

Abstract
The biological processes occurring in a cell are complex and dynamic, and to achieve a comprehensive understanding of the molecular mechanisms underlying these processes, both temporal and spatial information is required. While cryo-electron tomography (cryoET) provides three-dimensional (3D) still pictures of near-native state cells and organelles at molecular resolution, fluorescence light microscopy (fLM) offers movies of dynamic cellular processes in living cells. Combining and integrating these two commonly used imaging modalities (termed correlative microscopy) provides a powerful means to not only expand the imaging scale and resolution but also to complement the dynamic information available from optical microscopy with the molecular-level, 3D ultrastructure detail provided by cryoET. As such, a correlative approach performed on a given specimen can provide high resolution snapshots of dynamic cellular events. In this article, I review recent advances in correlative light microscopy and cryoET and discuss major findings made available by applying this method.
Introduction
Both fluorescence light microscopy and electron microscopy have been instrumental for understanding the structure and function of biological processes and have provided a wealth of information regarding protein localization and dynamics and the cellular ultrastructure. Cryo-electron tomography (cryoET) has emerged as a powerful imaging technique that allows three-dimensional (3D) visualization of the molecular landscape within cells, at 2–4 nm resolution, in a close-to physiological state [1]. By applying this method, significant structural detail has been obtained for bacterial cells [2, 3], cellular compartments [4], and purified or abundantly budding virus particles [5–7]. With recent developments in subvolume averaging from cellular tomograms, the structures of native protein assemblies, at resolutions below 2 nm, are now possible [8, 9]. However, several issues associated with cryoET have limited its potential in advancing cellular structural biology. First, the inherent low contrast of unstained frozen-hydrated specimen in cryoET, combined with specimen radiation sensitivity, makes it difficult to locate areas of interest inside a cell before cryoET analysis. Second, cryoET delivers only still pictures of cells. Third, although providing molecular resolution, cellular tomograms offer a very limited field of view, typically only ~10% of a cell’s area and restricted to very thin regions, typically those < 300nm thick. Thus, the vast majority of a eukaryotic cell is not accessible to cryoET alone. Finally, due to the crowded cellular environment, in the absence of EM labels, individual proteins and macromolecular complexes are not readily distinguishable at the current resolution, with the exception of a few, very large complexes [10, 11].
On the other hand, fluorescence light microscopy (fLM) offers a complementary set of imaging capabilities. It provides time-resolved, large scale (whole cell) visualization of the dynamics of individual fluorescent protein(s) of interest in living cells. Thus, fluorescence imaging has been employed to investigate the temporal and spatial distribution of specific molecular players in a variety of cellular processes. Recent advances in super-resolution fluorescence microscopy imaging, which apply switchable probes, as in PALM and STORM [12, 13], or elegant illumination strategies, as in STED and SIM [14–16], have further improved the resolution of fLM by an order of magnitude and have enabled a better understanding of many cellular processes [17, 18]. Yet, the limited spatial resolution of fLM often prevents definitive localization of proteins relative to the cellular ultrastructure or organelles. Further, only a few molecules can be visualized simultaneously with fLM, in contrast to the full repertoire of macromolecules that can be visualized with cryoET.
Therefore, a correlative approach that combines the strength of these two complimentary imaging modalities is highly desirable. In such a way, specific features highlighted by fluorescent signals are identified and located by fLM and then used to guide the acquisition of high resolution 3D structural data by cryoET, which in turn complement the dynamic information obtained by live-cell fLM. Using correlative microscopy, one can not only visualize the dynamics of the cellular process in a whole living cell but can also zoom in to observe the molecular details of the cellular context of the process to gain novel insights into the process.
Correlative light and electron microscopy (CLEM) has become a powerful approach for structural and functional analysis of cellular processes in a single experiment. CLEM was achieved primarily using either chemically or cryo-fixed, resin embedded and sectioned specimen at room temperature, typically after immunolabeling [19, 20] or photoconversion of diaminobenzadine into an electron-dense marker, such as miniSOG [21•]. Abundant application of this approach has yielded many significant findings and has provided insights into some fundamental biological processes and their underlying molecular mechanisms [19, 22•]. Here, I focus on CLEM studies performed at cryogenic temperature (cryo-CLEM), which is more challenging, but allows fluorescent signals to be examined in frozen-hydrated cells preserved at near-native condition for subsequent cryoET analysis [23]. In this review, I outline the workflow for cryo-CLEM, highlight recent technical advances and the major findings obtained using this approach, and discuss the future prospects for using correlative microscopy in conjunction with superresolution (see article by Moerner), cryoFIB milling (see article by Baumeister), and/or genetically expressed EM markers, to advance our imaging capability for investigating cell signaling and cellular processes.
