

Abstract
N-methyl-d-aspartate receptors (NMDARs) are glutamate-gated ion channels that are critical to the regulation of excitatory synaptic function in the CNS. NMDARs govern experience-dependent synaptic plasticity and have been implicated in the pathophysiology of various neuropsychiatric disorders including the cognitive deficits of schizophrenia and certain forms of autism. Certain neurosteroids modulate NMDARs experimentally but their low potency, poor selectivity, and very low brain concentrations make them poor candidates as endogenous ligands or therapeutic agents. Here we show that the major brain-derived cholesterol metabolite 24(S)-hydroxycholesterol (24(S)-HC) is a very potent, direct, and selective positive allosteric modulator of NMDARs with a mechanism that does not overlap that of other allosteric modulators. At submicromolar concentrations 24(S)-HC potentiates NMDAR-mediated EPSCs in rat hippocampal neurons but fails to affect AMPAR or GABAA receptors (GABAARs)-mediated responses. Cholesterol itself and other naturally occurring oxysterols present in brain do not modulate NMDARs at concentrations ≤10 μm. In hippocampal slices, 24(S)-HC enhances the ability of subthreshold stimuli to induce long-term potentiation (LTP). 24(S)-HC also reverses hippocampal LTP deficits induced by the NMDAR channel blocker ketamine. Finally, we show that synthetic drug-like derivatives of 24(S)-HC, which potently enhance NMDAR-mediated EPSCs and LTP, restore behavioral and cognitive deficits in rodents treated with NMDAR channel blockers. Thus, 24(S)-HC may function as an endogenous modulator of NMDARs acting at a novel oxysterol modulatory site that also represents a target for therapeutic drug development.
Introduction
N-methyl-d-aspartate receptors (NMDARs) are heterotetrameric ligand-gated ion channels implicated in forms of synaptic plasticity, such as long-term potentiation (LTP) and long-term depression (LTD), thought to underlie learning and memory (Bashir et al., 1991; Asztely et al., 1992; Cui et al., 2004; Traynelis et al., 2010). NMDARs have been implicated in the pathophysiology of several neuropsychiatric disorders including schizophrenia, depression, Alzheimer's disease, and epilepsy (Li and Tsien, 2009; Zorumski and Izumi, 2012) and many of these disorders are associated with deficits in cognitive function. Because NMDAR channel blockers, such as ketamine and phencyclidine (PCP) produce psychotic symptoms and cognitive deficits (Luby et al., 1959) mimicking schizophrenia in humans (for review, see Tamminga, 1998; Coyle, 2006), there has been considerable interest in discovering drugs that augment NMDAR function to treat the debilitating negative and cognitive symptoms of schizophrenia (Tamminga, 1998; Coyle, 2006). Recent clinical studies of NMDAR enhancers appear to support this strategy (Coyle, 2006).
NMDARs are regulated by various natural substances including neuroactive steroids (Williams, 2009). Neuroactive steroids are natural or synthetic steroids that directly and rapidly modulate ligand-gated ion channels, such as GABAA-type receptors (GABAARs) or NMDARs to alter inhibitory and excitatory neurotransmission (Majewska et al., 1986; Paul and Purdy, 1992). Among these, the neurosteroid pregnenolone sulfate (PREGS) has been studied as a potential endogenous NMDAR modulator (Belelli and Lambert, 2005). PREGS is a positive and negative allosteric modulator of NMDARs and GABA-ARs, respectively (Wu et al., 1991; Park-Chung et al., 1999; Horak et al., 2004). PREGS enhances LTP in hippocampal slices (Sliwinski et al., 2004), enhances memory, and reverses memory impairment induced by NMDAR blockers in rodents (Flood et al., 1992; Mathis et al., 1994). However, PREGS is a relatively weak NMDAR modulator in vitro (Wu et al., 1991) and is essentially undetectable in rodent brain (Liere et al., 2009). Nonetheless, these findings have prompted efforts to find other steroidal NMDAR modulators.
In a preliminary report, Madau et al. (2009) identified other steroid-like NMDAR-positive allosteric modulators (PAMs). The oxysterol derivative [Δ5,6-3β-oxy-nor-cholenyl]-dimethyl-carbinol (CAS No. 35882-85-0; SGE-201) and several structurally related oxysterol derivatives appeared to act as potent NMDAR PAMs in vitro (Madau et al., 2009) and in vivo (Connick et al., 2009), but the exact nature of this PAM activity and it relationship to activity observed for PREGS remain unclear. Given the structural similarity of these compounds to natural oxysterols and the recently appreciated role oxysterols play in cell signaling (Janowski et al., 1996; Corcoran and Scott, 2006), we screened a series of natural oxysterols and related compounds at NMDARs and now report that the major cholesterol metabolite in brain, 24(S)-hydroxycholesterol (24(S)-HC), is a highly selective and potent NMDAR PAM. We show that 24(S)-HC interacts with a privileged NMDAR binding site that is distinct from PREGS. We also identify synthetic derivatives of 24(S)-HC that selectively and potently modulate NMDAR function. These data suggest that 24(S)-HC may serve as a natural modulator of NMDARs, acting at a novel oxysterol regulatory site that is a potential target for therapeutic drug development.
