

Abstract
AIM: To investigate the effects of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) deficiency on the cytotoxicity of chemotherapeutic agents toward colorectal cancer cells.
METHODS: PTEN-deficient colorectal cancer (CRC) cells were generated by human somatic cell gene targeting using the adeno-associated virus system. The cytotoxic effects of compounds including curcumin, 5-fluorouracil (5-FU), dihydroartemisinin (DHA), irinotecan (CPT-11) and oxaliplatin (OXA) on cancer cells were determined using the MTT assay. Enhanced cytotoxicity of curcumin in PTEN-deficient CRC cells was observed, and this was confirmed using clonogenic assays. Apoptosis and cell cycle progression were analyzed by flow cytometry. Levels of apoptosis and cell cycle-related proteins were examined by Western blotting.
RESULTS: We developed an isogenic set of CRC cell lines that differed only in their PTEN status. Using this set of cell lines, we found that disruption of the PTEN gene had no effect on the sensitivity of CRC cells to 5-FU, CPT-11, DHA, or OXA, whereas PTEN disruption increased the sensitivity of CRC cells to curcumin. Loss of PTEN did not alter the curcumin-induced apoptosis in CRC cells. However, PTEN deficiency led to an altered pattern of curcumin-mediated cell cycle arrest. In HCT116 PTEN+/+ cells, curcumin caused a G2/M phase arrest, whereas it caused a G0/G1 phase arrest in HCT116 PTEN-/- cells. Levels of cell cycle-related proteins were consistent with these respective patterns of cell cycle arrest.
CONCLUSION: Curcumin shows enhanced cytotoxicity toward PTEN-deficient cancer cells, suggesting that it might be a potential chemotherapeutic agent for cancers harboring PTEN mutations.
Keywords: Phosphatase and tensin homolog deleted on chromosome 10, Curcumin, Chemotherapeutic agents, Cell cycle, AKT signaling
Core tip: Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) mutations lead to cancer progression and drug resistance. Chemotherapeutic agents with enhanced effectiveness against cancers with PTEN mutations are urgently required. In this study, we generated an isogenic set of human colorectal cancer cell lines that differed only in their PTEN status. We found that curcumin showed enhanced cytotoxicity in cancer cells deficient in PTEN. Importantly, PTEN deficiency led to an alteration in the pattern of curcumin-induced cell cycle arrest, which was associated with the PTEN/AKT/p21 pathway. Our findings suggest that curcumin is a potential chemotherapeutic agent for PTEN-mutant cancers.
INTRODUCTION
Cancer is a leading cause of death worldwide and increasing attention has been focused on the strategies to reduce its incidence[1]. Chemotherapy is one of the main means of treating cancers; however, resistance to chemotherapeutic drugs remains a major obstacle to effective cancer therapy[2,3]. Therefore, novel intervention strategies to enhance the effectiveness of chemotherapeutic drugs and reduce their resistance are urgently required.
Cancer is caused by a series of genetic changes, including mutations in oncogenes and tumor suppressor genes[4]. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is one of the most frequently mutated tumor suppressor genes in human cancers, after p53[5]. PTEN is involved in many important biological processes, including cell proliferation, growth, migration and death[6]. Germline mutations of PTEN result in several rare cancer predisposition syndromes, such as Cowden disease, Bannayan-Zonana syndrome, Proteus syndrome and Lhermitte-Duclos disease[7]. Mice heterozygous for PTEN develop spontaneous tumors in a number of organs[8]. Conditional deletion of the murine PTEN gene leads to tissue-specific tumorigenesis[9]. PTEN acts as a lipid phosphatase that antagonizes the phosphatidylinositide 3-kinase (PI3K) signaling by dephosphorylating phosphatidylinositol (3,4,5)-trisphosphate (PIP3) back to phosphatidylinositol (4,5)-bisphosphate (PIP2). Mutations in PTEN lead to constitutive activation of AKT kinase and other downstream effectors, and thus promote tumorigenesis[10].
PTEN mutations are found in a number of human cancers, including cancers of the colon, breast, lung, liver, and lymphatic system[11-15]. The fact that mutated PTEN is present only in the cancer cells makes PTEN an appealing drug target for cancer treatment[16]. However, mutational inactivation of PTEN leads to cancer progression and chemotherapy resistance. Loss of PTEN expression is frequently observed in trastuzumab or tamoxifen-resistant breast cancers[17,18]. Drug resistance induced by PTEN deficiency is also observed in colorectal cancer (CRC). Loss of PTEN occurs in 35% of CRC[19], and PTEN-deficient CRC cells are more resistant to cetuximab than PTEN-expressing cells[20]. In addition, loss of PTEN expression results in poor clinical outcome in CRCs treated with anti-EGFR therapy[21]. To identify chemotherapeutic agents that could overcome resistance in PTEN-mutant CRC, we developed an isogenic set of human CRC cell lines that differed only in their PTEN status. Cytotoxicity analysis of these cell lines with several anti-cancer agents showed that curcumin had enhanced cytotoxicity towards CRC cells deficient in PTEN. Moreover, loss of PTEN expression led to a change in curcumin-induced cell cycle arrest patterns, which might be associated with PTEN-regulated AKT/p21 signaling. Our findings suggest that curcumin might have potential in the treatment of cancers with PTEN mutations.