Cryo-CLEM implementation
Cryo-correlative light and electron microscopy (i.e. cryo-CLEM) is a multistep experiment involving protein fluorescent-labeling, live-cell confocal microscopy, sample vitrification, cryo-fluorescent light microscopy (cryo-fLM), and cryoET data collection and analysis. A typical workflow for cryo-CLEM is illustrated in Figure 1 (steps 1 through 6). Cells grown directly on indexed, gold EM grids (Fig. 1a) are typically labeled with one or more fluorescent dyes and examined with time lapse, live-cell confocal imaging (Fig. 1b) to assess the dynamic behavior of the labeled protein(s) of interest. At a specific dynamic stage of interest, the cells are preserved at near-native condition by rapid vitrification (Fig. 1c). Cryo-fLM is performed to identify the area of interest using a cryo-light microscopy (cryo-LM) stage that maintains the sample in the frozen-hydrated state (Fig. 1d). Specific features highlighted by fluorescent labeling are identified and located, and their coordinates are transferred to the electron microscope for acquisition of high resolution 3D structural data using cryoET (Fig. 1e). The final stage of cryo-CLEM (Fig. 1f) involves structural and functional analysis of the biological process of interest by integrating the 3D structure data with dynamics information obtained from the live-cell imaging (step 6). In many cases, when the structures of interest are stable, the fLM images recorded in live cultures (Fig. 1 b) can be used directly to guide cryoEM localization of the area of interest (step 2′). However, for highly dynamic features, a cryo-LM stage, to retain the vitrified state, is needed for direct correlation.
Figure 1.
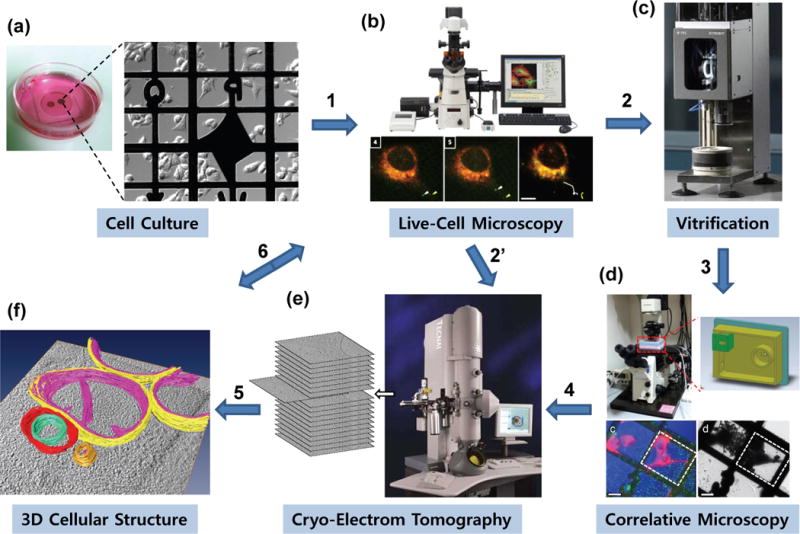
An illustrative workflow of correlative light microscopy and cryo-electron tomography of cells. The steps include: (a) culturing of eukaryotic cells on carbon-coated gold EM index grids in a glass-bottom petri dish; (b) time-lapse, confocal live-cell imaging of a dynamic cellular process, such as viral infection, directly performed on cells grown on EM grids; (c) arresting the cellular process at a particular state by vitrification (plunge-freezing); (d) localizing the target of interest using fluorescence signals by cryo-fLM for subsequent cryoET; (e) cryoET data acquisition and 3D reconstruction of the identified target; and (f) 3D structural analysis and interpretation, as well as correlating the high resolution cellular structure with the dynamics of the fluorescently labeled proteins (step 6). For stable and static structures, live cell fluorescence images can also be used to guide cryoEM localization (step 2′), without cryo-fLM.
Recent technical advances in cryo-CLEM
Cryo-CLEM holds great promise as a tool for structural and functional analysis of cellular processes under close-to-native conditions. But, it is technically challenging, particularly during the cryo-fLM imaging stage of the method. Although a number of cryo-LM stages have been developed and successfully applied [24–30], the approaches carried out with these stages have involved separate LM and EM instruments and, thus, required specimen transfer, making the specimen vulnerable to physical damage and contamination. To minimize this risk during grid transfer, various techniques have been applied, one of which involved the development and application of an integrated specimen cartridge holder that allows both cryo-fLM and cryoEM to be performed within a single specimen holder, eliminating the need for grid transfer [25••, 28]. Very recently, however, an integrated cryo-CLEM sample stage (ILEM2) became available to address this issue. Specifically, the ILEM2, combines cryo-fLM and cryoEM into a single instrument using a retractable optical element and a rotating specimen holder, to circumvent the need for specimen transfer [31••]. This “2 in 1” approach greatly simplifies sample handling and navigation between the two modalities, resulting in faster throughput and greater efficiency. The ILEM concept was first demonstrated with room temperature correlation [32] and has recently been commercialized (FEI’s Tecnai™ with iCorr™: FEI Company, USA). However, to image dynamic events, the two-step correlation approach, as illustrated in Fig. 1(b–d), is still required.