Materials and Methods
Chemicals and solutions
Oxysterols were prepared as concentrated stocks in 100% DMSO; working solutions contained ≤0.1% DMSO. SGE-201 (Madau et al., 2009), previously described as an intermediate in the synthesis of steroids, was synthesized as described previously (Plattner and Pataki, 1943; Mouriño et al., 1978). Briefly, SGE-201 was prepared in four steps from commercially available 3α, 6α-dihydroxy-5β-cholan-24-oic acid. First, the methyl ester was formed, followed by tosylation of the 3α- and 6α-hydroxyl groups. In a single pot under mild basic conditions, the C5/C6 double was formed after elimination of the 6-tosylate and the 3β-hydroxyl moiety was obtained after inversion of the configuration of the 3α-tosylate upon hydrolysis. In the last step, the dimethyl groups were installed at C-24 via methyl lithium addition to the ester. SGE-301 was synthesized from SGE-201 in a straightforward manner. Dess–Martin oxidation of the 3β-hydroxy to the ketone, followed by methyl Grignard addition yielded SGE-301 in two steps. SGE-201 and SGE-301 were characterized by liquid chromatography (LC)/MS and 1H-NMR as described in patent: WO2013/036835A1 and were ≥95% pure. 24(S)-HC and all other steroid derivatives were purchased from Avanti Polar Lipids or Steraloids.
Whole-cell recording
Hippocampal cultures were prepared using previously reported methods (Mennerick et al., 1995). Whole-cell and excised-patch recordings were made using an Axopatch 200B amplifier (Molecular Devices) at room temperature from primary dissociated cultures of mouse (see Fig. 1) or rat (see Figs. 2–5, 7) hippocampal neurons from either sex (days in vitro 5–13) grown as mass cultures or on substrate microdots to elicit recurrent EPSC/IPSCs (Mennerick et al., 1995). Bath solutions for the screening studies in Figure 1 contained the following (in mm): 140 NaCl, 3 CsCl, 0.2 CaCl2, 10 glucose, 10 HEPES, 4.5 sucrose, 0.0005 glycine, 0.00035 TTX, pH 7.4. Bath solution for subsequent studies in cultured neurons contained the following (in mm): 140 NaCl, 4 KCl, CaCl2 (1 for synaptic studies, 0.5 for exogenous NMDA applications), 10 glucose, 10 HEPES, pH 7.25. NBQX (1 μm), D-APV (25 μm), and gabazine (10 μm) were included as needed to isolate relevant currents. Membrane potential was typically clamped to −70 mV, and saline solution contained 0.5 μm glycine and was nominally Mg2+-free. Whole-cell pipette solutions for the screening studies in Figure 1 contained the following (in mm): 120 CsCl, 2 ATP, 0.2 CaCl2, 10 EGTA, 10 HEPES, 1 MgCl2, 20 TEA-Cl, 0.2 cAMP, pH 7.2. For subsequent studies in cultured neurons the whole-cell pipette solution contained the following (in mm): 140 cesium methanesulfonate, 4 NaCl, 0.5 CaCl2, 5 EGTA, 10 HEPES, pH 7.3, and the same solution was used for excised outside-out patch recordings. For evoked recurrent PSCs, potassium gluconate replaced cesium methanesulfonate. For application of drugs to whole cells and to excised patches, a multibarrel solution exchange system with common delivery tip was used (Warner Instruments). The common tip was placed 0.5 mm from the center of the microscope field. Solution exchange times were 120 ± 14 ms (10–90% rise) estimated from the rise of junction currents at the tip of an open patch pipette. Experiments were performed at room temperature, and quantification of whole-cell peak current response was used for all figure summaries.
Figure 1.

24(S)-HC and SGE-201 are potent oxysterol positive allosteric modulators of NMDA receptors. A, Natta projection structures for 24(S)-HC, SGE-201, and SGE-301. Note that the key hydroxyl group (denoted in red) in all three structures is on the same carbon relative to the cholesterol backbone. The similarity in the location of the hydroxyl group is further emphasized in the three dimensional stick and ball models below (D-ring and C-17 side chain shown). The 3-α methyl group that distinguishes SGE-201 and SGE-301 is circled in red. B, Effects of endogenous oxysterols and other cholesterol metabolites on NMDA receptor currents. Cultured primary hippocampal neurons were preincubated with 10 μm test compound in 0.5 μm glycine for 90 s, followed by 10 s NMDA (10 μm). The percentage change in NMDA current is plotted. C, Representative traces from B. The red lines represent application of test article (note that the red lines do not encompass the full 90 s of preincubation). The black lines represent the application of NMDA (10 μm, 10 s). Scale bar: vertical = 200 pA, horizontal = 10 s. D, Active oxysterols compared with pregnenolone metabolites. All compounds were tested at 10 μm, 90 s preincubation, with the exception of 24(S)-HC (10 μm, 360 s preincubation), and pregnenolone sulfate (10 and 50 μm, 90 s preincubation). Cholesterol was solubilized in ethanol rather than DMSO. Asterisk represents a significant difference from current induced by NMDA alone (p < 0.05).