MATERIALS AND METHODS
Cell culture
Human colon cancer HCT116 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, United States). HCT116 p53-/- cells were kindly provided by Dr. Bert Vogelstein (The Johns Hopkins University, Baltimore, MD, United States). The cells were maintained in McCoy’s 5A medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin solution in a humidified incubator at 37 °C with 5% CO2. Cells harboring the targeting vector were grown in medium containing 1000 μg/mL G418.
Reagents and antibodies
Curcumin, 5-fluorouracil (5-FU), dihydroartemisinin (DHA), irinotecan (CPT-11), and oxaliplatin (OXA) were purchased from Sigma-Aldrich. PTEN, AKT (pan), phosphorylated AKT (p-AKT), p53, caspase 3, caspase 9, GAPDH, and beta-actin antibodies were obtained from Cell Signaling Technology. Bcl2, p21, p27, Cyclin B1, Cdc2, Cyclin D1, PARP and peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology.
Gene targeting in HCT116 cells
Human somatic cell gene targeting was performed using adeno-associated virus vectors. A PTEN-targeting vector was constructed to delete exon 4 of the PTEN gene in HCT116 cells. To create this vector, homology arms were amplified from a human genomic DNA template by polymerase chain reaction (PCR), sequentially cloned into the pMD18-T vector (TaKaRa) and sequenced. The constructed targeting vector was co-transfected with pHelper and pAAV-RC plasmids into HEK293 cells to obtain a PTEN-AAV viral stock that was used to infect HCT116 cells. After infection with recombinant virus, HCT116 cells were seeded in 96-well plates and selected with 1000 μg/mL G418 for 2 wk. Individual clones were obtained, expanded, cryopreserved, and tested by PCR for the presence of a heterozygous knockout. Heterozygous clones were transfected with the pCX-CRE plasmid, and then plated at a density of 200 cells/well in a 96-well plate to obtain single cell-derived clones. These clones were then screened by PCR to identify those in which the PGK-neo cassette had been deleted. Then the PTEN+/- cells were re-transfected with the PTEN-AAV virus, to obtain clones in which both PTEN alleles had been targeted.
MTT assays
Cell viability was determined using the MTT assay, as described previously[22]. Briefly, PTEN+/+ and PTEN-/- cells were seeded in 96-well plates in triplicate. At the indicated time points, 10 μL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) stock solution (5 mg/mL) was added to each well. Plates were incubated at 37 °C for another 4 h. The medium was carefully removed and the formazan produced was dissolved in dimethyl sulfoxide (DMSO). The absorbance was measured at a wavelength of 570 nm on a microplate reader (BioRad Model 550). To test the compounds for cytotoxic effects, cells were seeded at a density of 1 × 104 cells/well in 96-well plates and cultured for 24 h. One-hundred microliters of medium supplemented with twice the desired concentration of compounds was then added to each well. After incubation for 72 h, MTT solution was added. Four hours later, formazan production was measured as described earlier. The viability was calculated as % viability = (OD of treated cells/OD of control cells) × 100.
Clonogenic assays
A total of 1000 cells were plated in 6-well plates, in triplicate, and incubated for 24 h to allow cells to adhere. After treatment with 10-μmol/L curcumin or 0.1% DMSO for 48 h, cells were carefully washed and drug-free medium was added. The plates were incubated at 37 °C for 10-12 d and stained with 0.5% (w/v) crystal violet. Colonies containing more than 50 cells were scored as positive. The experiment was performed at least three times using triplicate cultures.
Apoptosis analysis
Curcumin-induced apoptosis was quantified by flow cytometry using the Annexin V-FITC Apoptosis Detection kit according to the manufacturer’s instructions. Briefly, 5 × 105 cells were seeded in 6-well plates and treated with 10-μmol/L curcumin or 0.1% DMSO for 48 h prior to analysis. Floating and trypsinized adherent cells were collected, washed with PBS, and resuspended in cold binding buffer. After incubation for 15 min in the dark at 4 °C, 10 μL of Annexin V-FITC and 5 μL of propidium iodide (PI) solution were added. Samples were analyzed using an FACS-Calibur cytometer (Becton Dickinson).
Cell cycle analysis
A total of 5 × 105 cells were seeded in 6-well plates and treated with 10-μmol/L curcumin or 0.1% DMSO for 48 h. Then, the cells were harvested by centrifugation at 1000 rpm for 5 min. Cell pellets were washed twice with PBS, fixed with ice-cold 70% ethanol and stored at -20 °C overnight. Then, the pellets were washed with cold PBS, suspended in 500 mL PBS containing 50 mg/mL PI, 0.1 mg/mL RNase A and 0.05% Triton X-100, and incubated at 37 °C for 40 min in the dark. The cell cycle distribution was determined by flow cytometry (FACSCalibur; Becton Dickinson). The experiment was repeated thrice under the same conditions.
