

Abstract
Obesity due to excessive food intake and the lack of physical activity is becoming one of the most serious public health problems of the 21st century. With the increasing prevalence of obesity, non-alcoholic fatty liver disease is also emerging as a pandemic. While previously this pathophysiological condition was mainly attributed to triglyceride accumulation in hepatocytes, recent data show that the development of oxidative stress, lipid peroxidation, cell death, inflammation and fibrosis are mostly due to accumulation of fatty acids, and the altered composition of membrane phospholipids. In fact, triglyceride accumulation might play a protective role, and the higher toxicity of saturated or trans fatty acids seems to be the consequence of a blockade in triglyceride synthesis. Increased membrane saturation can profoundly disturb cellular homeostasis by impairing the function of membrane receptors, channels and transporters. However, it also induces endoplasmic reticulum stress via novel sensing mechanisms of the organelle’s stress receptors. The triggered signaling pathways in turn largely contribute to the development of insulin resistance and apoptosis. These findings have substantiated the lipotoxic liver injury hypothesis for the pathomechanism of hepatosteatosis. This minireview focuses on the metabolic and redox aspects of lipotoxicity and lipoapoptosis, with special regards on the involvement of endoplasmic reticulum stress responses.
Keywords: Saturated fatty acid, Lipotoxicity, Steatosis, Lipoapoptosis, Endoplasmic reticulum stress
Core tip: Surplus of free fatty acids contributes to hepatic injuries in obesity and type 2 diabetes. Intracellular accumulation of fatty acyl-CoA causes oxidative and endoplasmic reticulum (ER) stress, which lead to cell death, inflammation and fibrosis. Steatohepatosis is the consequence of an intensive fat synthesis, aiming to reduce the metabolic burden. The higher toxicity of saturated vs unsaturated fatty acids is partly due to a limited capacity of the liver cells to insert them into triglycerides. Moreover, increased membrane saturation triggers the ER stress response though a unique mechanism, which aggravates the metabolic derangements and liver injuries.
INTRODUCTION
Special metabolic features make hepatocytes play a central role in the metabolism of nutrients as well as endo- and xenobiotics. Liver can consume or produce various nutrients with a great compliance and adapts quickly to changing circumstances. However, the challenge raised by substantial alterations in modern life style and diet or the abuse of certain foods or drinks pushes the limits of adjustability, which leads to liver damage and metabolic disorders. The increasing prevalence of obesity, the metabolic syndrome and type 2 diabetes brings the pathological role of fatty acids into the focus of interest. This review is focused on the metabolic and redox bases of the fatty acid-induced cell injuries (lipotoxicity) and cell death (lipoapoptosis) in hepatocytes, with special regards to the contribution of oxidative and endoplasmic reticulum (ER) stress.
FATTY ACID METABOLISM IN THE LIVER
Liver plays a prominent role in maintaining a balanced nutrient supply in blood throughout the ever changing nutritional and metabolic conditions. Therefore, hepatocytes can shift from intensive fatty acid synthesis (in fed state) to rapid fatty acid breakdown (in starvation). Fatty acids, the most efficiently and economically storable fuel molecules are synthesized from the excess of carbohydrates and amino acids during the absorptive phase (Figure 1A). However, they are not released into the blood plasma as non-esterified or free fatty acids (NEFA or FFA) but incorporated into complex lipids such as triglycerides, phosphoglycerolipids or cholesterylesters in the ER membrane[1]. Microsomal triglyceride transfer protein (MTP) mediates the association of triglycerides with ApoB100 in the ER lumen and the assembled very low density lipoprotein (VLDL) lipoprotein particles finally leave the cell through exocytosis[2] (Figure 1A). Fatty acid components of ingested lipids are absorbed predominantly as reconstructed complex lipids packed in chylomicrons. These lipoproteins reach the systemic circulation through lymph vessels rather than portal veins and deliver triglycerides in the adipose tissue for storage.
Figure 1.
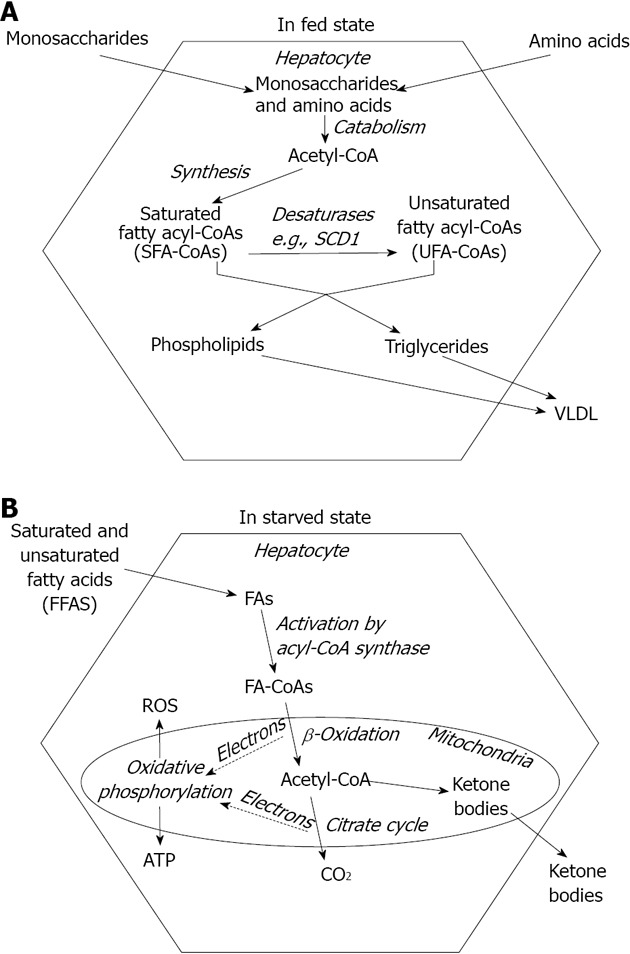
Fatty acid metabolism in the liver. A: In fed state, monosaccharides and amino acids are taken up from the portal blood and converted to fatty acyl-CoAs through acetyl-CoA intermediate; The primary endogenous fatty acids (e.g., palmitate) are saturated. Formation of mono- or polyunsaturated fatty acids involves modification(s) by desaturase enzymes; Newly synthesized fatty acids are inserted into complex (saponifiable) lipids, which in turn are packed in very low density lipoprotein (VLDL) particles and so secreted from the hepatocytes; B: In starvation, free fatty acids (FFAs) derive from triglyceride mobilization in the adipose tissue. After a protein-mediated uptake across the plasma membrane, they are activated to acyl-CoA, and catabolized in the mitochondria through β-oxidation and citrate cycle. These metabolic pathways provide electrons to oxidative phosphorylation, the major source of ATP and ROS in aerobic cells. Shortage of citrate cycle intermediates leads to accumulation of acetyl-CoA, and thence enhances the synthesis of ketone bodies.