Figure 2.
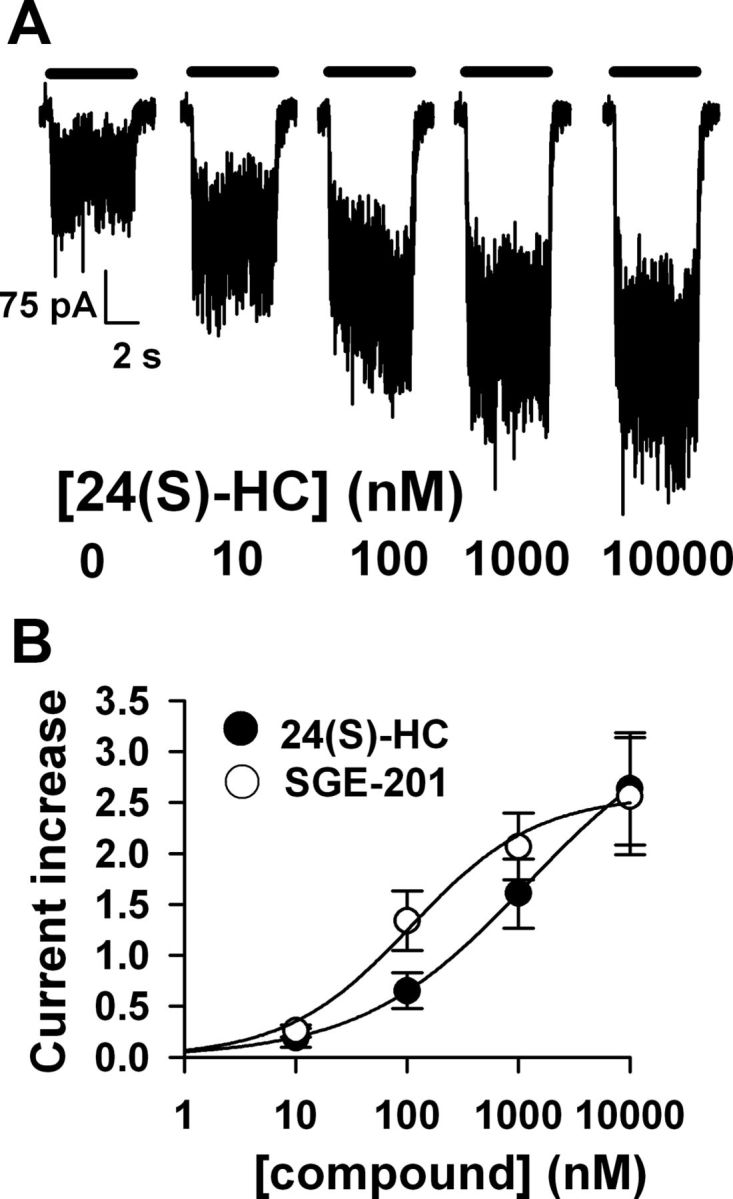
24(S)-HC and SGE-201 are effective at submicromolar concentrations. A, Potentiation of 10 μm NMDA (0.5 μm glycine) by increasing 24(S)-HC concentrations with 40 s oxysterol preapplication in a DIV5 rat hippocampal neuron. B, Potentiation values for 24(S)-HC and for SGE-201 were fit with the Hill equation (solid lines). EC50 estimates were 0.11 μm for SGE-201 (N = 7 cells), and 1.2 μm for 24(S)-HC (N = 5 cells).
Figure 3.