Western blotting analysis
Cells were grown to 90% confluence in a 6-well plate. After washing twice with PBS, cells were resuspended and lysed in cold lysis buffer (50 mol/L Tris/HCl, pH 7.4, 5 mol/L EDTA, 0.5% NP-40, 150 mol/L NaCl) supplemented with the Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology). After incubation on ice for 20 min, the lysate was centrifuged at 12000 rpm for 20 min at 4 °C, and the supernatant was collected. Protein concentration was determined using the Bradford assay (Bio-Rad, Hercules, CA, United States). Equal amounts of total protein were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, United States). After blocking in 5% milk in TBST [10 mol/L Tris/HCl pH 7.4, 150 mol/L NaCl, and 0.1% (v/v) Tween-20], the membranes were incubated with primary antibodies, followed by secondary antibodies. Protein bands were visualized using an ECL system (Amersham Biosciences, United States).
Statistical analysis
Data were summarized using means ± SD. Statistical comparisons were made using the Student’s t-test. P-values of less than 0.05 were considered statistically significant.
RESULTS
Targeted deletion of PTEN in colorectal cancer cells
Recombinant adeno-associated virus vectors were used to disrupt the endogenous PTEN gene in a near-diploid colon cancer cell line, HCT116, containing two wild-type alleles of PTEN. Targeting was directed to the exon 4 of PTEN, which resulted in a frame-shift mutation and thus a loss of PTEN expression. The detailed strategy is depicted in Figure 1A. The first allele of PTEN was disrupted by homologous recombination with the PTEN-targeting vector. The PTEN+/- clones were confirmed by PCR and transiently exposed to Cre recombinase, mediating the excision of the internal neomycin selection cassette flanked by loxP sites. A PTEN+/- clone was then used to generate PTEN-/- cells via a second round of recombination with the PTEN-targeting vector. The absence of PTEN mRNA in PTEN-/- clones was verified by PCR (Figure 1B). Western blotting analysis confirmed a decrease in PTEN levels in PTEN+/- cells and a complete loss of PTEN expression in PTEN-/- clones, which was accompanied by increased AKT phosphorylation (Figure 1C). Deletion of exon 4 of the PTEN gene was further verified by sequencing (Figure 1D). The PTEN-/- cells displayed a similar morphology to the parental PTEN+/+ cell line (Figure 1E). The effect of PTEN deficiency on the viability of HCT116 cells was assessed using the MTT assay. However, no significant difference in cell growth characteristics was observed between PTEN+/+ and PTEN-/- cells (Figure 1F).
Figure 1.
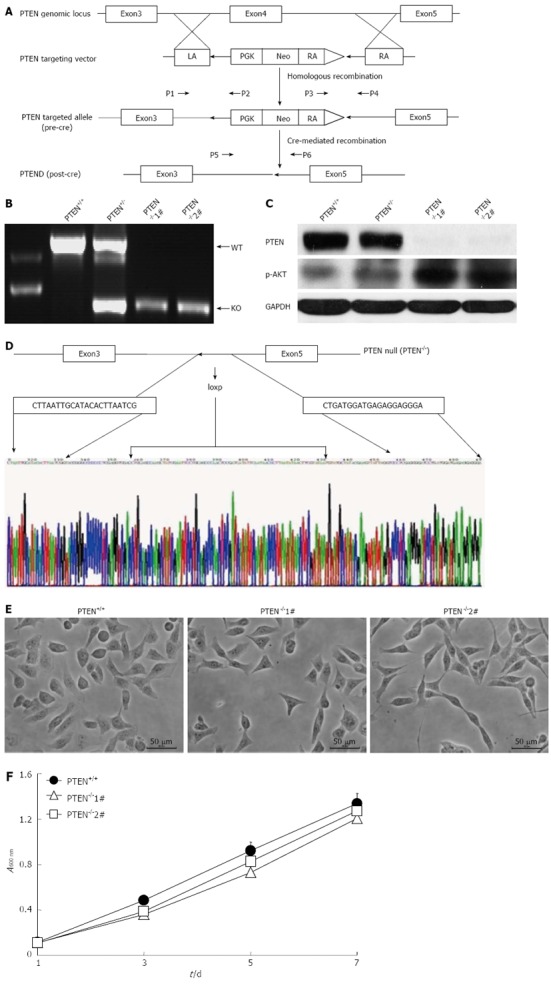
Gene targeting at the PTEN locus. A: Schematic illustration of the strategy used to inactivate phosphatase and tensin homolog deleted on chromosome 10 (PTEN) by somatic cell gene targeting. The primers (P1, P2, P3, P4, P5, and P6) used for polymerase chain reaction (PCR)-based genotyping are indicated; B: Identification of the desired targeted clones by PCR. Primers P5 and P6 were used to amplify a 1,155-bp product from the wild-type PTEN allele and a 522-bp product from the final knockout allele, respectively; C: Western blotting analysis of PTEN expression; D: Confirmation of PTEN exon 4 deletion in PTEN-deficient cells by sequencing; E: Morphology of PTEN+/+ and PTEN-/- cells. F: Determination of cell viability for PTEN+/+ and PTEN-/- cell lines using the MTT assay. PTEN-/-1# and PTEN-/-2# are two PTEN knockout clones. LA: Left arm; RA: Right arm; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