Along with these instrumentation and grid transfer issues, correlating fLM images and EM images is a non-trivial task, due to the vastly different image intensities, contrast, magnification, and resolution. Because correlation is commonly done manually, with the aid of finder grids, the accuracy of the correlation is susceptible to error. For this reason, along with the ILEM2, Faas and colleagues developed an integrated software, which applies a coordinate-based, pre-calibrated relocation system [30], making navigation between the two imaging modalities much easier [31••]. The alignment can be further simplified and improved by including fiducial markers that provide contrast for both fLM and cryoEM [33, 34].
Correlating structure, function and dynamics of cellular processes
As an emerging technique, correlative cryoET and fLM has been successfully applied to investigate cell adhesion [35•], mitosis [36], neuronal synapses [30], and the cytoplasmic microtubule network [27]. In these cases, correlation experiments were primarily carried out to facilitate cryoET data acquisition; the structure of interest was localized using cryo-fLM to allow meaningful reconstruction of cellular macromolecular structures. More recent efforts, as exemplified in the studies described below, have focused on using cryo-CLEM to connect the cellular ultrastructure to the function and/or dynamics of a biological process, providing new insights into how molecular composition and dynamics are related to cellular structure and function.
Cryo-CLEM provides a revised view of a prokaryotic structure
A large body of fLM studies employing fluorescent protein fusion tags had shown that the bacterial actin homolog, MreB, polymerizes into long helical filaments that encircle diverse rod-shaped bacteria. These studies led to a predominate view in the field that a helical cytoskeleton plays a central role not only in cell shape determination but also in chromosome segregation, cell polarity, motility, and growth [37]. Jensen’s group recently applied cryo-CLEM to understand the function and structural organization of MreB filaments in a number of rod-shaped bacteria [38•]. Cells overexpressing GFP-MreB were labeled with the membrane dye FM4-64, plunge frozen on an EM finder-grid, and imaged by cryo-fLM, then by cryoET. Through this correlative approach, they confirmed that MreB forms large bundles of filaments in the cytoplasm (Fig. 2a). However, in contrast to the vast number of previous fLM observations, no MreB helical filaments were seen encircling the cells. Instead, the extended helical patterns consistently seen in the previous fLM studies were observed to be artifacts due to overexpression of the MreB fusion protein (Fig. 2b) [39]. More recent live-cell optical microscopy studies also indicate that MreB does not form extended helices but rather forms discrete patches that move circumferentially around the cell [40–43]. Taken together, these results shed new light on the structure and function of MreB, which is now understood to be a dynamic entity that moves around the cell envelope as part of the cell wall synthetic machinery, instead of a large, rigid helical scaffold.
Figure 2.

Visualizing bacterial cytoplasmic MreB filaments. (a) Correlated cryo-fLM (inset) and cryoET of a V. cholerae cell overexpressing GFP-MreB. Red is the membrane-dye FM4-64 and green is GFP-MreB (inset) with the dashed lines (red and green) representing the thresholded boundaries of each signal, respectively. Cryo-electron tomogram, acquired from the same cell, display a large filament bundle running through the cytoplasm (arrows), which corresponds to the GFP-MreB signal. Scale bars, 1 μm in the fLM inset and 200 nm in the cryoET slice [38•]. (b) MreB helices are an artifact of the YFP tag when overexpressed in E. coli cells. No filaments were seen near the membranes of wild-type cells (WT) or cells overexpressing native MreB (MreB OE), but many helical and other filaments were found just inside the membranes of cells overexpressing YFP-MreB (YFP-MreB OE, YFP-MreB hyperOE). Inset, 3D model of a YFP-MreB OE cell’s inner membrane (blue) and YFP-MreB filaments [39].
Cryo-CLEM to understand viral dynamics in cells
Application of cryo-CLEM to dynamic processes, such as viral entry, egress, and budding during infection, in cells arrested during a particular functional state of interest, can provide high-resolution still pictures of specific stages of dynamic processes. For example, Ibiricu and colleagues applied live-cell imaging along with cryoET to understand the dynamics and structure of herpes simplex virus 1 (HSV1) in neurons during egress. Specifically, they first determined the peak for anterograde virus transport by live-cell imaging, then used cryoET structural analysis to determine the structure of viruses during this peak [44••] (Fig. 3). Their analysis revealed that HSV1 capsids were transported independently of the viral envelope and that the secondary capsid envelopment occurs in axon terminals. Further, using cellular cryoET combined with subvolume averaging [8, 45], they were able to visualize the tegument density that is exclusively associated with the capsid vertices on intra-axonal capsids and to determine that both DNA-packed capsids and empty capsids undergo active axonal transport. Yet, due to technical difficulties, the live-cell imaging and cryoET were not performed on the same specimen, thus the structure and function correlation was indirect.