FFAs normally derive from triglyceride breakdown in the adipocytes, and hence their abundance occurs in starvation when store mobilization is stimulated by hormones (e.g., glucagon, adrenalin and glucocorticoids)[3]. FFAs are utilized as energy source in the liver cells. After a protein-mediated uptake across the plasma membrane[4], they are activated to acyl-CoA, transported into the mitochondrial matrix via carnitine shuttle and catabolized through β-oxidation and citrate cycle[5]. Long term starvation can deprive citrate cycle of intermediates and make it unable to keep pace with acetyl-CoA production. Accumulating acetyl-CoA is diverted toward the synthesis of β-ketobutyrate (acetoacetate) and β-hydroxybutyrate[3]. These ketone bodies are secreted from the hepatocytes into the blood plasma and serve as alternative fuels to most aerobic tissues including the brain (Figure 1B).
Fatty acids, either endogenous or exogenous, are handled in an activated form, i.e., attached to CoA through a thioester bond. Carnitine shuttle leading to degradation as well as acyl transferase enzymes incorporating fatty acids in complex lipids require the formation acyl-CoA in the cytosolic compartment[6]. Fatty acyl-CoA molecules can also be modified by ER-associated enzymes to reach the species-specific composition of triglycerides and phospholipids. These modifications include chain elongation and desaturation (Figure 1A). Monounsaturated fatty acids typically contain a cis double bond at the Δ9 position, which is inserted into fatty acyl-CoAs by stearyl-CoA desaturase 1 (SCD1) in humans. SCD1 converts palmitoyl (C16:0)-CoA into palmitoleoyl (C16:1)-CoA and stearoyl (C18:0)-CoA to oleoyl (C18:1)-CoA. It is an iron-containing enzyme that receives electrons from NAD(P)H through cytochrome b5 reductase and cytochrome b5 in the ER membrane[7]. Ncb5or, a novel soluble oxidoreductase contains both cytochrome b5 reductase like and cytochrome b5 like domains and hence it is also proposed to participate in fatty acid desaturation[8].
Membrane lipid saturation is regulated through the activity of various lysophospolipid acyltransferase enzymes exhibiting different acyl-CoA specificities. For example, among the enzymes involved in phosphatidylcholine remodeling, lysophosphatidylcholine acyltransferase 1 (LPCAT1) preferentially incorporates saturated fatty acids (e.g., palmitate)[9] while LPCAT3 favors polyunsaturated fatty acids (e.g., arachidonic acid)[10], which allows modulation of phospholipid fatty acid composition through altered expression of these isoenzymes[11].
HEPATOSTEATOSIS
Triglyceride deposition within the hepatocytes is the hallmark of both alcoholic and nonalcoholic fatty liver diseases (AFLD and NAFLD)[12]. It occurs when VLDL assembly and secretion cannot keep pace with lipid synthesis. This hepatosteatosis can be caused by various conditions including stimulated hepatic fatty acid synthesis due to overfeeding and high fructose consumption[13] and inhibition of MTP[14,15]. Ethanol, one of the best known inducers of fatty liver both enhances fatty acid synthesis and hinders VLDL secretion[16]. It has been revealed that elevated FFA level should also be considered as a major cause of hepatosteatosis. Combination of genetic predisposition with certain environmental factors, such as sedentary life style, overfeeding and obesity can lead to a surplus of FFA due to deregulation of fatty acid storage and mobilization. The metabolic stress caused by excessive supply of nutrients in various tissues comprises oxidative and organelle stress components. The consequent stress response and inflammatory reaction can lead to insulin resistance, which favors triglyceride mobilization and decreases FFA uptake in the adipocytes[17]. Untrained skeletal muscle of low metabolic rate and of small relative mass has a limited contribution to fuel consumption, which further increases both insulin resistance and the strain on the adipose tissue. Moreover, the increased adipocyte mass is associated with an elevated FFA release. Enlargement of visceral adipose tissue is especially important because it secretes FFA into the portal circulation, i.e., directly to the liver. In addition to the mounting FFA supply, the compensatory hyperinsulinemia stimulates fatty acid synthesis and inhibits fatty acid catabolism in the hepatocytes. When fatty acid input overcomes the capacity of β-oxidation, accumulating acyl-CoA is drained by triglyceride synthesis, which leads to steatosis in the liver (Figure 2A).
Figure 2.
