain uptake in our study, however, they may still can used as CNS drugs. In this paper, PET imaging results with [11C]CW4 showed the PK in baboon brain and would a valuable guide for the diazepane-based orexin receptor antagonists development when the brain as the target.
General methods and materials
All reagents and solvents were of ACS-grade purity or higher and used without further purification. NMR data were recorded on a Varian 500 MHz magnet and were reported in ppm units downfield from trimethylsilane. Analytical separation was conducted on an Agilent 1100 series HPLC fitted with a diode-array detector, quaternary pump, vacuum degasser, and autosampler. Mass spectrometry data were recorded on an Agilent 6310 ion trap mass spectrometer (ESI source) connected to an Agilent 1200 series HPLC with quaternary pump, vacuum degasser, diode-array detector, and autosampler. 11CO2 (1.2 Ci) was obtained via the 14N(p, α)11C reaction on nitrogen with 2.5% oxygen, with 11 MeV protons (Siemens Eclipse cyclotron), and trapped on molecular sieves in a TRACERlab FX-MeI synthesizer (General Electric). 11CH4 was obtained by the reduction of 11CO2 in the presence of Ni/hydrogen at 350 °C and recirculated through an oven containing I2 to produce 11CH3I via a radical reaction.
All animal studies were carried out at Massachusetts General Hospital (PHS Assurance of Compliance No. A3596-01). The Subcommittee on Research Animal Care (SRAC) serves as the Institutional Animal Care and Use Committee (IACUC) for the Massachusetts General Hospital (MGH). SRAC reviewed and approved all procedures detailed in this paper.
MR-PET imaging was performed in anesthetized (ketamine, isoflurane) baboon (papioanubis) to minimize discomfort. Highly-trained animal technicians monitored animal safety throughout all procedures and veterinary staff were responsible for daily care. All animals were socially housed in cages appropriate for the physical and behavioral health of the individual animal. Animals were fed thrice per diem, with additional nutritional supplements provided as prescribed by the attending veterinarian. Audio, video and tactile enrichment was provided on a daily basis to promote psychological well-being. No non-human primates were euthanized to accomplish the research presented.
PET/CT imaging was performed in anesthetized (isoflurane) rats (Sprague Dawley) to minimize discomfort. Highly-trained animal technicians monitored animal safety throughout all procedures and veterinary staff were responsible for daily care. All animals were socially housed in cages appropriate for the physical and behavioral health of the individual animal. Animals were given unlimited access to food and water, with additional nutritional supplements provided as prescribed by the attending veterinary staff. Animals were euthanized at the end of the study using sodium pentobarbital (200 mg/kg, IP).
LogD Determination
An aliquot (~50 μL) of the formulated radiotracer was added to a test tube containing 2.5 mL of octanol and 2.5 mL of phosphate buffer solution (pH 7.4).The test tube was mixed by vortex for 2 min and then centrifuged for 2 min to fully separate the aqueous and organic phase. A sample taken from the octanol layer (0.1 mL) and the aqueous layer (1.0 mL) was saved for radioactivity measurement. An additional aliquot of the octanol layer (2.0 mL) was carefully transferred to a new test tube containing 0.5 mL of octanol and 2.5 mL of phosphate buffer solution (pH 7.4). The previous procedure (vortex mixing, centrifugation, sampling, and transfer to the next test tube) was repeated until six sets of aliquot samples had been prepared. The radioactivity of each sample was measured in a well counter (Perkin-Elmer, Waltham, MA).The log D of each set of samples was derived by the following equation: log D = log (decay-corrected radioactivity in octanol sample x 10/decay-corrected radioactivity in phosphate buffer sample).
Radiosynthesis of [11C]CW3, [11C]CW4 and [11C]CW6
11CH3I (300-400 mCi) was trapped in a TRACERlab FX-M synthesizer reactor (General Electric) preloaded with a solution of precursors (4, 8 or 10) (1.0 mg) and Cs2CO3 (6.0 mg) in dry DMSO (300 μL). The solution was stirred at 50 °C for 3 min and water (1.2 mL) was added. The reaction mixture was purified by reverse phase semi-preparative HPLC and the desired fraction was collected. The final product was reformulated by loading onto a solid-phase exchange (SPE) C-18 cartridge, rinsing with H2O (5 mL), eluting with EtOH (1 mL), and diluting with saline (0.9%, 9 mL). The chemical and radiochemical purity of the final product was tested by analytical HPLC. The identity of the product was confirmed by analytical HPLC with additional co-injection of reference standards.