Figure 3.
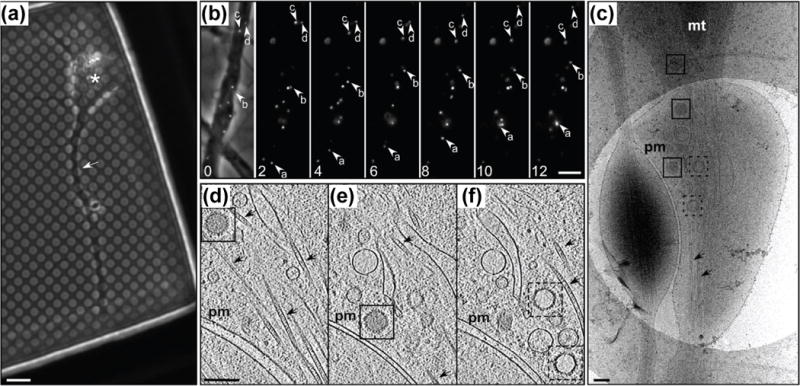
Transport of HSV1 capsids in neurons during egress. (a) Phase contrast image of a hippocampal neuron grown on a holey carbon support film for 7 days. A neurite (white arrow) and cell body (asterisk) are indicated. (b) Time-lapse fluorescence images of a mid-axon region of a HSV1(KOS)-GFPVP26 infected neuron at 16 hours postinfection, recorded at the indicated time (seconds). The left image shows an overlay of the fluorescence signal with the bright field image. Arrows indicate the positions of individual viral particles. (c) CryoEM projection image of an intact axon at 16 hours post-infection, showing cytosolic C- and A-capsids (boxed with solid and dashed lines, respectively). (d–f) Slices through the reconstructed tomographic volume obtained from the area of interest in (c). pm: plasma membrane; mt: mitochondria; black arrows: microtubules. Scale bars: 6 μm in (a), 5 μm in (b), and 200 nm in (c–f) [44••].
A direct correlation between the time-lapse, live-cell imaging and high resolution cryoET at the level of individual viral particles was first accomplished by Jun et al. during visualization of single native HIV-1 cores within cells at an early stage of infection [25••]. Studying early HIV-1 infection events in the context of the dynamical interplay between virus and host cell components is particularly challenging [46], due to the small size of the viral particle (diffraction limited), the rarity of infection, and the highly dynamic nature of infection events. Catching a few single infecting viral particles (100 nm) in a vast host cell (50 μm) at the right moment for cryoET analysis would be nearly impossible without correlative live-cell imaging. Using high-speed confocal live-cell light microscopy followed by cryo-fLM and cryoET on the exact same viral particle (see Fig. 1 for workflow), Jun et al., for the first time, directly detected and imaged individual HIV-1 particles at different infection stages by high resolution cryoET [25••]. While a majority of stationary HIV-1 particles retained viral envelope and were either attached to the cell surface or internalized in vesicular compartments, fast moving particles appeared to be released into the cytoplasm after membrane fusion and to be en route to the nucleus (Fig. 4). Thus, with correlative imaging, Jun et al. were able to connect viral dynamics in a live cell to the structure of viral components in the cellular context during infection. The group further obtained direct evidence to suggest that a hyperstable HIV-1 capsid mutation, E45A, delayed capsid uncoating compared to the wild-type capsid. Together, these results demonstrate the importance of correlating 3D live-cell imaging with cryoET when rare and dynamic cellular processes are to be analyzed or when cryoET data will be more meaningful with information about the dynamics or a functional state of cell under investigation.
Figure 4.

Correlation between HIV-1 dynamics and 3D structure of host cell complexes during HIV infection by combining confocal live cell microscopy, cryo-fLM, and cryoET. (a–c) Direct visualization of a “trafficking” native HIV-1 core within the infected cell. The HIV-1 particle was tracked in 3D for 40 min (yellow trace) with time-lapse live cell confocal imaging (a), followed by plunge-freezing and cryo-fLM imaging to localize the particle, and subsequent low-dose cryoEM imaging of the correlated target region (marked with “*”) at low magnification (b) and cryoET (c). A 4 nm thick tomographic slice from the 3D tomographic volume acquired from the correlated cellular region (* in b) shows an intact intracellular HIV-1 core (boxed) situated next to a vesicle and in the vicinity of cellular filaments and microtubules. Scale bars: 10 μm in (a) and (b), 100 nm in (c). (d–f) Direct visualization of a “stationary” native HIV-1 virion (circled in d) bound to a HeLa cell by live cell imaging with 3D tracking (d, red trace, stationary, blue trace, dynamic, 30 min), low (e) and high (f) magnification cryoEM imaging of the correlated area (boxed in e). The enveloped, static HIV-1 particle was observed bound to a cell protrusion (arrow). Scale bars, 5 μm in (d) and (e), and 100 nm in (f) [25••].