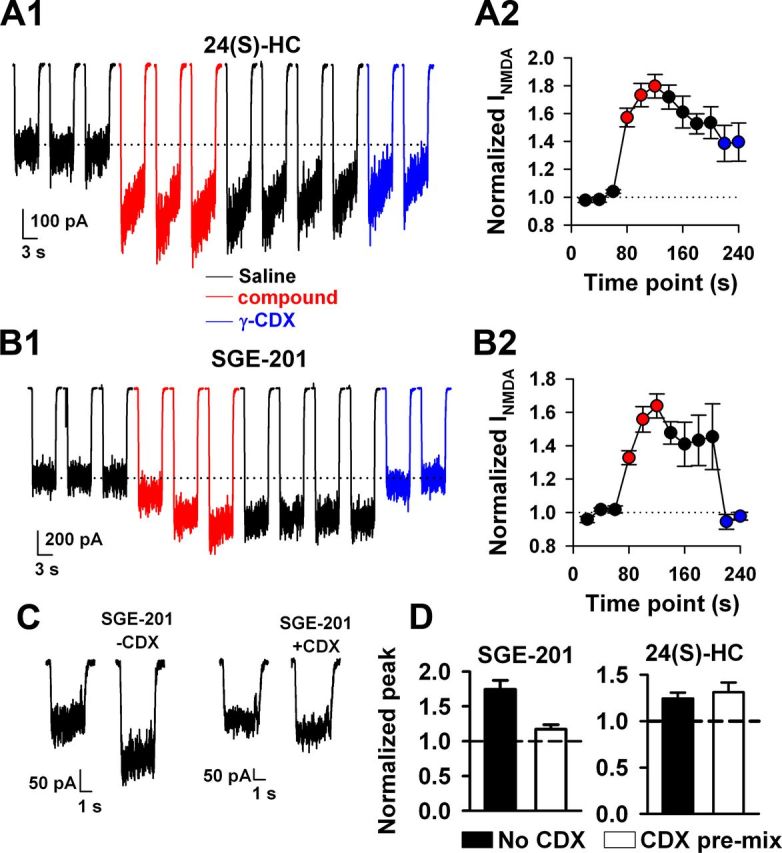
Slow oxysterol reversibility. A, B, NMDA (10 μm) currents were potentiated with 24(S)-HC (2 μm) or with SGE-201 (0.2 μm, red traces) with repeated 20 s preapplication of potentiator between successive NMDA challenges. Following 60 s total oxysterol exposure (three red traces), cells were challenged with saline wash for 80 s (four black traces). There was little reversibility for either drug. Cells were then challenged with γ-cyclodextrin (CDX; 500 μm) wash, before NMDA application in the absence of γ-cyclodextrin (blue traces). A1, B1, Traces show results from representative neurons. A2, B2, Summary plots for five and six cells, respectively. γ-Cyclodextrin extracted SGE-201 potentiation but not 24(S) potentiation. C, D, Oxysterols were premixed with γ-CDX in solution at subsaturating oxysterol concentrations (0.5 μm for each compound mixed with 1 mm γ-cyclodextrin). γ-Cyclodextrin effectively reduced the free concentration of SGE-201, indicated by reduced potentiation, but failed to affect 24(S)-HC potentiation.
Figure 4.

Occlusion studies suggest a unique mechanism for oxysterols versus other lipophilic positive modulators. A, B, Examples of occlusion protocol. Cells were preincubated for >5 min in 10 μm 24(S)-HC then challenged with 10 μm NMDA plus 50 μm PREGS (A) as a representative nonoccluding potentiator or 0.2 μm SGE-201 (B) as a representative occluding potentiator. C, Summary of results from 6 to 10 cells per bar for the four indicated modulators. Arachidonic acid was used at 5 μm. SGE-301 was used at 0.5 μm. Asterisks indicate a significant reduction in potentiation (p < 0.05).
Figure 5.

Potentiation in excised outside-out membrane patches. A, Baseline NMDAR channel activity (black trace) in 300 μm NMDA in an excised outside-out patch from a DIV2 hippocampal neuron was augmented following SGE-201 incubation for 60 s (0.2 μm, red trace). The patch was excised before drug applications. B, The all-points histograms represent 30 s of NMDA-induced channel activity before (black) and following (red) SGE-201 application from the patch represented in A. C, Summary of NPo analysis from eight excised outside-out patches.
Figure 7.

Selective potentiation of NMDAR EPSCs. A, Potentiation by 1 μm 24(S)-HC of evoked NMDAR EPSCs isolated with 1 μm NBQX and 10 μm gabazine. Slow onset, slow reversibility, and relative γ-CDX insensitivity paralleled effects on responses to exogenous NMDA. B, Pharmacologically isolated AMPAR EPSCs (10 μm gabazine and 25 μm D-APV) were unaffected. C, SGE-201 (0.2 μm; 90 s) also potentiated peak NMDAR EPSCs with little effect on decay time course of the EPSCs (inset). D, Summary of effects of 1 μm 24(S)-HC on NMDAR EPSCs and AMPAR EPSCs (N = 8 and 7, respectively). AMPARs were statistically unaltered. A summary of 0.2 μm SGE-201 effects is also shown (N = 7 and 6). E, GABAAR IPSCs were statistically unaltered by prolonged 24(S)-HC application (N = 11 for1 μm 24(S)-HC and N = 6 for 1 μm SGE-201).
Outside-out membrane patches were excised from DIV2-5 hippocampal neurons. Currents were sampled at 8 kHz and filtered at 1 kHz. NPo values were calculated by idealization of traces using QUB software (University of Buffalo). Perfusion buffer for patch experiments included 1 μm NBQX, 0.5 μm tetrodotoxin, and 10 μm gabazine.