For [11C]CW3
Reverse phase semi-preparative HPLC condition: Phenomenex Gemini NX-C18, 250 mm x 10 mm, 5 μm, 4.0 mL/min, 65% 0.1M AMF/ 35% CH3CN. Analytical HPLC condition: Agilent Eclipse XDB-C18, 150 mm x 4.6 mm, 1.5 mL/min, 40% 0.1M AMF/ 60% CH3CN. The average time required for the synthesis from end of cyclotron bombardment to end of synthesis was 40 min. The radiochemical yield was 7.5% (non-decay corrected relative to trapped [11C]CH3I). Chemical and radiochemical purities were ≥ 95 % with a specific activity 0.6 Ci/μmol (EOS).
For [11C]CW4
Reverse phase semi-preparative HPLC condition: Phenomenex Luna 5u C8(2), 250 mm × 10 mm, 5 μm, 4.0 mL/min, 45% H2O + TFA (0.1% v/v)/ 55% CH3CN + TFA (0.1% v/v). Analytical HPLC condition: Agilent Eclipse XDB-C18, 150 mm × 4.6 mm, 2.0 mL/min, 30% H2O + TFA (0.1% v/v)/ 70% CH3CN + TFA (0.1% v/v). The average time required for the synthesis from end of cyclotron bombardment to end of synthesis was 35 min. The average radiochemical yield was 16-21% (non-decay corrected relative to trapped [11C]CH3I; n = 4). Chemical and radiochemical purities were ≥ 95 % with a specific activity 1.5 ± 0.2 Ci/μmol (EOS).
For [11C]CW6
Reverse phase semi-preparative HPLC condition: Phenomenex Luna 5u C8 (2), 250 mm × 10 mm, 5 μm, 4.0 mL/min, 45% H2O + TFA (0.1% v/v)/ 55% CH3CN + TFA (0.1% v/v). Analytical HPLC condition: Agilent Eclipse XDB-C18, 150 mm × 4.6 mm, 2.0 mL/min, 30% H2O + TFA (0.1% v/v)/ 70% CH3CN + TFA (0.1% v/v). The time required for the synthesis from end of cyclotron bombardment to end of synthesis was 35 min. The radiochemical yield was 15.9% (non-decay corrected relative to trapped [11C]CH3I). Chemical and radiochemical purities were ≥ 95 % with a specific activity 1.8 Ci/μmol (EOS).
Rodent PET/CT Acquisition and Post Processing
Male Sprague-Dawley rats were utilized in pairs, anesthetized with inhalational isoflurane (Forane) at 3% in a carrier of 2 L/min medical oxygen and maintained at 2% isoflurane for the duration of the scan. The rats were arranged head-to-head in a Triumph TrimodalityPET/CT/SPECT scanner (Gamma Medica, Northridge, CA). Rats were injected standard references or vehicle via a lateral tail vein catheterization at the start of PET acquisition. Dynamic PET acquisition lasted for 60 min and was followed by computed tomography (CT) for anatomic coregistration. PET data were reconstructed using a 3D-MLEM method resulting in a full width at half-maximum resolution of 1 mm. Reconstructed images were exported from the scanner in DICOM format along with an anatomic CT for rodent studies. These files were imported to PMOD (PMOD Technologies, Ltd.) and manually coregistered using six degrees of freedom.
Rodent PET/CT Image Analysis
Volumes of interest (VOIs) were drawn manually as spheres in brain regions guided by high resolution CT structural images and summed PET data, with a radius no less than 1mm to minimize partial volume effects. Time-activity curves (TACs) were exported in terms of decay corrected activity per unit volume at specified time points with gradually increasing intervals. The TACs were expressed as percent injected dose per unit volume for analysis.