Concluding remarks
Correlative cryoET and optical microscopy is an emerging powerful approach to visualize dynamic cellular processes. Recent technical advances in cryoEM and cryoET, including direct electron detectors and phase plates, provide a positive prospect on achieving multiscale andhigh resolution in analyzing biological systems in the coming years. However, cryoET, like other TEM techniques, suffers from a basic limitation: sample thickness, which needs to be <300 nm. Given this limitation, cryoET and correlative imaging have been more successfully applied to small prokaryotes and viruses. Many eukaryotic cells are too thick to be imaged by cryoET, and currently, only the peripheral regions of the cell are suitable for 3D structural analysis by cryoET. Reducing the thickness of frozen-hydrated specimens, to allow access to the vast interior of the cell, by vitreous sectioning [47–49] or by cryo-focused ion beam (FIB) milling [50–52] (see review article by Baumeister) would greatly expand the capability of cryoET and correlative imaging of eukaryotic cells.
CryoET offers the potential to visualize the full repertoire of macromolecular complexes in a cellular landscape at 2–4 nm resolution. However, discerning these complexes in a crowded cellular environment [53], possibly with a template-matching approach [54], is a challenge. Nonetheless, with the advancements in super-resolution optical imaging, identification of supramolecular complexes (10–20 nm) in correlated cellular tomograms may become possible [22•]. Dual labels which are both fluorescent and electron dense are highly desirable to achieve correlation at the single molecule level. Future development in this area, in particular genetically encoded GFP-equivalent analogues for cellular cryoET, such as tetracysteine-ReAsh [55] and metallothionein [56], will greatly enhance the ability to dissect the molecular mechanisms of cellular events.
TEM with a liquid cell technology, which potentially allows unconventional live-cell imaging by TEM [57], combined with fLM, could offer new possibilities for time-resolved high resolution views of cellular processes, including viral infection, cell signaling, and transcription/translation processes. It is also expected that integrating additional imaging modules, such as atomic force microscopy [58], x-ray tomography [59, 60], and scanning electron microscopy [61], combined with molecular and cell biology methods, will provide complementary spatial, temporal, structural, biochemical and biophysical details that allow a thorough and comprehensive understanding of cellular and molecular phenomena.
Highlights.
Describe a workflow for correlative fluorescent microscopy and cryo-electron tomography
Highlight technical advances in cryo-correlative microscopy (cryo-CLEM)
Correlate structure, function and dynamics of cellular processes with cryo-CLEM
Acknowledgments
The author thanks Dr. Teresa Brosenitsch for critical reading of the manuscript. This study was supported by the National Institutes of Health Grant GM082251 and GM085043 to P.Z.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Leis A, Rockel B, Andrees L, Baumeister W. Visualizing cells at the nanoscale. Trends Biochem Sci. 2009;34:60–70. doi: 10.1016/j.tibs.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Li Z, Jensen GJ. Electron cryotomography: a new view into microbial ultrastructure. Curr Opin Microbiol. 2009;12:333–340. doi: 10.1016/j.mib.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milne JL, Subramaniam S. Cryo-electron tomography of bacteria: progress, challenges and future prospects. Nat Rev Microbiol. 2009;7:666–675. doi: 10.1038/nrmicro2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Harush K, Maimon T, Patla I, Villa E, Medalia O. Visualizing cellular processes at the molecular level by cryo-electron tomography. J Cell Sci. 2010;123:7–12. doi: 10.1242/jcs.060111. [DOI] [PubMed] [Google Scholar]
- 5.Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. Structure and assembly of immature HIV. Proc Natl Acad Sci USA. 2009;106:11090–11095. doi: 10.1073/pnas.0903535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlson LA, de Marco A, Oberwinkler H, Habermann A, Briggs JA, Krausslich HG, Grunewald K. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 2010;6:e1001173. doi: 10.1371/journal.ppat.1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 2007;26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyerson JR, Tran EE, Kuybeda O, Chen W, Dimitrov DS, Gorlani A, Verrips T, Lifson JD, Subramaniam S. Molecular structures of trimeric HIV-1 Env in complex with small antibody derivatives. Proc Natl Acad Sci USA. 2013;110:513–518. doi: 10.1073/pnas.1214810110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck M, Forster F, Ecke M, Plitzko JM, Melchior F, Gerisch G, Baumeister W, Medalia O. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science. 2004;306:1387–1390. doi: 10.1126/science.1104808. [DOI] [PubMed] [Google Scholar]