Electrophysiological recordings obtained from heterologous cells expressing NMDA receptors were obtained as follows. HEK 293 cells stably expressing human GluN1 (transcript variant NR1-3, RefSeq: NM_007327.1, NP_015566) were transiently transfected with a plasmid encoding human GluN2A (RefSeq: NM_000833.2, NP_000824). In subsequent experiments (see Fig. 6C) constructs used for transient HEK cell transfections included rat GluN1A splice variant (accession numbers U11418, U08261), mouse GluN2A (NM008170), rat GluN2B (M91562), rat GluN2C (M91563), and rat GluN2D (L31611). Constructs were validated by selective restriction digests and functional testing, including verification of high Zn2+ sensitivity of GluN2A vs. GluN2B subunits and low Mg2+ sensitivity of GluN2C and GluN2D subunits. Manual patch-clamp electrophysiology recordings were obtained with an EPC-7/EPC-10, HEKA Electronics Amplifier. Electrical signals were captured and analyzed with PatchMaster Software (HEKA Electronics). Each compound was tested at 0.1 μm or 1.0 μm in the presence of NMDA (30 μm) and glycine (5.0 μm). NMDA and glycine induced currents in the presence of test compound were compared with current induced by NMDA and glycine alone. Fifty micrometers PREGS served as the positive control. 0.1% DMSO was used as the vehicle.
Figure 6.

Potentiation of recombinant receptors suggests little or no subunit selectivity. A, HEK-293 cells stably expressing human GluN1–3 and transiently expressing human GluN2A were activated with NMDA (30 μm) and glycine (5.0 μm) (small gray bars). After determining the baseline response to NMDA and glycine, a test compound (as indicated) was added at 0.1 μm (short white bars) or 1 μm (tall white bars). B, The mean (± SEM) percentage potentiation (by 1 μm test compound) above NMDA and glycine alone is plotted. Asterisks denote a significant difference from baseline (p < 0.05). C, Sample traces from HEK cells transiently transfected with GluN1a plus each of the indicated GluN2 subunits. Potentiation of 10 μm NMDA currents (0.5 μm glycine) by 0.2 μm SGE-201 is shown (gray traces of each pair). D, Each subunit combination exhibited significant potentiation by SGE-201 above baseline (asterisks), but no significant difference in potentiation values among subunits was detected.
Hippocampal slice recordings
Hippocampal slices were prepared from juvenile [postnatal day (P)30–32] or adult (P120) Sprague Dawley rats purchased from Harlan (Izumi and Zorumski, 1999). Rats were anesthetized with isoflurane and decapitated. Slices were cut transversely into 500 μm slices using a rotary slicer in artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 5 KCl, 2 MgSO4, 2CaCl2, 1.25 NaH2PO4, 22 NaHCO3, 10 glucose, bubbled with 95% O2/5% CO2 at 4–6°C. Acutely prepared slices were placed on mesh in 10 ml beakers containing gassed ACSF and maintained for at least 1 h at 30°C before experiments (Tokuda et al., 2010).
At the time of study, slices were transferred individually to a submerged recording chamber. Experiments were performed at 30°C with continuous bath perfusion of ACSF at 2 ml/min. Extracellular recordings were obtained from the apical dendritic layer of the CA1 region (stratum radiatum) for analysis of population EPSPs. EPSPs were measured by their maximal slopes and were monitored by applying single stimuli to the Schaffer collateral pathway every 60 s at half-maximal intensity (estimated from baseline input-output curves). After establishing stable responses for at least 10 min, LTP was induced using a standard 100 Hz × 1 s high-frequency stimulus (HFS). This HFS produces reliable LTP at P30, but is subthreshold for LTP at P120 resulting only in short-term potentiation (STP) under the ionic conditions used (Izumi and Zorumski, 1999). The magnitude of LTP was determined 60 min following HFS. Signals were digitized and analyzed using PCLAMP software (Molecular Devices).
Slice data are expressed as percentage of baseline control responses (set at 100%). In these studies, n represents the number of slices studied in a given condition, and, unless stated otherwise, data were normalized with respect to initial control responses. Points in the graphs without error bars have SEM smaller than the symbol size. Statistical comparisons are based on analysis of input/output curves at baseline and 60 min following HFS, and represent the degree of change at the half-maximal point on the input/output curves compared with baseline responses. Slice statistical analyses were performed using commercial software (SigmaStat 3.11; Systat Software). For slice studies, if a test of equal variance failed, the nonparametric Mann–Whitney rank sum test was applied.