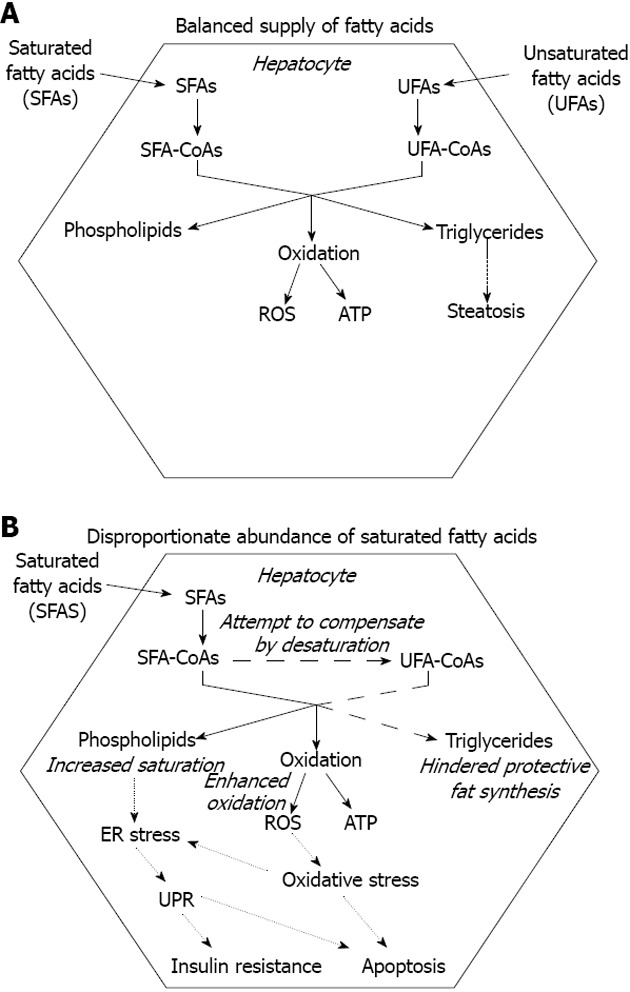
Fatty acid toxicity in the liver. A: Balanced (over) supply of saturated and unsaturated fatty acids allows the hepatocyte to enhance triglyceride synthesis and deposition. Storage of excess fatty acids prevents superfluous fatty acid oxidation and thereby protects against oxidative stress; B: Disproportionate abundance of saturated fatty acids might not be compensated for by fatty acid desaturation (dashed lines). Hindrance of protective fat synthesis favors fatty acid oxidation and leads to excessive ROS generation; The emerging oxidative stress and the increased saturation of membrane lipids trigger the endoplasmic reticulum (ER) stress response, which contributes to insulin resistance and further enhances cell death (dotted lines).
Accumulation of fatty acids or fatty acyl-CoAs, however, may be more harmful to hepatocytes than deposition of triglycerides. It has been demonstrated in HepG2 and Huh7 human liver cell lines that lipotoxic effect of the saturated stearate is initiated by interruption of triacylglycerol synthesis. Retained capacity to synthesize and accumulate triglycerides in the presence of unsaturated oleate, in fact, turned out to be protective[18].
TOXICITY OF SATURATED FATTY ACIDS AND TRANS FATTY ACIDS
The typical dietary pattern of western populations leads to high intake of saturated fatty acids (SFAs) derived from animal sources. Their metabolic and health effects are similar to those of trans fatty acids (TFAs), i.e., unsaturated fatty acids containing at least one non-conjugated double bond in the trans configuration. TFAs are also found in foods of natural origin because ruminant animals obtain these lipids from bacterial hydrogenation of unsaturated fatty acids in the rumen[19]. In addition, TFAs are also formed during the industrial production of certain foods, and major food sources of dietary TFA in the United States. population were found to be cakes, cookies, pies and pastries[20]. The proinflammatory status favored by both SFAs and TFAs plays an important role in several diseases[21].
The greater hepatotoxicity of saturated vs unsaturated fatty acids has been demonstrated in several studies[18,22-25]. Moreover, the unsaturated fatty acids often prove to be protective against SFA-induced toxicity[18,24]. Interruption of triglyceride synthesis, apparently because of the formation of a pool of oversaturated intermediates, seems to be a key event in SFA-induced lipotoxicity. Accumulation of stearoyl-CoA and a decreased capacity of triglyceride production have been found in HepG2 and Huh7 human hepatoma cells exposed to stearate[18]. In accordance with its central role in converting SFAs to monounsaturated fatty acids (MUFAs), SCD1 proved to be protective against SFA-induced hepatocyte lipoapoptosis (Figure 2B). Studies on SCD1 knockout mice lead to the conclusion that prevention of steatohepatitis involves partitioning excess SFAs into MUFAs by hepatic SCD1[26]. Hepatocytes of Ncb5or knockout mice have an increased sensitivity to SFA-induced damage, which can be at least partly attributed to a lower SCD-specific activity in these cells[27]. This observation strongly supports the hypothesized functional link between SCD1 and Ncb5or[8]. Some natural human mutations of NCB5OR have been reported to remarkably decrease the level of active enzyme due to enhanced proteasomal degradation[28]; therefore, the potential role of this gene in human pathology deserves further investigation.
Controversial data have been published regarding the control of autophagy by unsaturated and saturated fatty acids in primary hepatocytes and hepatoma cells[29,30]. Although oleic acid has been found to induce autophagy-mediated apoptosis in mild but not in severe steatosis in HepG2 cells[31], it seems plausible that fatty acid-induced autophagy generally plays a pro-survival role in lipotoxicity[29,30,32]. However, hindrance of the autophagic flux by oxidative stress and/or advanced apoptosis[29,32] is more likely to occur in SFA-induced lipotoxicity. Therefore, the greater toxicity of saturated vs unsaturated fatty acids might be also attributed to differential regulation of autophagy.