- 11.Brandt F, Carlson LA, Hartl FU, Baumeister W, Grunewald K. The three-dimensional organization of polyribosomes in intact human cells. Mol Cell. 2010;39:560–569. doi: 10.1016/j.molcel.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 13.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gustafsson MG. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hell SW. Toward fluorescence nanoscopy. Nat Biotechnol. 2003;21:1347–1355. doi: 10.1038/nbt895. [DOI] [PubMed] [Google Scholar]
- 16.Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, Kner P, Burke B, Cardoso MC, Agard DA, Gustafsson MG, et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008;320:1332–1336. doi: 10.1126/science.1156947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Annu Rev Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol. 2010;190:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Rijnsoever C, Oorschot V, Klumperman J. Correlative light-electron microscopy (CLEM) combining live-cell imaging and immunolabeling of ultrathin cryosections. Nat Methods. 2008;5:973–980. doi: 10.1038/nmeth.1263. [DOI] [PubMed] [Google Scholar]
- 20.Spiegelhalter C, Tosch V, Hentsch D, Koch M, Kessler P, Schwab Y, Laporte J. From dynamic live cell imaging to 3D ultrastructure: novel integrated methods for high pressure freezing and correlative light-electron microscopy. PLoS One. 2010;5:e9014. doi: 10.1371/journal.pone.0009014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. • “MiniSOG” was introduced as a genetic fusion tag to efficiently convert singlet oxygen generated by light illumination to electron-dense markers via diaminobenzadine, thus permitting a direct correlation between light and electron microscopy. The authors demonstrated its application as a versatile label for correlative microscopy of genetically tagged proteins in cells and tissues using miniSOG-tagged presynaptic SynCAM1 and postsynaptic miniSOG-tagged SynCAM2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Watanabe S, Punge A, Hollopeter G, Willig KI, Hobson RJ, Davis MW, Hell SW, Jorgensen EM. Protein localization in electron micrographs using fluorescence nanoscopy. Nat Methods. 2011;8:80–84. doi: 10.1038/nmeth.1537. • In this study, super-resolution microscopy methods, STED and PALM, were combined with conventional electron microscopy for correlated, nanoscopic localization of proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plitzko JM, Rigort A, Leis A. Correlative cryo-light microscopy and cryo-electron tomography: from cellular territories to molecular landscapes. Curr Opin Biotechnol. 2009;20:83–89. doi: 10.1016/j.copbio.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Carlson DB, Evans JE. Low-cost cryo-light microscopy stage fabrication for correlated light/electron microscopy. J Vis Exp. 2011 doi: 10.3791/2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25••.Jun S, Ke D, Debiec K, Zhao G, Meng X, Ambrose Z, Gibson GA, Watkins SC, Zhang P. Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure. 2011;19:1573–1581. doi: 10.1016/j.str.2011.09.006. •• Correlative high-speed confocal live-cell microscopy and cryo-electron tomography was employed for imaging rare and dynamic cellular processes, such as viral infection. Using this approach, the authors directly visualized intact HIV-1 cores released into the cytoplasm during early stages of infection for the first time. They further linked viral dynamics to ultrastructure and function and provided direct evidence for the behavior of a hyperstable HIV-1 capsid mutant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sartori A, Gatz R, Beck F, Rigort A, Baumeister W, Plitzko JM. Correlative microscopy: bridging the gap between fluorescence light microscopy and cryo-electron tomography. J Struct Biol. 2007;160:135–145. doi: 10.1016/j.jsb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz CL, Sarbash VI, Ataullakhanov FI, McIntosh JR, Nicastro D. Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. J Microsc. 2007;227:98–109. doi: 10.1111/j.1365-2818.2007.01794.x. [DOI] [PubMed] [Google Scholar]