Baboon PET/MR Acquisition and Post Processing
A female Papio Anubis baboon, deprived of food for 12 h prior to the study, was administered intramuscular ketamine (10 mg/kg) and intubated. For maintenance of anesthesia throughout the study, the baboon was provided 1%-4% isoflurane (Forane) in a mixture of medical oxygen and nitrogen. The baboon was catheterized antecubitally for radiotracer injection and a radial arterial line was placed for metabolite analysis. MR-PET images were acquired in a BiographmMR scanner (Siemens, Munich, Germany), and PET compatible 8-channel coil arrays for non-human primate brain imaging with a PET resolution of 5 mm and field of view of 59.4 cm and 25.8 cm (transaxial and axial, respectively). Dynamic PET image acquisition was initiated followed by administration of the radiotracer in a homogenous solution of 10% ethanol and 90% isotonic saline. An MEMPRAGE sequence began after 30 min of the baseline scan for anatomic coregistration. To characterize the specific binding of [11C]CW4, a second imaging experiment was carried out in which ketanserin (1 mg/kg) was co-administered intravenously at the start of acquisition. Both scans were carried out in the same animal on the same day, separated by 2.5 h. In both scans, 4-5 mCi of [11C]CW4 was administered to the baboon. Dynamic data from the PET scans were recorded in list mode and corrected for attenuation. Baboon data were reconstructed using a 3D-OSEM method resulting in a full width at half-maximum resolution of 4 mm.
Baboon PET/MR Image Analysis
Volumes of interest (VOIs) were drawn manually as spheres in brain regions guided by high resolution MR structural images and summed PET data, with a radius no less than 4mm. A common VOI mask was applied to both baboon scans. Time-activity curves (TACs) were exported in terms of decay corrected activity per unit volume at specified time points with gradually increasing intervals. The TACs were expressed as percent injected dose per unit volume for analysis.
Chemical synthesis
Synthesis of (2-hydroxy-6-methoxyphenyl)(4-(quinolin-2-yl)-1,4-diazepan-1-yl)methanone (4). To a solution of 2-chloroquinoline (1) (4.0 g, 24 mmol) and 1, 4-diazepane (2) (7.2 g, 72 mmol) in 20 mL DMSO, sodium carbonate (3.8 g, 36 mmol) was added. The mixture was stirred at 120 °C for 10 h. After cooling down to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate (30 mL x 3), the combined organic phase was washed with brine, dried over magnesium sulfate, filtered, and concentrated. Crude intermediate 3 was used for next step without purification. Solid 3 (1.0 g, 4.4 mmol) was dissolved in DCM, 2-hydroxy-6-methoxybenzoic acid (0.74 g, 4.4 mmol) and EDC·HCl (0.92 g, 4.8 mmol) were added into the solution. The mixture was stirred at room temperature for 1 h, then water was added. The mixture was extracted with DCM (20 mL x 3). The organic layer was separated and combined, dried over magnesium sulfate, filtered, and the solvent was evaporated to dryness. The product was purified by chromatography (hexane/ethyl acetate = 1:3) and afforded white solid (0.66 g, 24 % for two steps).1H-NMR (500 MHz, DMSO-d6): δ 9.66 (s, 1H), 8.01 (d, J = 9.0 Hz, 1H), 7.67 (m, 1H), 7.48 (m, 2H), 7.11 (m, 3H), 6.49 (m, 2H), 3.93 (m, 6H), 3.53 (m, 2H), 3.44 (s, 3H), 1.99 (m, 1H), 1.68 (m, 1H). 13C-NMR (125 MHz, DMSO-d6): δ 156.8, 156.0, 154.5, 148.2, 137.7, 130.0, 129.6, 127.8, 126.3, 122.9, 121.9, 114.1, 109.9, 108.9, 102.3, 55.6, 49.3, 47.8, 45.7, 27.0, 25.4. LC-MS calculated for C22H23N3O3 expected [M]: 377.2; found [M+H]+: 378.2. HPLC purity: 96.1%.