The health risk raised by the increasing ingestion of TFAs has recently got into the focus of scientific interest. The relationship between dietary fat induced obesity and TFAs is not unequivocally elucidated. Nevertheless, dietary intake of high TFAs increases liver weight and hepatic triglyceride content, and causes deleterious alterations in serum cholesterol levels in rats[33]. TFAs have been shown to be a primary factor responsible for the development of NAFLD in an animal model of American lifestyle induced obesity syndrome either with[34] or without inclusion of high fructose corn syrup[35,36] in the diet. Increased hepatic lipid peroxidation as an indicator of oxidative stress has also been demonstrated in the livers of high TFA-fed rats, and it might underlie the development of NAFLD[37]. Long term ingestion of TFAs has been shown to increase liver triglycerides due to enhanced activity and expression of lipogenic enzymes and elevated expression of sterol regulatory element-binding protein SREBP-1a in mice. However these effects largely depend on the dietary fatty acid composition[38]. The extent and severity of health injuries caused by TFAs as well as the mechanisms of TFA-induced hepatotoxicity remain to be clarified.
LIPOTOXIC OXIDATIVE STRESS
Several observations suggest the role of increased reactive oxygen species (ROS) generation in hepatic lipotoxicity. Growing oxidative challenge is well demonstrated by increased serum levels of ox-LDL and higher serum lipid peroxidation in nonalcoholic steatohepatitis[39]. Palmitate-induced oxidative stress has been reported to contribute to insulin resistance in H4IIEC3 rat hepatocytes[40]. Fatty acid accumulation stimulates ROS generation in the liver presumably due to enhanced β-oxidation and to the consequent electron overflow in the mitochondrial electron transfer chain[41] (Figure 2B). However, a decrease in mitochondrial quinone pool and a related inhibition of mitochondrial oxidative metabolism were also suggested to underlie the increased mitochondrial ROS production in high fat diet[42]. Mitochondrial depolarization, cytochrome c release, and increased ROS production were detected in HepG2 and McNtcp.24 liver cells exposed to saturated FFAs, and the role of lysosomal disruption was also suggested[43]. Increased expression[44] and activity[45,46] of cytochrome P450 2E1 (CYP2E1) monooxygenase likely contributes to the oxidative stress in lipotoxicity. CYP2E1 is an integral membrane protein of the ER and a significant source of oxidative intermediates including free radicals. It is involved in the biotransformation of several endo/xenobiotics, and it carries out omega hydroxylation of fatty acids. CYP2E1 activity was shown to positively correlate with body mass index and with the degree of steatosis[47]. The role of omega hydroxylating CYP4A isoenzymes was also suggested in animal models of nonalcoholic steatohepatitis[48]. CYP4A enzymes are under control of peroxisome proliferator activated receptors, which also induce peroxisomal fatty acid beta oxidation, another prominent ROS generating pathway[49]. Besides the enhancement of oxidative fatty acid metabolism, palmitate-mediated up-regulation of direct ROS production by NADPH oxidase 3 has been also revealed recently in db/db mice and HepG2 cells[50].
Excessive ROS production is directly deleterious by injuring DNA, proteins and lipids. It also favors cell death through activation of certain stress-sensitive signaling pathways, such as nuclear factor κB, p38MAPK, and c-Jun N-terminal kinase (JNK)[51,52]. In addition, a general cellular oxidative stress inevitably affects the intricate redox homeostasis in the ER lumen[53-55]. The controlled maintenance of an oxidized thiol-disulfide redox system in this compartment is the prerequisite of appropriate protein maturation[56]. On the other hand, the sufficiently reduced pyridine nucleotide pool in the ER lumen provides a metabolic basis for prereceptor glucocorticoid hormone activation[57,58].
LIPOTOXIC ER STRESS
Protein processing, one of the major functions of the ER[56,59], is necessarily affected by any severe dysfunction of the organelle, i.e., the ER stress[60]. Lowered protein folding capacity of the ER renders it unable to keep pace with protein load. Accumulation of immature proteins in the ER lumen triggers a complex primarily adaptive signaling network called the unfolded protein response (UPR) in order to restore ER homeostasis. Unfolded proteins recruit increasing amounts of BiP, ER chaperone, which is thus detached from the luminal domains of three transmembrane ER stress sensor proteins: RNA-dependent protein kinase-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). They initiate the three main branches of the UPR collectively inducing attenuation of protein synthesis, expression of ER chaperones, and degradation of misfolded proteins[61].
Phosphorylation of eukaryotic initiation factor eIF2α by PERK lowers the ER protein load by attenuating general translation, and also enhances the synthesis of ATF4 transcription factor. IRE1-mediated specific mRNA splicing allows the synthesis of X-box-binding protein 1 (XBP1) transcription factor, which enhances the ATF6-dependent adaptive transcriptional alterations (e.g., induction of ER chaperones). When the UPR fails to restore the ER functions, the ER-derived death signals may take effect. Apoptosis is stimulated mostly by induction of CCAAT/enhancer binding protein homologous protein (CHOP) and activation of JNK. It should be noted that JNK activation is also triggered by the oxidative stress in lipotoxicity[50]. In addition to its pro-apoptotic activity, JNK also interferes with insulin signaling by phosphorylating insulin receptor substrate-1 (IRS-1) at serine (307)[62], which represents a key link between ER-stress and insulin resistance.