Bioanalytical methods
Plasma and brain concentrations of SGE-201 and SGE-301 were determined by LC-MS/MS. Briefly, brain samples were homogenized with 3 volumes (v/w) of PBS, pH 7.4, before analysis. One-hundred milliliters internal standard (diclofenac 200 ng/ml) was added to aliquots of plasma (30 μl) or brain (200 μl) and extracted with 1 ml of methyl tert-butyl ether. The extracts were centrifuged and a portion of the organic phase was removed and evaporated to dryness under nitrogen. The extracts were reconstituted in 60 μl of methanol/water for analysis. A 20 μl aliquot was chromatographed on a Waters Atlantis C18 column eluting with a gradient of methanol/water/0.2% formic acid. SGE-201 was detected using positive ion electrospray ionization and mass transitions of 371.3/353.3 on a Sciex API 4000 Q-Trap. The limit of quantitation was typically 1 ng/ml for plasma and 4 ng/g for brain. SGE-301 was quantitated following the same tissue extraction procedure. The reconstituted samples were chromatographed on a Waters Acquity BEH C18 column with a methanol/ water/0.2% formic acid solvent gradient. SGE-301 was detected using positive ion electrospray ionization with mass transitions of 385.3/95.0. The limit of quantitation was typically 2 ng/ml for plasma and 40 ng/g for brain.
Behavioral methods
MK-801 impaired Y-maze spontaneous alternation task.
Male Swiss CD-1 mice (7–8 weeks, N = 17–19 per treatment group) were used to assess spontaneous alternation behavior in the Y-maze test (Hefco et al., 2003). The maze was constructed from clear Plexiglas with arms at 120° angles and with dimensions in cm: 57 long, 17 wide, and 35 high. Each mouse was placed in the center of the maze at the start of the study and allowed to move freely for 10 min. An alternation is defined as sequentially entering all three arms without reentry into a previously visited arm. Percentage alternation was calculated as follows: (alternations/total entries − 2) × 100. One hour before testing mice received an intraperitoneal injection of vehicle (25% hydroxypropyl-β-cyclodextrin in PBS) or SGE-201 (3, 10, and 30 mg/kg) in a volume of 10 ml/kg. MK-801 was administered 30 min before testing (0.25 mg/kg in water, i.p.). Data are reported as the mean ± SEM and data were analyzed using one-way ANOVA with Bonferroni post hoc comparisons.
Subchronic PCP model of schizophrenia.
Male Long–Evans rats (160–220 g, N = 12–15 per treatment group) were group housed in a climate controlled environment (22 ± 2°C, 12 h light/dark cycle) with ad libitum access to food and water. PCP (5 mg/kg; Sigma-Aldrich) or vehicle (0.9% saline) was administered twice daily for 7 d (intraperitoneally) followed by a 7 d washout period (Grayson et al., 2007; Snigdha and Neill, 2008). On day 13, rats were habituated to the test chamber (90 × 90 × 40 cm square arena; 60 lux) for 10 min. On day 14, rats received acute intraperitoneal administration of SGE-301 (3, 10, or 30 mg/kg) or vehicle (30% Captisol+ 0.01% Tween 80) 60 min before testing social interaction, or risperidone as a positive control (0.2 mg/kg, i.p.; 60 min pretreatment). Social interaction (SI) was tested during the light phase by placing each rat into the test chamber with an unfamiliar naive male rat (Snigdha and Neill, 2008). Spontaneous SI was monitored by an observer blind to treatment for 10 min. The time spent engaged in active nonaggressive social behavior (defined as sniffing, following, grooming, kicking, mounting, wrestling, boxing or crawling over/under) was recorded. After SI testing, rats received an additional 7 d washout period before novel object recognition (NOR) testing. On day 20 the rats were habituated to the NOR chamber (60 × 60 × 40 cm; 60 lux) for 10 min (Grayson et al., 2007). On day 21, rats received acute intraperitoneal administration of SGE-301 (1, 3, or 10 mg/kg), vehicle (30% Captisol+ 0.01% Tween 80) or risperidone as a positive control (0.2 mg/kg, i.p.) 60 min before NOR training, where each rat is allowed a maximum of 6 min to accumulate 8 s of exploration on each of two identical objects (familiar objects). After a 30 min retention interval, rats were placed back into the arena with one familiar and one novel object and the duration of object exploration was scored manually by an observer blind to treatment for 3 min. For NOR, the discrimination ratio was calculated by dividing the time spent exploring the novel object by the total object exploration time in the test session. This measure corrects for variance in total exploration. With this ratio, 0.5 = chance performance (equal exploration of novel and familiar objects); 1= only novel object exploration, and 0 = only familiar object exploration. All behavioral data are reported as the mean ± SEM and were analyzed with ANOVA and Bonferroni post hoc comparisons.