Curcumin shows enhanced cytotoxicity toward PTEN-deficient cancer cells
PTEN acts as a tumor suppressor and its status is associated with sensitivity to chemotherapeutic agents[23,24]. Therefore, we investigated whether PTEN disruption affected the cytotoxicity of several clinical drugs and natural anti-cancer compounds. The PTEN-deficient cells and isogenic PTEN positive cells were exposed to increasing concentrations of anti-cancer compounds and their viability was determined. Disruption of the PTEN gene had no effect on the sensitivity of HCT116 cells to 5-FU, CPT-11, DHA, or OXA (Figure 2A-D). Surprisingly, PTEN-deficient cells were more sensitive to curcumin as compared with the parental PTEN+/+ cells. The IC50 value of curcumin for HCT116 PTEN-/- cells was approximately 2-fold lower than that for PTEN+/+ cells (Figure 2E). Next, we determined whether curcumin also showed increased cytotoxicity toward HCT116 cells deficient in p53. However, we found that disruption of p53 had no effect on the sensitivity of HCT116 cells to curcumin (Figure 2F). The fact that PTEN deficiency resulted in increased sensitivity of HCT116 cells to curcumin was further confirmed using a clonogenic assay. Following curcumin exposure, a significantly smaller number of colonies was observed with HCT116 PTEN-/- cells than with PTEN+/+ cells (Figure 3). Together, these results suggest that curcumin exhibits enhanced cytotoxicity toward HCT116 cells deficient in PTEN.
Figure 2.
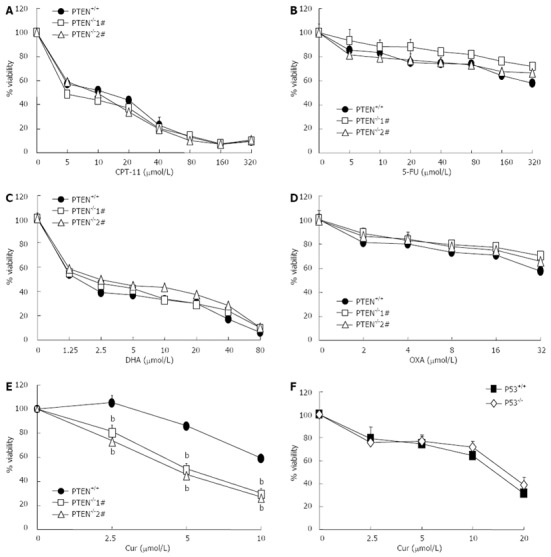
Curcumin shows enhanced cytotoxicity toward PTEN-deficient cells. A-E: Cytotoxic effects of irinotecan (CPT-11), 5-fluorouracil (5-FU), dihydroartemisinin (DHA), oxaliplatin (OXA) and curcumin (Cur) on cell viability of HCT116 phosphatase and tensin homolog deleted on chromosome 10 (PTEN)+/+ and PTEN-/- cells were determined using the MTT assay; F: Cytotoxic effects of curcumin (Cur) on HCT116 p53+/+ and p53-/- cells ( bP < 0.01 vs control group).
Figure 3.
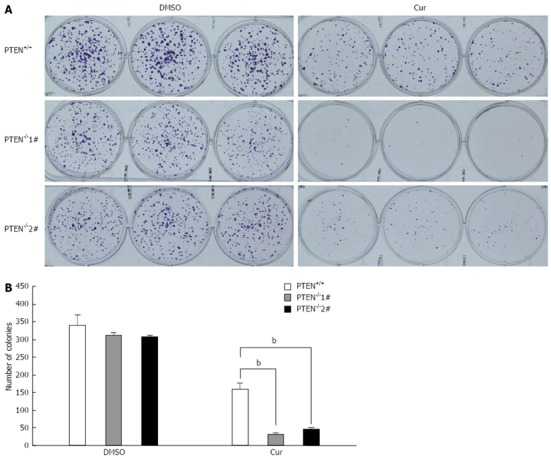
Curcumin has an increased growth-inhibitory effect on PTEN-deficient cells in clonogenic assays. A: Photographs of clonogenic assay plates; B: Histograms showing clonogenic assay results (bP < 0.01 vs control group). Cur: Curcumin.
PTEN deficiency does not increase curcumin-induced apoptosis
Curcumin treatment induces cell apoptosis[25]. To understand the molecular mechanism underlying the increased cytotoxicity of curcumin toward PTEN-deficient CRC cells, we tested whether PTEN loss resulted in increased curcumin-mediated apoptosis. Using flow cytometric analysis with FITC-labeled annexin V and propidium iodide staining, we observed significantly increased apoptosis in both HCT116 PTEN+/+ and PTEN-/- cells after curcumin exposure. However, the apoptotic index was not affected by disruption of the PTEN gene, being similar in PTEN+/+ and PTEN-/- cells (Figure 4A and B). In accordance with these results, the expression patterns of apoptosis-related proteins, such as Bcl-2, procaspase 3 and 9, p53 and PARP were similar in both cell lines following curcumin exposure (Figure 4C). Therefore, these data suggest that the enhanced cytotoxicity of curcumin toward PTEN-/- colon cancer cells is not due to an increase in curcumin-induced apoptosis.