ER stress contributes to the pathology of several liver diseases associated with steatosis. Activation of the UPR is involved in fatty liver disease either alcoholic or non-alcoholic as well as viral hepatitis. Enhanced apoptosis of liver cells in these diseases may be at least partly due to unresolved ER stress[63]. Mounting evidence support the involvement of ER dysfunctions in the toxicity of fatty acids, especially long chain, saturated ones. The programmed cell death, at least partly due to ER stress, caused by fatty acids is referred to as lipoapoptosis. The role of PERK/ATF4/CHOP signaling pathway has been demonstrated in saturated fatty acid-induced ER stress and lipoapoptosis in L02 and HepG2 human liver cell lines[64]. Elevated exogenous pamitate has been shown to disrupt ER homeostasis by reducing the expression of Bip in HepG2 cells. Overexpression of Bip attenuated ER stress, reduced CHOP expression and protected the cells from palmitate-induced apoptosis[22]. Lipotoxic derangement of ER functions is likely due to the above mentioned oxidative stress, disturbed calcium homeostasis and altered membrane saturation. Membrane bound and luminal oxidase enzymes maintain a thiol oxidizing environment in the ER, a prerequisite of oxidative folding[65]. It is therefore not surprising that the protein processing machinery of the organelle is sensitive to any disturbance of the redox and antioxidant status of the cell[66]. Inhibition of fatty acid oxidation has been shown to protect hepatocytes from ER stress, and this was accompanied by an increased cellular redox potential as judged by an elevated ratio of oxidized to reduced glutathione and enhanced oxidative folding in the ER[67]. Saturated, long chain fatty acids have been shown to reduce thapsigargin-sensitive ER calcium stores and increase biochemical markers of ER stress in H4IIE liver cells and primary hepatocytes[24]. Palmitate- but not linoleate-induced ER stress was prevented by depletion of intracellular calcium flux in the same cell line[68].
It has been demonstrated that insufficient desaturase activity (SCD1 knockdown) or decreased incorporation of unsaturated fatty acids in membrane phospholipids (LPCAT3 knockdown) can synergistically induce the UPR. The effect is likely due to a decrease in membrane phospholipids unsaturation as it is further enhanced by saturated while it is rescued by unsaturated fatty acids[69] (Figure 2B). Recent findings suggest that ER lipid perturbation can trigger the UPR directly and independently of luminal accumulation of unfolded proteins. The mechanisms through which the UPR is activated by accumulating unfolded proteins and membrane lipid saturation turned out to be different as the latter does not involve large protein cluster formation in the ER membrane[70]. Two of the ER stress receptors, IRE1α and PERK have been revealed to respond to increased lipid saturation. Their lipid sensitivity was retained when the luminal unfolded protein sensing domains were removed and was associated to their transmembrane domains[71]. This novel mechanism is in accordance with the greater toxicity of saturated vs unsaturated fatty acids as well as with the protective effect of unsaturated fatty acids against saturated fatty acid induced toxicity[18,22-25].
The thapsigargin- and tunicamycin-induced UPR was found to increase the expression of sterol regulatory element-binding protein (SREBP-1c) transcription factor in HepG2 cells, through the cap-independent translation mediated by an internal ribosome entry site[72]. Thapsigargin-induced SREBP-1c activation and the consequent stimulation of fatty acid synthesis were also confirmed in the normal human L02 cell line[73]. This effect might create a positive feed back loop and further aggravate the metabolic disorder in lipotoxic ER stress. On the other hand, several genes related to lipoprotein secretion are controlled by IRE1α, and hence induction of the UPR upon membrane lipid perturbation might contribute to the prevention of hepatic steatosis[74]. Interestingly, mice with hepatocyte-specific IRE1α deletion without ER stress display modest hepatosteatosis, and this is aggravated after induction of ER stress[74]. Therefore, disturbed lipid metabolism can lead to ER stress and trigger the UPR, and the ER stress-dependent alteration in lipid homeostasis might underlie the hepatic steatosis.
CONCLUSION
The unhealthy combination of modern diet and lifestyle often leads to nutrient surplus and a consequent fat deposition in various non-adipose tissues including the liver. This hepatosteatosis has long been considered as the fundamental cause of hepatic injuries characterizing both alcoholic and non-alcoholic fatty liver diseases, i.e. oxidative and ER stress, cell death, inflammation and fibrosis. In light of recent studies, however, fat accumulation is considered rather protective as the hepatocyte damage is mostly caused by fatty acyl-CoA (Figure 1A). Enhanced triglyceride synthesis decreases the oxidative challenge by lowering fatty acid oxidation. The higher toxicity of SFAs vs unsaturated fatty acids seems to be at least partly due to an increased tendency of the SFAs to accumulate because of a limited capacity of the liver cells to insert them into triglycerides (Figure 2B). SFA-induced toxicity is indeed enhanced by insufficient activity of the enzymes involved in fatty acid desaturation and triglyceride synthesis while attenuated by simultaneous administration of unsaturated fatty acids. Moreover, growing evidence indicates that increased saturation of phospholipid membranes can trigger the ER stress response though a unique mechanism. The activated signaling pathways lead to insulin resistance and hepatocyte apoptosis, significantly contributing to aggravation of the metabolic derangements and liver injuries caused by SFAs (Figure 2B).