- 28.Rigort A, Bauerlein FJ, Leis A, Gruska M, Hoffmann C, Laugks T, Bohm U, Eibauer M, Gnaegi H, Baumeister W, et al. Micromachining tools and correlative approaches for cellular cryo-electron tomography. J Struct Biol. 2010;172:169–179. doi: 10.1016/j.jsb.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 29.van Driel LF, Valentijn JA, Valentijn KM, Koning RI, Koster AJ. Tools for correlative cryo-fluorescence microscopy and cryo-electron tomography applied to whole mitochondria in human endothelial cells. Eur J Cell Biol. 2009;88:669–684. doi: 10.1016/j.ejcb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Lucic V, Kossel AH, Yang T, Bonhoeffer T, Baumeister W, Sartori A. Multiscale imaging of neurons grown in culture: from light microscopy to cryo-electron tomography. J Struct Biol. 2007;160:146–156. doi: 10.1016/j.jsb.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 31••.Faas FG, Barcena M, Agronskaia AV, Gerritsen HC, Moscicka KB, Diebolder CA, van Driel LF, Limpens RW, Bos E, Ravelli RB, et al. Localization of fluorescently labeled structures in frozen-hydrated samples using integrated light electron microscopy. J Struct Biol. 2013;181:283–290. doi: 10.1016/j.jsb.2012.12.004. •• In this article, the authors describe an integrated correlative microscopy instrumentation that combines cryo-light and cryo-electron microscopy in a single platform to eliminate specimen transfer and facilitate correlation, thus improving throughput and efficiency. They demonstrated its utility for cryo-correlative microscopy by detecting viable bacteria, polymerized microtubules, mitochondria and viruses in cells and/or in vitrious sections. Such a technical advancement will become very useful in future cryo-correlative studies. [DOI] [PubMed] [Google Scholar]
- 32.Agronskaia AV, Valentijn JA, van Driel LF, Schneijdenberg CT, Humbel BM, van Bergen en Henegouwen PM, Verkleij AJ, Koster AJ, Gerritsen HC. Integrated fluorescence and transmission electron microscopy. J Struct Biol. 2008;164:183–189. doi: 10.1016/j.jsb.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Preibisch S, Saalfeld S, Schindelin J, Tomancak P. Software for bead-based registration of selective plane illumination microscopy data. Nat Methods. 2010;7:418–419. doi: 10.1038/nmeth0610-418. [DOI] [PubMed] [Google Scholar]
- 34.Jun S, Zhao G, Ning J, Gibson GA, Watkins SC, Zhang P. Correlative Microscopy for 3D Structural Analysis of Dynamic Interactions. J Vis Exp. 2013;76:e50386. doi: 10.3791/50386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.Patla I, Volberg T, Elad N, Hirschfeld-Warneken V, Grashoff C, Fassler R, Spatz JP, Geiger B, Medalia O. Dissecting the molecular architecture of integrin adhesion sites by cryo-electron tomography. Nat Cell Biol. 2010;12:909–915. doi: 10.1038/ncb2095. • A correlative cryo-fluorescent and cryoET approach was used to identify integrin-mediated focal adhesion sites for high resolution structural analysis. The study provides the molecular architecture of focal adhesions in a close-to-native enviroment. [DOI] [PubMed] [Google Scholar]
- 36.Elad N, Abramovitch S, Sabanay H, Medalia O. Microtubule organization in the final stages of cytokinesis as revealed by cryo-electron tomography. J Cell Sci. 2011;124:207–215. doi: 10.1242/jcs.073486. [DOI] [PubMed] [Google Scholar]
- 37.Chastanet A, Carballido-Lopez R. The actin-like MreB proteins in Bacillus subtilis: a new turn. Front Biosci (Schol Ed) 2012;4:1582–1606. doi: 10.2741/s354. [DOI] [PubMed] [Google Scholar]
- 38•.Swulius MT, Chen S, Jane Ding H, Li Z, Briegel A, Pilhofer M, Tocheva EI, Lybarger SR, Johnson TL, Sandkvist M, et al. Long helical filaments are not seen encircling cells in electron cryotomograms of rod-shaped bacteria. Biochem Biophys Res Commun. 2011;407:650–655. doi: 10.1016/j.bbrc.2011.03.062. • Correlating the fluorescent signal of MreB with the filamentous structures observed with cryoET allowed the authors to question a widely accepted model for the role of MreB in cell shape determination, chromosome segregation, cell polarity, motility, and growth. This study also draws caution to the artifact introduced by overexpressing fluorescent-fusion proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swulius MT, Jensen GJ. The helical MreB cytoskeleton in Escherichia coli MC1000/pLE7 is an artifact of the N-Terminal yellow fluorescent protein tag. J Bacteriol. 2012;194:6382–6386. doi: 10.1128/JB.00505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominguez-Escobar J, Chastanet A, Crevenna AH, Fromion V, Wedlich-Soldner R, Carballido-Lopez R. Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science. 2011;333:225–228. doi: 10.1126/science.1203466. [DOI] [PubMed] [Google Scholar]