Results
24(S)-Hydroxycholesterol is a potent NMDAR modulator
To determine whether endogenous oxysterols modulate NMDARs, we initially screened a series of naturally occurring oxysterols (and structurally related sterols and steroids; Fig. 1A,B) at 10 μm in neonatal mouse hippocampal neurons using whole-cell patch-clamp electrophysiology (Fig. 1B, C). Comparators were SGE-201 (10 μm) and PREGS (50 μm) (Fig. 1D). Remarkably, 24(S)-HC, one of the most abundant endogenous cholesterol metabolites in brain, significantly potentiated NMDAR-mediated currents (Fig. 1B, C). 24(S)-HC did not alter membrane current on its own in the absence of NMDA. None of the other oxysterol liver X receptor (LXR) ligands, including 22(R)-HC and 20(S)-HC, affected NMDARs at concentrations ≤10 μm (Fig. 1B–D). Cholesterol itself was also inactive at concentrations ≤10 μm (Fig. 1B).
The concentration-response relationship of NMDAR modulation by 24(S)-HC and SGE-201 was explored by preincubating hippocampal neurons for 40 s in increasing concentrations of compound before agonist application (Fig. 2). Both oxysterols potentiated NMDA-induced currents at submicromolar concentrations (Fig. 2A,B). Interestingly, 24(S)-HC was active at concentrations well below those measured in the CNS (Meljon et al., 2012). SGE-201 was ∼5- to 10-fold more potent than 24(S)-HC (Fig. 2B). We synthesized another synthetic analog, SGE-301, designed to be more bioavailable than 24(S)-HC or SGE-201 by introduction of a 3α-methyl group (Fig. 1A), and evaluated its activity in subsequent experiments (see below).
Although we did not fully explore the pharmacological mechanisms of oxysterol modulation, an interesting aspect of potentiation is that it did not appear to depend strongly on agonist or coagonist concentration. Normalized to baseline responses, SGE-201 potentiation of responses of hippocampal neurons to a near saturating concentration of 300 μm NMDA was 2.8 ± 0.6 (N = 5). Under comparable conditions, the potentiation of responses to 10 μm NMDA (below the EC50; Patneau and Mayer, 1990) was 2.5 ± 0.8 (N = 5, p > 0.05). Similarly, potentiation did not strongly depend on coagonist concentration. Potentiation of responses to 10 μm NMDA in 0.5 μm glycine was similar to potentiation in 10 μm glycine (2.1 ± 0.2 vs 2.4 ± 0.3, p > 0.05, N = 7).
Oxysterols modulate NMDARs with slow kinetics
In pilot experiments we found that 24(S)-HC and SGE-201 required preincubations >20 s to reach maximal effect, and once established, potentiation was poorly reversible. It is possible that the slow onset of potentiation and slow reversibility observed for these ligands could result from a direct receptor interaction that involves membrane partitioning or cell permeation (Akk et al., 2005). Alternatively, potentiation could involve indirect modulation by second messengers or transcriptional regulation. Both of these alternative mechanisms have precedent in explaining the effects of oxysterols in other signaling pathways (Kalaany and Mangelsdorf, 2006; Nachtergaele et al., 2012). Furthermore, it appeared that macroscopic desensitization was augmented in the presence of oxysterol (Fig. 3A1), a phenomenon that represents additional kinetic complexity and could reflect a mixed potentiating/inhibiting action similar to PREGS (Horak et al., 2004). If the slow oxysterol actions require rate-limiting partitioning that equilibrates with a receptor binding site, potentiation might be rapidly reversed by applying γ-cyclodextrin, a scavenger of membrane neurosteroids and sterols (Ohtani et al., 1989; Akk et al., 2005). We found that γ-cyclodextrin effectively reversed potentiation of NMDARs by SGE-201 but much less so for 24(S)-HC (Fig. 3A,B). Although this result could suggest distinct mechanisms of NMDAR potentiation by SGE-201 and 24(S)-HC, it is also possible that the natural oxysterol may not as efficiently form inclusion complexes with γ-cyclodextrin, or may partition at a site inaccessible to γ-cyclodextrin.
To test the former possibility, we prepared solutions of 0.5 μm 24(s)-HC or 0.5 μm SGE-201 with 1 mm γ-cyclodextrin. If oxysterols effectively bind γ-cyclodextrin, the mixture should result in a lower free oxysterol concentration, and brief exposures to the mixtures should reduce potentiation compared with control exposures in the absence of γ-cyclodextrin. Consistent with this expectation, SGE-201 potentiation was reduced compared with a control solution containing no γ-cyclodextrin (Fig. 3C,D). By contrast, the effect of a subsaturating 24(S)-HC concentration was unaffected by γ-cyclodextrin premixing (Fig. 3D). These results are consistent with the idea that 24(S)-HC fails to effectively bind γ-cyclodextrin, explaining the resistance to cyclodextrin extraction of 24(S)-HC.