Figure 4.
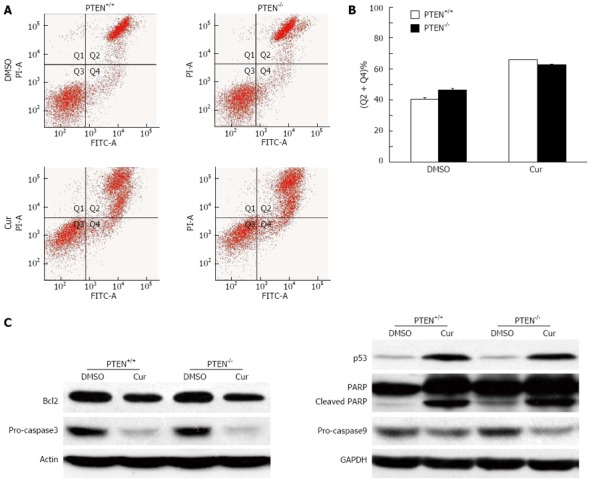
PTEN deficiency does not enhance curcumin-induced apoptosis. A: Images showing flow cytometric analysis of apoptosis. Apoptosis was analyzed by flow cytometry with the Annexin V-FITC kit; B: Histograms showing apoptosis assay results. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) deletion did not result in increased apoptosis following curcumin treatment; C: Western blotting analysis. Cell lysates were separated on SDS-PAGE, transferred to polyvinylidene difluoride membranes, and incubated with indicated antibodies. Cur: Curcumin; PI: Propidium iodide; FITC: Fluorescein isothiocyanate; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
PTEN deficiency results in altered curcumin-induced cell cycle arrest
Next, we tested whether the enhanced cytotoxicity of curcumin to PTEN-deficient cells was caused by altered cell cycle progression. Consistent with a previous report[26], we found that curcumin exposure led to a marked G2/M phase cell cycle arrest in HCT116 cells with wild-type PTEN (Figure 5A). Surprisingly, in PTEN-/- cells, curcumin treatment resulted in a significant accumulation of cells in G0/G1 phase, characteristic of a G0/G1 phase arrest (Figure 5A). To explore the mechanism underlying the altered cell cycle arrest pattern, we investigated the expression of proteins related to AKT signaling and the cell cycle. Curcumin exposure marginally reduced AKT phosphorylation and significantly induced p21 expression in HCT116 cells harboring wild-type PTEN. Interestingly, the opposite results were observed in HCT116 PTEN-/- cells, which showed a significant induction of AKT phosphorylation. Curcumin exposure further elevated this increase in AKT phosphorylation, which was accompanied by a significant reduction in p21 expression (Figure 5B). In agreement with the differential expression of p21 in curcumin-treated PTEN+/+ and PTEN-/- cells, we observed a down-regulation of Cyclin B1 and Cdc2 in PTEN+/+ cells, whereas decreased expression of Cyclin D1 was observed in PTEN-/- cells (Figure 5B), consistent with their respective patterns of cell cycle arrest. p27 is another known regulator of G0/G1 phase; however, we observed no difference in its expression levels between the PTEN+/+ and PTEN-/- cells following curcumin exposure (Figure 5B). Together, these data suggest that the enhanced cytotoxicity of curcumin caused by PTEN deficiency might be due to an altered pattern of cell cycle arrest that is related to the PTEN/AKT/p21 pathway.
Figure 5.
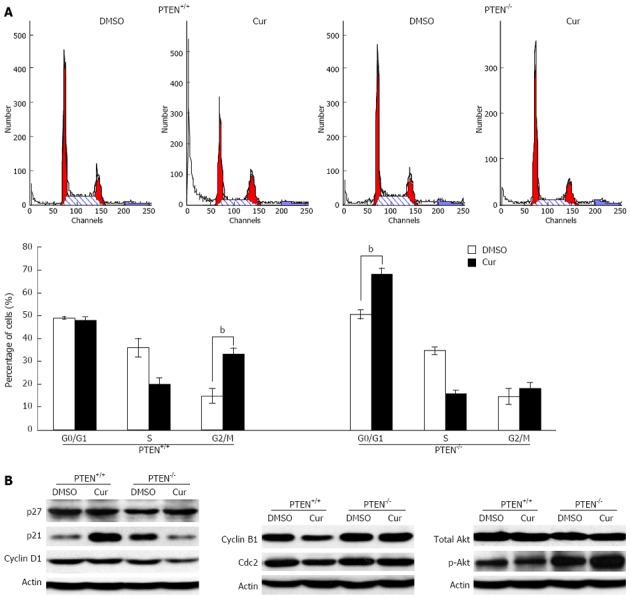
Loss of PTEN alters curcumin-mediated cell cycle arrest. A: Cell cycle distributions of phosphatase and tensin homolog deleted on chromosome 10 (PTEN)+/+ (left) and PTEN-/- (right) cell samples were analyzed by flow cytometry (bP < 0.01 vs control group); B: Western blotting analysis. Whole cell lysate isolated from PTEN+/+ and PTEN-/- cells were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and incubated with the indicated antibodies. Cur: Curcumin.