ACKNOWLEDGEMENTS
Éva Kereszturi is a grantee of the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Footnotes
Supported by Hungarian Scientific Research Fund (OTKA 104113 and 106060) and the Hungarian Research and Technological Innovation Fund (KMR_12-1-2012-0074)
P- Reviewers Fan XM, Pai CG, Sheng L, Zhong ZH S- Editor Qi Y L- Editor A E- Editor Yan JL
References
- 1.Stein O, Stein Y. Lipid synthesis, intracellular transport, storage, and secretion. I. Electron microscopic radioautographic study of liver after injection of tritiated palmitate or glycerol in fasted and ethanol-treated rats. J Cell Biol. 1967;33:319–339. doi: 10.1083/jcb.33.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khatun I, Zeissig S, Iqbal J, Wang M, Curiel D, Shelness GS, Blumberg RS, Hussain MM. Phospholipid transfer activity of microsomal triglyceride transfer protein produces apolipoprotein B and reduces hepatosteatosis while maintaining low plasma lipids in mice. Hepatology. 2012;55:1356–1368. doi: 10.1002/hep.25504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grey NJ, Karl I, Kipnis DM. Physiologic mechanisms in the development of starvation ketosis in man. Diabetes. 1975;24:10–16. doi: 10.2337/diab.24.1.10. [DOI] [PubMed] [Google Scholar]
- 4.Kazantzis M, Stahl A. Fatty acid transport proteins, implications in physiology and disease. Biochim Biophys Acta. 2012;1821:852–857. doi: 10.1016/j.bbalip.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bremer J. Carnitine--metabolism and functions. Physiol Rev. 1983;63:1420–1480. doi: 10.1152/physrev.1983.63.4.1420. [DOI] [PubMed] [Google Scholar]
- 6.Coleman RA, Lewin TM, Van Horn CG, Gonzalez-Baró MR. Do long-chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr. 2002;132:2123–2126. doi: 10.1093/jn/132.8.2123. [DOI] [PubMed] [Google Scholar]
- 7.Man WC, Miyazaki M, Chu K, Ntambi JM. Membrane topology of mouse stearoyl-CoA desaturase 1. J Biol Chem. 2006;281:1251–1260. doi: 10.1074/jbc.M508733200. [DOI] [PubMed] [Google Scholar]
- 8.Deng B, Parthasarathy S, Wang W, Gibney BR, Battaile KP, Lovell S, Benson DR, Zhu H. Study of the individual cytochrome b5 and cytochrome b5 reductase domains of Ncb5or reveals a unique heme pocket and a possible role of the CS domain. J Biol Chem. 2010;285:30181–30191. doi: 10.1074/jbc.M110.120329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakanishi H, Shindou H, Hishikawa D, Harayama T, Ogasawara R, Suwabe A, Taguchi R, Shimizu T. Cloning and characterization of mouse lung-type acyl-CoA: lysophosphatidylcholine acyltransferase 1 (LPCAT1). Expression in alveolar type II cells and possible involvement in surfactant production. J Biol Chem. 2006;281:20140–20147. doi: 10.1074/jbc.M600225200. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Chen YQ, Bonacci TM, Bredt DS, Li S, Bensch WR, Moller DE, Kowala M, Konrad RJ, Cao G. Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J Biol Chem. 2008;283:8258–8265. doi: 10.1074/jbc.M710422200. [DOI] [PubMed] [Google Scholar]
- 11.Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, Shimizu T. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci USA. 2008;105:2830–2835. doi: 10.1073/pnas.0712245105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsukamoto H. Fat paradox in liver disease. Keio J Med. 2005;54:190–192. doi: 10.2302/kjm.54.190. [DOI] [PubMed] [Google Scholar]
- 13.Gaby AR. Adverse effects of dietary fructose. Altern Med Rev. 2005;10:294–306. [PubMed] [Google Scholar]
- 14.Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–156. doi: 10.1056/NEJMoa061189. [DOI] [PubMed] [Google Scholar]
- 15.Lee RG, Fu W, Graham MJ, Mullick AE, Sipe D, Gattis D, Bell TA, Booten S, Crooke RM. Comparison of the pharmacological profiles of murine antisense oligonucleotides targeting apolipoprotein B and microsomal triglyceride transfer protein. J Lipid Res. 2013;54:602–614. doi: 10.1194/jlr.M029215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasineni K, Casey CA. Molecular mechanism of alcoholic fatty liver. Indian J Pharmacol. 2012;44:299–303. doi: 10.4103/0253-7613.96297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18:139–143. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mantzaris MD, Tsianos EV, Galaris D. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS J. 2011;278:519–530. doi: 10.1111/j.1742-4658.2010.07972.x. [DOI] [PubMed] [Google Scholar]
- 19.Bauman DE, Mather IH, Wall RJ, Lock AL. Major advances associated with the biosynthesis of milk. J Dairy Sci. 2006;89:1235–1243. doi: 10.3168/jds.S0022-0302(06)72192-0. [DOI] [PubMed] [Google Scholar]
- 20.Kris-Etherton PM, Lefevre M, Mensink RP, Petersen B, Fleming J, Flickinger BD. Trans fatty acid intakes and food sources in the U.S. population: NHANES 1999-2002. Lipids. 2012;47:931–940. doi: 10.1007/s11745-012-3704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estadella D, da Penha Oller do Nascimento CM, Oyama LM, Ribeiro EB, Dâmaso AR, de Piano A. Lipotoxicity: effects of dietary saturated and transfatty acids. Mediators Inflamm. 2013;2013:137579. doi: 10.1155/2013/137579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu X, Li K, Laybutt DR, He ML, Zhao HL, Chan JC, Xu G. Bip overexpression, but not CHOP inhibition, attenuates fatty-acid-induced endoplasmic reticulum stress and apoptosis in HepG2 liver cells. Life Sci. 2010;87:724–732. doi: 10.1016/j.lfs.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 23.Nivala AM, Reese L, Frye M, Gentile CL, Pagliassotti MJ. Fatty acid-mediated endoplasmic reticulum stress in vivo: differential response to the infusion of Soybean and Lard Oil in rats. Metabolism. 