Synthesis of (2,6-dimethoxyphenyl)(4-(quinolin-2-yl)-1,4-diazepan-1-yl)methanone (5, CW3). Solid 3 (1.0 g, 4.4 mmol) was dissolved in DCM, 2,6-dimethoxybenzoic acid (0.80 g, 4.4 mmol) and EDC·HCl (0.92 g, 4.8 mmol) were added into the solution. The mixture was stirred at room temperature for 1 h, then water was added. The mixture was extracted with DCM (20 mL x 3). The organic layer was separated and combined, dried over magnesium sulfate, filtered, and the solvent was evaporated to dryness. The product was purified by chromatography (hexane/ethyl acetate = 1:2) and afforded white solid (0.86 g, 30 % for two steps).1H-NMR (500 MHz, DMSO-d6): δ 8.00 (m, 1H), 7.67 (m, 1H), 7.50 (m, 2H), 7.27 (m, 1H), 7.18 (m, 1H), 7.10 (m, 1H), 6.64 (d, J = 8.5 Hz, 1H), 6.60 (d, J = 8.5 Hz, 1H), 3.93 (m, 1H), 3.81 (m, 6H), 3.58 (s, 3H), 3.50 (s, 3H), 3.13 (m, 1H), 1.95 (m, 1H), 1.60 (m, 1H). 13C-NMR (125 MHz, DMSO-d6): δ 165.7, 156.3, 148.2, 137.7, 130.5, 129.6, 127.8, 126.3, 122.9, 122.8, 121.8, 115.0, 109.9, 109.7, 104.5, 104.4, 55.9, 48.5, 47.5, 45.9, 45.5, 27.1, 26.0. LC-MS calculated for C23H25N3O3 expected [M]: 391.2; found [M+H]+: 392.4. HPLC purity: 97.2%.
Synthesis of (4-(6-chlorobenzo[d]thiazol-2-yl)-1, 4-diazepan-1-yl)(2-hydroxyphenyl)methanone (8)
To a solution of 2, 6-dichlorobenzo[d]thiazole (4.9 g, 24 mmol) and 1, 4-diazepane (7.2 g, 72 mmol) in 20 mL DMSO, sodium carbonate (3.8 g, 36 mmol) was added. The mixture was stirred at 120 °C for 10 h. After cooling down to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate (30 mL x 3), the combined organic phase was washed with brine, dried over magnesium sulfate, filtered, and concentrated. Crude intermediate 7 was used for next step without purification. Solid 7 (1.0 g, 3.8 mmol) was then dissolved in DCM, 2-hydroxybenzoic acid (0.53 g, 3.8 mmol) and EDC.HCl (0.73 g, 3.8 mmol) were added into the solution. The mixture was stirred at room for 1h, then water was added. The mixture was extracted with DCM (20 mLx3). The organic layer was separated and combined, dried over magnesium sulfate, filtered, and the solvent was evaporated to dryness. The product was purified by chromatography (hexane/ethyl acetate = 1:3) and afforded white solid (1.0 g, 34 % for two steps).).1H-NMR (500 MHz, DMSO-d6): δ 9.75 (s, 1H), 7.89 (s, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 2.0 Hz, 1H), 7.17 (m, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.78 (m, 1H), 6.60 (m, 1H), 3.76 (m, 4H), 3.51 (m, 2H), 1.98 (m, 2H), 1.70 (m, 2H). 13C-NMR (125 MHz, DMSO-d6): δ 169.0, 153.3, 149.8, 130.3, 126.4, 121.2, 120.5, 120.3, 119.7, 119.2, 116.1, 115.6, 110.0, 109.8, 50.3, 47.5, 43.1, 25.4, 22.1. LC-MS calculated for C19H18ClN3O2S expected [M]: 387.1; found [M+H]+: 388.5. HPLC purity: 98.6%.