DISCUSSION
PTEN is a tumor suppressor gene that is frequently mutated in human colorectal cancers. PTEN functions as a lipid phosphatase that negatively regulates PI3K/AKT signaling, which is critical for cell proliferation, apoptosis, and cell cycle progression[27]. PTEN mutations result in aberrant activation of the AKT pathway, facilitating cancer progression and causing drug resistance in CRC[28,29]. Therefore, chemotherapeutic agents, such as natural products and synthetic compounds, that can enhance therapeutic efficacy for CRC with PTEN mutations, are urgently needed. In the present study, we found that curcumin showed enhanced cytotoxicity toward CRC cells deficient in PTEN. This intriguing finding indicates that curcumin might be applied alone or in combination with other chemotherapeutic agents for the therapy of PTEN-mutant cancers.
Curcumin is a natural pigment extracted from the roots of the turmeric plant (Curcuma longa). In addition to its anti-oxidative and anti-inflammatory properties, curcumin inhibits cell proliferation in a number of cancer cell lines, including those derived from colon, breast, lung and bladder cancers[30-33]. Moreover, curcumin inhibits tumorigenesis in vivo. It has been shown that curcumin effectively prevents tumor implantation and growth in mice, and suppresses the development of bladder cancer in a rat model[34,35]. Mechanistic studies demonstrated that curcumin targets a number of molecules, including apoptosis-related proteins such as Bcl-2, caspase 3 and 9, as well as cell cycle regulators[36,37]. In this study, we report for the first time that the enhanced cytotoxicity of curcumin toward PTEN-deficient cells is not due to an increase in apoptosis, but rather to altered cell cycle arrest patterns, from the G2/M phase arrest seen in PTEN+/+ cells to a G0/G1 phase arrest in PTEN-/- cells.
The PTEN+/+ and PTEN-/- cells used in this study differ only in their PTEN status. Since PTEN is an upstream regulator of AKT signaling, the differential effects of curcumin on cells with and without PTEN might be associated with altered AKT signaling. Consistent with this hypothesis, we observed differences in curcumin-induced AKT phosphorylation in PTEN+/+ and PTEN-/- cells. In contrast to the slight decrease observed in p-AKT in PTEN+/+ cells, p-AKT was significantly increased in PTEN-/- cells after curcumin exposure. p21 is a well-known downstream effector of AKT signaling. p-AKT can phosphorylate p21 and restrict it to the cytoplasm for degradation[38]. In accordance with this, p21 expression was significantly increased in PTEN+/+ cells, but markedly decreased in PTEN-/- cells following curcumin exposure. Consequently, the increased expression of p21 led to a down-regulation of Cyclin B1 and Cdc2 and thus a G2/M phase arrest in PTEN+/+ cells, which is consistent with a previous study[39]. Despite its function as a negative regulator of the cell cycle, p21 can also positively regulate cell cycle progression by serving as an assembly factor for the Cyclin D/Cdk4 complex and facilitating the transition from G1 phase to S phase[40-42]. Consistent with this role, we observed a decrease in p21 expression, accompanied by a reduced level of Cyclin D1 and G0/G1 arrest in PTEN-/- cells after curcumin exposure. Based on our findings, we speculate that PTEN deficiency alters AKT signaling and thus the expression of p21 induced by curcumin, and that this alteration results in an increased G0/G1 phase arrest, which may account for the enhanced curcumin sensitivity of PTEN-deficient cells. Our results suggest that p21 is a potential regulator in the enhanced cytotoxicity of curcumin toward PTEN-deficient cells; however, the detailed mechanism remains to be elucidated.
In conclusion, we have shown that curcumin exhibits increased cytotoxicity toward PTEN-deficient cancer cells, and the underlying mechanism might involve cell cycle arrest alterations that are associated with the PTEN/AKT/p21 pathway. These findings also suggest that curcumin might potentially contribute to the therapy of PTEN-mutated cancers.
ACKNOWLEDGMENTS
We thank Dr. Yue-Zhen Deng (Chinese Academy of Sciences) for technical assistance.
COMMENTS
Background
Resistance to chemotherapeutic drugs remains a major obstacle to the effective treatment of cancers. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is one of the most frequently mutated tumor suppressor genes in human cancers. Accordingly, mutational inactivation of PTEN leads to cancer progression and chemotherapy resistance. Therefore, chemotherapeutic agents with greater efficacy for cancer with PTEN mutations are urgently required.
Research frontiers
Curcumin is a major yellow-colored dietary pigment from Curcuma longa. Curcumin has demonstrated anticancer properties in various cancers, including colon cancer. This study analyzes the possible effects of curcumin on colorectal cancer cells with PTEN deficiency.