- 41.Garner EC, Bernard R, Wang W, Zhuang X, Rudner DZ, Mitchison T. Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science. 2011;333:222–225. doi: 10.1126/science.1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Teeffelen S, Gitai Z. Rotate into shape: MreB and bacterial morphogenesis. EMBO J. 2011;30:4856–4857. doi: 10.1038/emboj.2011.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Teeffelen S, Wang S, Furchtgott L, Huang KC, Wingreen NS, Shaevitz JW, Gitai Z. The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc Natl Acad Sci USA. 2011;108:15822–15827. doi: 10.1073/pnas.1108999108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44••.Ibiricu I, Huiskonen JT, Dohner K, Bradke F, Sodeik B, Grunewald K. Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 2011;7:e1002406. doi: 10.1371/journal.ppat.1002406. •• Live cell imaging was used to identify the optimal time point of herpes simplex virus axonal transport during egress. High resolution snapshots taken at specified time points allowed detailed visualization of the tegument density on different types of viral capsids and identification of the secondary envelopment of capsids at nerve terminals. The authors also applied 3D subtomogram averaging method to analyze the viral capsids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beck M, Lucic V, Forster F, Baumeister W, Medalia O. Snapshots of nuclear pore complexes in action captured by cryo-electron tomography. Nature. 2007;449:611–615. doi: 10.1038/nature06170. [DOI] [PubMed] [Google Scholar]
- 46.Campbell EM, Hope TJ. Live cell imaging of the HIV-1 life cycle. Trends Microbiol. 2008;16:580–587. doi: 10.1016/j.tim.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al-Amoudi A, Chang JJ, Leforestier A, McDowall A, Salamin LM, Norlen LP, Richter K, Blanc NS, Studer D, Dubochet J. Cryo-electron microscopy of vitreous sections. EMBO J. 2004;23:3583–3588. doi: 10.1038/sj.emboj.7600366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gruska M, Medalia O, Baumeister W, Leis A. Electron tomography of vitreous sections from cultured mammalian cells. J Struct Biol. 2008;161:384–392. doi: 10.1016/j.jsb.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 49.Zhang P, Bos E, Heymann J, Gnaegi H, Kessel M, Peters PJ, Subramaniam S. Direct visualization of receptor arrays in frozen-hydrated sections and plunge-frozen specimens of E. coli engineered to overproduce the chemotaxis receptor Tsr. J Microsc. 2004;216:76–83. doi: 10.1111/j.0022-2720.2004.01395.x. [DOI] [PubMed] [Google Scholar]
- 50.Marko M, Hsieh C, Schalek R, Frank J, Mannella C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat Methods. 2007;4:215–217. doi: 10.1038/nmeth1014. [DOI] [PubMed] [Google Scholar]
- 51.Rigort A, Bauerlein FJ, Villa E, Eibauer M, Laugks T, Baumeister W, Plitzko JM. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proc Natl Acad Sci USA. 2012;109:4449–4454. doi: 10.1073/pnas.1201333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang K, Strunk K, Zhao G, Gray JL, Zhang P. 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. J Struct Biol. 2012;180:318–326. doi: 10.1016/j.jsb.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellis RJ. Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr Opin Struct Biol. 2001;11:114–119. doi: 10.1016/s0959-440x(00)00172-x. [DOI] [PubMed] [Google Scholar]
- 54.Bohm J, Frangakis AS, Hegerl R, Nickell S, Typke D, Baumeister W. Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms. Proc Natl Acad Sci USA. 2000;97:14245–14250. doi: 10.1073/pnas.230282097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- 56.Mercogliano CP, DeRosier DJ. Concatenated metallothionein as a clonable gold label for electron microscopy. J Struct Biol. 2007;160:70–82. doi: 10.1016/j.jsb.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peckys DB, Mazur P, Gould KL, de Jonge N. Fully hydrated yeast cells imaged with electron microscopy. Biophys J. 2011;100:2522–2529. doi: 10.1016/j.bpj.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Labernadie A, Thibault C, Vieu C, Maridonneau-Parini I, Charriere GM. Dynamics of podosome stiffness revealed by atomic force microscopy. Proc Natl Acad Sci USA. 2010;107:21016–21021. doi: 10.1073/pnas.1007835107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hagen C, Guttmann P, Klupp B, Werner S, Rehbein S, Mettenleiter TC, Schneider G, Grunewald K. Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells. J Struct Biol. 2012;177:193–201. doi: 10.1016/j.jsb.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith EA, Cinquin BP, McDermott G, Le Gros MA, Parkinson DY, Kim HT, Larabell CA. Correlative microscopy methods that maximize specimen fidelity and data completeness, and improve molecular localization capabilities. J Struct Biol. 2013 doi: 10.1016/j.jsb.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murphy GE, Narayan K, Lowekamp BC, Hartnell LM, Heymann JA, Fu J, Subramaniam S. Correlative 3D imaging of whole mammalian cells with light and electron microscopy. J Struct Biol. 2011;176:268–278. doi: 10.1016/j.jsb.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]