Synthesis of (4-(6-chlorobenzo[d]thiazol-2-yl)-1, 4-diazepan-1-yl)(2-methoxyphenyl)methanone (9, CW4)
Solid 7 (1.0 g, 3.8 mmol) was dissolved in DCM, 2-methoxybenzoic acid (0.53 g, 3.8 mmol) and EDC·HCl (0.73 g, 3.8 mmol) were added into the solution. The mixture was stirred at room temperature for 1 h, then water was added. The mixture was extracted with DCM (20 mL x 3). The organic layer was separated and combined, dried over magnesium sulfate, filtered, and the solvent was evaporated to dryness. The product was purified by chromatography (hexane/ethyl acetate = 1:2) and afforded white solid (1.2 g, 40 % for two steps).1H-NMR (500 MHz, DMSO-d6): δ 7.90 (s, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.33 (M, 1H), 7.29 (d, J = 2.0 Hz, 1H), 7.02 (m, 1H), 6.94 (m, 1H), 6.73 (m, 1H), 3.76 (m, 6H), 3.60 (s, 3H), 1.90 (m, 2H), 1.70 (m, 2H). 13C-NMR (125 MHz, DMSO-d6): δ 168.3, 155.1, 152.2, 132.4, 130.5, 127.7, 127.4, 126.3, 125.0, 121.2, 120.7, 119.6, 111.8, 110.0, 55.6, 47.2, 45.0, 43.7, 27.2, 25.8. LC-MS calculated for C20H20ClN3O2S expected [M]: 401.1; found [M+H]+: 402.5. HPLC purity: 97.5%.
Synthesis of (4-(6-chlorobenzo[d]thiazol-2-yl)-1, 4-diazepan-1-yl)(2-hydroxy-6-methoxyphenyl) methanone (10)
Solid 7 (1.0 g, 3.8 mmol) was dissolved in DCM, 2-hydroxy-6-methoxybenzoic acid (0.74g, 4.4 mmol) and EDC.HCl (0.92g, 4.8 mmol) were added into the solution. The mixture was stirred at room temperature for 1 h, then water was added. The mixture was extracted with DCM (20 mLx3). The organic layer was separated and combined, dried over magnesium sulfate, filtered, and the solvent was evaporated to dryness. The product was purified by chromatography (hexane/ethyl acetate = 1:3) and afforded white solid (0.86 g, 27 % for two steps).1H-NMR (500 MHz, DMSO-d6): δ 9.62 (s, 1H), 7.89 (d, J = 2.0 Hz, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.27 (m, 1H), 7.11 (m, 1H), 6.47 (m, 2H), 3.80 (m, 6H), 3.72 (m, 2H), 3.46 (s, 3H), 1.97 (m, 1H), 1.65 (m, 1H). 13C-NMR (125 MHz, DMSO-d6): δ 168.0, 166.2, 156.8, 154.5, 152.2, 132.4, 130.2, 126.4, 124.9, 121.2, 119.5, 113.7, 108.8, 102.3, 55.6, 48.1, 44.8, 43.5, 27.3, 25.8. LC-MS calculated for C20H20ClN3O3S expected [M]: 417.1; found [M+H]+: 418.5. HPLC purity: 98.6%; tR: 1.7 min; condition: Agilent Eclipse XDB-C8, 150 mm x 4.6 mm, 2.0 mL/min, 30% H2O + TFA (0.01% v/v)/ 70% CH3CN + TFA (0.01% v/v). HPLC purity: 94.3%.
Synthesis of (4-(6-chlorobenzo[d]thiazol-2-yl)-1, 4-diazepan-1-yl)(2, 6-dimethoxyphenyl) methanone (11, CW6)
Solid 7 (1.0 g, 3.8 mmol) was dissolved in DCM, 2,6-dimethoxybenzoic acid (0.80 g, 4.4 mmol) and EDC·HCl (0.92 g, 4.8 mmol) were added into the solution. The mixture was stirred at room temperature for 1 h, and then water was added. The mixture was extracted with DCM (30 mL × 3). The organic layer was separated and combined, dried over magnesium sulfate, filtered, and the solvent was evaporated to dryness. The product was purified by chromatography (hexane/ethyl acetate = 1:2) and afforded white solid (1.1 g, 33 % for two steps).1H-NMR (500 MHz, DMSO-d6): δ 7.90 (d, J = 2.0 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.28 (m, 3H), 6.63 (d, J = 8.5 Hz, 1H), 3.70 (m, 4H), 3.52 (s, 3H), 3.49 (s, 3H), 3.40 (m, 1H), 3.21 (m, 1H), 1.92 (m, 1H), 1.62 (m, 1H). 13C-NMR (125 MHz, DMSO-d6): δ 167.3, 156.3, 152.3, 144.0, 141.7, 132.4, 130.6, 130.0, 129.8, 126.1, 124.6, 119.5, 104.5, 104.4, 55.8, 50.4, 48.3, 45.0, 43.5, 27.3, 25.1. LC-MS calculated for C21H22ClN3O3S expected [M]: 431.1; found [M+H]+: 432.5. HPLC purity: 95.1%.