Innovations and breakthroughs
By gene targeting PTEN in HCT116 cells we created PTEN null HCT116 cells, which are isogenic to the PTEN positive HCT116 parental cell line. The authors found that PTEN null cells exhibited greater sensitivity to curcumin than PTEN positive cells. The authors further established that this difference was not due to an increase in apoptosis, but the result of altered cell cycle arrest induced by curcumin in PTEN null cells.
Applications
Curcumin is a potential chemotherapeutic agent for PTEN mutant cancers, and could be used in individualized cancer therapy. The PTEN-deficient cell line might be a useful tool for screening chemotherapeutic agents against PTEN-mutant cancers.
Terminology
The PI3K/AktKT pathway is an intracellular signaling pathway important for cell growth, survival, cell cycle progression, and apoptosis.
Peer review
This is the first report of the effects of PTEN deficiency on the cytotoxicity of curcumin for colorectal cancer cells. The authors found that PTEN deficiency affects the cytotoxicity of curcumin on HCT116 cells. Upon further investigation they found that this effect is related to increased G0/G1 arrest but not to increased apoptosis. The manuscript is consistent and well structured.
Footnotes
Supported by The National Natural Science Foundation of China, No. 81101472 to Liao WQ; No. 31071086 to Lu XC
P- Reviewers Ceelen W, Wang YP S- Editor Zhai HH L- Editor Webster JR E- Editor Zhang DN
References
- 1.Kanavos P. The rising burden of cancer in the developing world. Ann Oncol. 2006;17 Suppl 8:viii15–viii23. doi: 10.1093/annonc/mdl983. [DOI] [PubMed] [Google Scholar]
- 2.De Angelis PM, Svendsrud DH, Kravik KL, Stokke T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Cancer. 2006;5:20. doi: 10.1186/1476-4598-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–235. [PubMed] [Google Scholar]
- 4.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Jiang X. PTEN: a default gate-keeping tumor suppressor with a versatile tail. Cell Res. 2008;18:807–816. doi: 10.1038/cr.2008.83. [DOI] [PubMed] [Google Scholar]
- 7.Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41:323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 2000;60:3605–3611. [PubMed] [Google Scholar]
- 9.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 10.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 11.Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997;57:4183–4186. [PubMed] [Google Scholar]
- 12.Forgacs E, Biesterveld EJ, Sekido Y, Fong K, Muneer S, Wistuba II, Milchgrub S, Brezinschek R, Virmani A, Gazdar AF, et al. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 1998;17:1557–1565. doi: 10.1038/sj.onc.1202070. [DOI] [PubMed] [Google Scholar]
- 13.Grønbaek K, Zeuthen J, Guldberg P, Ralfkiaer E, Hou-Jensen K. Alterations of the MMAC1/PTEN gene in lymphoid malignancies. Blood. 1998;91:4388–4390. [PubMed] [Google Scholar]
- 14.Chen ST, Yu SY, Tsai M, Yeh KT, Wang JC, Kao MC, Shih MC, Chang JG. Mutation analysis of the putative tumor suppression gene PTEN/MMAC1 in sporadic breast cancer. Breast Cancer Res Treat. 1999;55:85–89. doi: 10.1023/a:1006142919428. [DOI] [PubMed] [Google Scholar]
- 15.Yeh KT, Chang JG, Chen YJ, Chen ST, Yu SY, Shih MC, Perng LI, Wang JC, Tsai M, Chang CP. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in hepatocellular carcinoma. Cancer Invest. 2000;18:123–129. doi: 10.3109/07357900009038243. [DOI] [PubMed] [Google Scholar]
- 16.Li HF, Keeton A, Vitolo M, Maddox C, Rasmussen L, Hobrath J, White EL, Park BH, Piazza GA, Kim JS, et al. A high-throughput screen with isogenic PTEN+/+ and PTEN-/- cells identifies CID1340132 as a novel compound that induces apoptosis in PTEN and PIK3CA mutant human cancer cells. J Biomol Screen. 2011;16:383–393. doi: 10.1177/1087057110397357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanic N, Milovanovic Z, Tanic N, Dzodic R, Juranic Z, Susnjar S, Plesinac-Karapandzic V, Tatic S, Dramicanin T, Davidovic R, et al. The impact of PTEN tumor suppressor gene on acquiring resistance to tamoxifen treatment in breast cancer patients. Cancer Biol Ther. 2012;13:1165–1174. doi: 10.4161/cbt.21346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C, et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012;18:6784–6791. doi: 10.1158/1078-0432.CCR-12-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naguib A, Cooke JC, Happerfield L, Kerr L, Gay LJ, Luben RN, Ball RY, Mitrou PN, McTaggart A, Arends MJ. Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study: associations with clinicopathological and dietary factors. BMC Cancer. 2011;11:123. doi: 10.1186/1471-2407-11-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS, Nasser S, Arango D, Shin J, Klampfer L, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68:1953–1961. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohn BA, Mina S, Krohn A, Simon R, Kluth M, Harasimowicz S, Quaas A, Bockhorn M, Izbicki JR, Sauter G, et al. Altered PTEN function caused by deletion or gene disruption is associated with poor prognosis in rectal but not in colon cancer. Hum Pathol. 2013;44:1524–1533. doi: 10.1016/j.humpath.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Wang O, Liu S, Zou J, Lu L, Chen L, Qiu S, Li H, Lu X. Anticancer activity of 2α, 3α, 19β, 23β-Tetrahydroxyurs-12-en-28-oic acid (THA), a novel triterpenoid isolated from Sinojackia sarcocarpa. PLoS One. 2011;6:e21130. doi: 10.1371/journal.pone.0021130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabian P, Berkovcová J. [Molecular predictive markers of EGFR-targeted therapy in metastatic colorectal cancer] Cesk Patol. 2011;47:154–158. [PubMed] [Google Scholar]
- 25.Karunagaran D, Rashmi R, Kumar TR. Induction of apoptosis by curcumin and its implications for cancer therapy. Curr Cancer Drug Targets. 2005;5:117–129. doi: 10.2174/1568009053202081. [DOI] [PubMed] [Google Scholar]
- 26.Lu JJ, Cai YJ, Ding J. Curcumin induces DNA damage and caffeine-insensitive cell cycle arrest in colorectal carcinoma HCT116 cells. Mol Cell Biochem. 2011;354:247–252. doi: 10.1007/s11010-011-0824-3. [DOI] [PubMed] [Google Scholar]
- 27.Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70:829–844. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 29.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 30.Lin L, Liu Y, Li H, Li PK, Fuchs J, Shibata H, Iwabuchi Y, Lin J. Targeting colon cancer stem cells using a new curcumin analogue, GO-Y030. Br J Cancer. 2011;105:212–220. doi: 10.1038/bjc.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamat AM, Tharakan ST, Sung B, Aggarwal BB. Curcumin potentiates the antitumor effects of Bacillus Calmette-Guerin against bladder cancer through the downregulation of NF-kappaB and upregulation of TRAIL receptors. Cancer Res. 2009;69:8958–8966. doi: 10.1158/0008-5472.CAN-09-2045. [DOI] [PubMed] [Google Scholar]
- 32.Yang CL, Liu YY, Ma YG, Xue YX, Liu DG, Ren Y, Liu XB, Li Y, Li Z. Curcumin blocks small cell lung cancer cells migration, invasion, angiogenesis, cell cycle and neoplasia through Janus kinase-STAT3 signalling pathway. PLoS One. 2012;7:e37960. doi: 10.1371/journal.pone.0037960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang T, Chen Z, Fang L. Curcumin inhibits LPS-induced EMT through downregulation of NF-κB-Snail signaling in breast cancer cells. Oncol Rep. 2013;29:117–124. doi: 10.3892/or.2012.2080. [DOI] [PubMed] [Google Scholar]
- 34.Sindhwani P, Hampton JA, Baig MM, Keck R, Selman SH. Curcumin prevents intravesical tumor implantation of the MBT-2 tumor cell line in C3H mice. J Urol. 2001;166:1498–1501. [PubMed] [Google Scholar]
- 35.Tian B, Wang Z, Zhao Y, Wang D, Li Y, Ma L, Li X, Li J, Xiao N, Tian J, et al. Effects of curcumin on bladder cancer cells and development of urothelial tumors in a rat bladder carcinogenesis model. Cancer Lett. 2008;264:299–308. doi: 10.1016/j.canlet.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 36.Shishodia S. Molecular mechanisms of curcumin action: gene expression. Biofactors. 2013;39:37–55. doi: 10.1002/biof.1041. [DOI] [PubMed] [Google Scholar]
- 37.Shishodia S, Amin HM, Lai R, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem Pharmacol. 2005;70:700–713. doi: 10.1016/j.bcp.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 38.Han CT, Schoene NW, Lei KY. Influence of zinc deficiency on Akt-Mdm2-p53 and Akt-p21 signaling axes in normal and malignant human prostate cells. Am J Physiol Cell Physiol. 2009;297:C1188–C1199. doi: 10.1152/ajpcell.00042.2009. [DOI] [PubMed] [Google Scholar]
- 39.Dvory-Sobol H, Cohen-Noyman E, Kazanov D, Figer A, Birkenfeld S, Madar-Shapiro L, Benamouzig R, Arber N. Celecoxib leads to G2/M arrest by induction of p21 and down-regulation of cyclin B1 expression in a p53-independent manner. Eur J Cancer. 2006;42:422–426. doi: 10.1016/j.ejca.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Kavurma MM, Khachigian LM. Sp1 inhibits proliferation and induces apoptosis in vascular smooth muscle cells by repressing p21WAF1/Cip1 transcription and cyclin D1-Cdk4-p21WAF1/Cip1 complex formation. J Biol Chem. 2003;278:32537–32543. doi: 10.1074/jbc.M305650200. [DOI] [PubMed] [Google Scholar]
- 41.Weiss RH, Joo A, Randour C. p21Waf1/Cip1 is an assembly factor required for platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem. 2000;275:28340. [PubMed] [Google Scholar]
- 42.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]