2013;62:753–760. doi: 10.1016/j.metabol.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei Y, Wang D, Gentile CL, Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem. 2009;331:31–40. doi: 10.1007/s11010-009-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Dong L, Yang X, Shi H, Zhang L. α-Linolenic acid prevents endoplasmic reticulum stress-mediated apoptosis of stearic acid lipotoxicity on primary rat hepatocytes. Lipids Health Dis. 2011;10:81. doi: 10.1186/1476-511X-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem. 2009;284:5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu M, Wang W, Frontera JR, Neely MC, Lu J, Aires D, Hsu FF, Turk J, Swerdlow RH, Carlson SE, et al. Ncb5or deficiency increases fatty acid catabolism and oxidative stress. J Biol Chem. 2011;286:11141–11154. doi: 10.1074/jbc.M110.196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kálmán FS, Lizák B, Nagy SK, Mészáros T, Zámbó V, Mandl J, Csala M, Kereszturi E. Natural mutations lead to enhanced proteasomal degradation of human Ncb5or, a novel flavoheme reductase. Biochimie. 2013;95:1403–1410. doi: 10.1016/j.biochi.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 29.Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan SH, Shui G, Zhou J, Li JJ, Bay BH, Wenk MR, Shen HM. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin) J Biol Chem. 2012;287:14364–14376. doi: 10.1074/jbc.M111.294157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu K, Lou J, Wen T, Yin J, Xu B, Ding W, Wang A, Liu D, Zhang C, Chen D, et al. Depending on the stage of hepatosteatosis, p53 causes apoptosis primarily through either DRAM-induced autophagy or BAX. Liver Int. 2013:Jun 16; Epub ahead of print. doi: 10.1111/liv.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang P, Huang Z, Zhao H, Wei T. Hydrogen peroxide impairs autophagic flux in a cell model of nonalcoholic fatty liver disease. Biochem Biophys Res Commun. 2013;433:408–414. doi: 10.1016/j.bbrc.2013.02.118. [DOI] [PubMed] [Google Scholar]
- 33.Ochiai M, Fujii K, Takeuchi H, Matsuo T. Effects of dietary trans fatty acids on fat accumulation and metabolic rate in rat. J Oleo Sci. 2013;62:57–64. doi: 10.5650/jos.62.57. [DOI] [PubMed] [Google Scholar]
- 34.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–G995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obara N, Fukushima K, Ueno Y, Wakui Y, Kimura O, Tamai K, Kakazu E, Inoue J, Kondo Y, Ogawa N, et al. Possible involvement and the mechanisms of excess trans-fatty acid consumption in severe NAFLD in mice. J Hepatol. 2010;53:326–334. doi: 10.1016/j.jhep.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 36.Machado RM, Stefano JT, Oliveira CP, Mello ES, Ferreira FD, Nunes VS, de Lima VM, Quintão EC, Catanozi S, Nakandakare ER, et al. Intake of trans fatty acids causes nonalcoholic steatohepatitis and reduces adipose tissue fat content. J Nutr. 2010;140:1127–1132. doi: 10.3945/jn.109.117937. [DOI] [PubMed] [Google Scholar]
- 37.Dhibi M, Brahmi F, Mnari A, Houas Z, Chargui I, Bchir L, Gazzah N, Alsaif MA, Hammami M. The intake of high fat diet with different trans fatty acid levels differentially induces oxidative stress and non alcoholic fatty liver disease (NAFLD) in rats. Nutr Metab (Lond) 2011;8:65. doi: 10.1186/1743-7075-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saín J, González MA, Lasa A, Scalerandi MV, Bernal CA, Portillo MP. Effects of trans-fatty acids on liver lipid metabolism in mice fed on diets showing different fatty acid composition. Ann Nutr Metab. 2013;62:242–249. doi: 10.1159/000339453. [DOI] [PubMed] [Google Scholar]
- 39.Chalasani N, Deeg MA, Crabb DW. Systemic levels of lipid peroxidation and its metabolic and dietary correlates in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:1497–1502. doi: 10.1111/j.1572-0241.2004.30159.x. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura S, Takamura T, Matsuzawa-Nagata N, Takayama H, Misu H, Noda H, Nabemoto S, Kurita S, Ota T, Ando H, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284:14809–14818. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seifert EL, Estey C, Xuan JY, Harper ME. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J Biol Chem. 2010;285:5748–5758. doi: 10.1074/jbc.M109.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vial G, Dubouchaud H, Couturier K, Cottet-Rousselle C, Taleux N, Athias A, Galinier A, Casteilla L, Leverve XM. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J Hepatol. 2011;54:348–356. doi: 10.1016/j.jhep.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 43.Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology. 2008;47:1495–1503. doi: 10.1002/hep.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128–133. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 45.Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quiñones L, Varela N, Contreras J, Lazarte R, Csendes A, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci (Lond) 2004;106:261–268. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 46.Orellana M, Rodrigo R, Varela N, Araya J, Poniachik J, Csendes A, Smok G, Videla LA. Relationship between in vivo chlorzoxazone hydroxylation, hepatic cytochrome P450 2E1 content and liver injury in obese non-alcoholic fatty liver disease patients. Hepatol Res. 2006;34:57–63. doi: 10.1016/j.hepres.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Chtioui H, Semela D, Ledermann M, Zimmermann A, Dufour JF. Expression and activity of the cytochrome P450 2E1 in patients with nonalcoholic steatosis and steatohepatitis. Liver Int. 2007;27:764–771. doi: 10.1111/j.1478-3231.2007.01524.x. [DOI] [PubMed] [Google Scholar]