Supplementary Material
Acknowledgements
This research was carried out at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Regional Resource supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program and/or High-End Instrumentation Grant Program; specifically, grant numbers: S10RR017208, S10RR026666, S10RR022976, S10RR019933, S10RR029495. We would like to thank members of the Hooker laboratory for helpful discussions and thanks to Dr. Ramesh Neelamegam for revising the paper. The authors are grateful to Joseph Mandeville and Helen Deng for assistance during NHP imaging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Lecea L, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95(1):322–7. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel JM. Hypocretin (orexin): role in normal behavior and neuropathology. Annu Rev Psychol. 2004;55:125–48. doi: 10.1146/annurev.psych.55.090902.141545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marcus JN, et al. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435(1):6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 5.Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785–97. doi: 10.1517/13543784.16.11.1785. [DOI] [PubMed] [Google Scholar]
- 6.Hirose M, et al. N-acyl 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline: the first orexin-2 receptor selective non-peptidic antagonist. Bioorg Med Chem Lett. 2003;13(24):4497–9. doi: 10.1016/j.bmcl.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 7.Brisbare-Roch C, et al. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med. 2007;13(2):150–5. doi: 10.1038/nm1544. [DOI] [PubMed] [Google Scholar]
- 8.Winrow CJ, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52–61. doi: 10.3109/01677063.2011.566953. [DOI] [PubMed] [Google Scholar]
- 9.Herring WJ, et al. Orexin receptor antagonism for treatment of insomnia: A randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265–74. doi: 10.1212/WNL.0b013e31827688ee. [DOI] [PubMed] [Google Scholar]
- 10.Bettica P, et al. The orexin antagonist SB-649868 promotes and maintains sleep in men with primary insomnia. Sleep. 2012;35(8):1097–104. doi: 10.5665/sleep.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bettica P, et al. Differential effects of a dual orexin receptor antagonist (SB-649868) and zolpidem on sleep initiation and consolidation, SWS, REM sleep, and EEG power spectra in a model of situational insomnia. Neuropsychopharmacology. 2012;37(5):1224–33. doi: 10.1038/npp.2011.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bettica P, et al. Phase I studies on the safety, tolerability, pharmacokinetics and pharmacodynamics of SB-649868, a novel dual orexin receptor antagonist. J Psychopharmacol. 2012;26(8):1058–70. doi: 10.1177/0269881111408954. [DOI] [PubMed] [Google Scholar]
- 13.Malherbe P, et al. Biochemical and behavioural characterization of EMPA, a novel high-affinity, selective antagonist for the OX(2) receptor. Br J Pharmacol. 2009;156(8):1326–41. doi: 10.1111/j.1476-5381.2009.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu F, et al. Radiosynthesis of [11C]BBAC and [11C]BBPC as potential PET tracers for orexin2 receptors. Bioorg Med Chem Lett. 2012;22(6):2172–4. doi: 10.1016/j.bmcl.2012.01.114. [DOI] [PubMed] [Google Scholar]
- 15.Ghose AK, et al. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem Neurosci. 2012;3(1):50–68. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitman DB, et al. Discovery of a potent, CNS-penetrant orexin receptor antagonist based on an n,n-disubstituted-1,4-diazepane scaffold that promotes sleep in rats. ChemMedChem. 2009;4(7):1069–74. doi: 10.1002/cmdc.200900069. [DOI] [PubMed] [Google Scholar]
- 17.Constantinescu CC, et al. Striatal and extrastriatal microPET imaging of D2/D3 dopamine receptors in rat brain with [(1)(8)F]fallypride and [(1)(8)F]desmethoxyfallypride. Synapse. 2011;65(8):778–87. doi: 10.1002/syn.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.