- 48.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardwick JP, Osei-Hyiaman D, Wiland H, Abdelmegeed MA, Song BJ. PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid omega-Hydroxylase (CYP4) Isozymes: Implications for Prevention of Lipotoxicity in Fatty Liver Disease. PPAR Res. 2009;2009:952734. doi: 10.1155/2009/952734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao D, Nong S, Huang X, Lu Y, Zhao H, Lin Y, Man Y, Wang S, Yang J, Li J. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J Biol Chem. 2010;285:29965–29973. doi: 10.1074/jbc.M110.128694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 52.Ghosh J, Das J, Manna P, Sil PC. Taurine prevents arsenic-induced cardiac oxidative stress and apoptotic damage: role of NF-kappa B, p38 and JNK MAPK pathway. Toxicol Appl Pharmacol. 2009;240:73–87. doi: 10.1016/j.taap.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 53.Bánhegyi G, Margittai E, Szarka A, Mandl J, Csala M. Crosstalk and barriers between the electron carriers of the endoplasmic reticulum. Antioxid Redox Signal. 2012;16:772–780. doi: 10.1089/ars.2011.4437. [DOI] [PubMed] [Google Scholar]
- 54.Bánhegyi G, Benedetti A, Csala M, Mandl J. Stress on redox. FEBS Lett. 2007;581:3634–3640. doi: 10.1016/j.febslet.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 55.Piccirella S, Czegle I, Lizák B, Margittai E, Senesi S, Papp E, Csala M, Fulceri R, Csermely P, Mandl J, et al. Uncoupled redox systems in the lumen of the endoplasmic reticulum. Pyridine nucleotides stay reduced in an oxidative environment. J Biol Chem. 2006;281:4671–4677. doi: 10.1074/jbc.M509406200. [DOI] [PubMed] [Google Scholar]
- 56.Csala M, Kereszturi É, Mandl J, Bánhegyi G. The endoplasmic reticulum as the extracellular space inside the cell: role in protein folding and glycosylation. Antioxid Redox Signal. 2012;16:1100–1108. doi: 10.1089/ars.2011.4227. [DOI] [PubMed] [Google Scholar]
- 57.Czegle I, Csala M, Mandl J, Benedetti A, Karádi I, Bánhegyi G. G6PT-H6PDH-11βHSD1 triad in the liver and its implication in the pathomechanism of the metabolic syndrome. World J Hepatol. 2012;4:129–138. doi: 10.4254/wjh.v4.i4.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bánhegyi G, Csala M, Benedetti A. Hexose-6-phosphate dehydrogenase: linking endocrinology and metabolism in the endoplasmic reticulum. J Mol Endocrinol. 2009;42:283–289. doi: 10.1677/JME-08-0156. [DOI] [PubMed] [Google Scholar]
- 59.Csala M, Bánhegyi G, Benedetti A. Endoplasmic reticulum: a metabolic compartment. FEBS Lett. 2006;580:2160–2165. doi: 10.1016/j.febslet.2006.03.050. [DOI] [PubMed] [Google Scholar]
- 60.Mandl J, Mészáros T, Bánhegyi G, Csala M. Minireview: endoplasmic reticulum stress: control in protein, lipid, and signal homeostasis. Mol Endocrinol. 2013;27:384–393. doi: 10.1210/me.2012-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mandl J, Mészáros T, Bánhegyi G, Hunyady L, Csala M. Endoplasmic reticulum: nutrient sensor in physiology and pathology. Trends Endocrinol Metab. 2009;20:194–201. doi: 10.1016/j.tem.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 62.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 63.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao J, Dai DL, Yao L, Yu HH, Ning B, Zhang Q, Chen J, Cheng WH, Shen W, Yang ZX. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell Biochem. 2012;364:115–129. doi: 10.1007/s11010-011-1211-9. [DOI] [PubMed] [Google Scholar]
- 65.Csala M, Margittai E, Bánhegyi G. Redox control of endoplasmic reticulum function. Antioxid Redox Signal. 2010;13:77–108. doi: 10.1089/ars.2009.2529. [DOI] [PubMed] [Google Scholar]
- 66.Bánhegyi G, Mandl J, Csala M. Redox-based endoplasmic reticulum dysfunction in neurological diseases. J Neurochem. 2008;107:20–34. doi: 10.1111/j.1471-4159.2008.05571.x. [DOI] [PubMed] [Google Scholar]
- 67.Tyra HM, Spitz DR, Rutkowski DT. Inhibition of fatty acid oxidation enhances oxidative protein folding and protects hepatocytes from endoplasmic reticulum stress. Mol Biol Cell. 2012;23:811–819. doi: 10.1091/mbc.E11-12-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Xue R, Zhang Z, Yang X, Shi H. Palmitic and linoleic acids induce ER stress and apoptosis in hepatoma cells. Lipids Health Dis. 2012;11:1. doi: 10.1186/1476-511X-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem. 2010;285:22027–22035. doi: 10.1074/jbc.M110.126870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kitai Y, Ariyama H, Kono N, Oikawa D, Iwawaki T, Arai H. Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells. 2013;18:798–809. doi: 10.1111/gtc.12074. [DOI] [PubMed] [Google Scholar]
- 71.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. 2013;110:4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Damiano F, Alemanno S, Gnoni GV, Siculella L. Translational control of the sterol-regulatory transcription factor SREBP-1 mRNA in response to serum starvation or ER stress is mediated by an internal ribosome entry site. Biochem J. 2010;429:603–612. doi: 10.1042/BJ20091827. [DOI] [PubMed] [Google Scholar]
- 73.Fang DL, Wan Y, Shen W, Cao J, Sun ZX, Yu HH, Zhang Q, Cheng WH, Chen J, Ning B. Endoplasmic reticulum stress leads to lipid accumulation through upregulation of SREBP-1c in normal hepatic and hepatoma cells. Mol Cell Biochem. 2013;381:127–137. doi: 10.1007/s11010-013-1694-7. [DOI] [PubMed] [Google Scholar]
- 74.Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, Chang L, Xu W, Miao H, Leonardi R, et al. The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30:1357–1375. doi: 10.1038/emboj.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]