24(S)-Hydroxycholesterol shares a common mechanism of action with SGE-201 and SGE-301 but not with known lipophilic positive modulators
To test more directly whether 24(S)-HC shares a common mechanism of action with synthetic oxysterol analogues and (or) shares a mechanism with the known lipophilic modulators PREGS or arachidonic acid, we incubated neurons for >5 min in 10 μm 24(S)-HC, and then challenged cells with agonist plus another modulator to probe occlusion (Fig. 4). 24(S)-HC preincubation did not significantly occlude PREGS or arachidonic acid potentiation but prevented further SGE-201 potentiation and potentiation by the 3α-methyl analog SGE-301. These latter two results demonstrate a shared mechanism of action between natural 24(S)-HC and the synthetic analogues and also demonstrate that preincubation in 10 μm 24(S)-HC was saturating, important for the interpretation of the PREGS and arachidonic acid results. Thus, 24(S)-HC likely acts via distinct modulatory sites from known modulators, but 24(S)-HC, SGE-201, and SGE-301 likely share a common or overlapping binding site.
Oxysterols show robust NMDAR PAM activity in excised membrane patches
Recently PREGS was found to induce NMDAR receptor insertion into the plasma membrane as part of its mechanism of potentiation (Kostakis et al., 2013), although PREGS also modulates NMDARs in excised membrane patches where insertion is unlikely (Bowlby, 1993; Abdrachmanova et al., 2001). To determine whether oxysterol modulation primarily involves receptor insertion, potentially explaining its slow onset and poor reversibility, we explored the effects of SGE-201 in excised outside-out membrane patches from hippocampal neurons. In support of direct allosteric modulation, but contrary to expectations for receptor insertion, we found that SGE-201 robustly potentiated NMDAR channel activity in excised outside-out membrane patches (Fig. 5). All-point histograms revealed that potentiation increased channel number and (or) open probability but did not discernibly increase single-channel current (Fig. 5B). Evaluation of NPo values from eight patches showed quantitative increases consistent with potentiation values for whole-cell NMDA currents. Given that the potentiation in outside-out patches, where receptor insertion is extremely unlikely, is quantitatively similar to whole-cell potentiation, it appears that 24(S)-HC mainly modulates NMDARs through a mechanism that does not involve receptor insertion or transcription. Although the results do not completely exclude a membrane-delimited second messenger mechanism, the data are most consistent with binding to a direct modulatory site.
Oxysterols modulate recombinant NMDARs
Because native cells are heterogeneous in their NMDAR subunit combination, we examined oxysterol modulation of recombinant receptors to gain additional insight into selectivity. We first evaluated 24(S)-HC and the synthetic analogues SGE-201 and SGE-301 on GluN2A-containing receptors expressed in HEK cells (Fig. 6A,B). Each of the three analogues (Fig. 6A; 0.1 and 1 μm) significantly potentiated NMDA responses, with the synthetic analogues exhibiting stronger effects than the natural oxysterol (Fig. 6B).
We also used recombinant receptors transiently expressed in HEK cells to evaluate subunit dependence of oxysterol effects. PREGS exhibits strong GluN2 subunit selectivity, potentiating GluN2A and GluN2B responses, and inhibiting responses from GluN2C and GluN2D-containing NMDARs (Malayev et al., 2002; Horak et al., 2006). By contrast, we found no qualitative difference in subunit dependence of oxysterol potentiation. SGE-201 significantly potentiated responses from all subunit combinations (Fig. 6C,D), and potentiation values did not differ significantly among the GluN2 subunits. Thus, these results further distinguish the actions of oxysterols from the known modulator PREGS.
24(S)-Hydroxycholesterol is a selective NMDAR modulator
The selectivity of 24(S)-HC for NMDARs was evaluated by examining evoked EPSCs and IPSCs in cultured hippocampal neurons. 24(S)-HC potentiated pharmacologically isolated, evoked NMDAR EPSCs with characteristic slow onset and offset (Fig. 7A). Unlike neurosteroid GABAergic PAMs (Harrison et al., 1987) or AMPAR PAMs (Nagarajan et al., 2001), oxysterol analogues primarily augmented NMDAR EPSC peak amplitude, rather than decay kinetics (Fig. 7C). On average 10–90% decay values were increased by 32 ± 11% and 33 ± 10% for 24(S)-HC and SGE-201, respectively (p = 0.02 and 0.06). By comparison, neither AMPAR EPSCs nor GABAAR PSCs were significantly affected by 24(S)-HC or by SGE-201 (Fig. 7B,D,E). Insensitivity of GABAARs to 24(S)-HC is instructive because GABAARs are exquisitely sensitive to other neurosteroid-like molecules, including PREGS (Akk et al., 2001; Wang et al., 2002).
24(S)-Hydroxycholesterol and SGE-201 enhance synaptic plasticity
Do the potentiating effects of 24(S)-HC and SGE-201 on NMDAR function at synapses influence synaptic plasticity? Consistent with evoked EPSC results in Figure 7, we found no effect of oxysterols on baseline AMPAR field fEPSPs from adult (P120) rat hippocampal slices. However, both SGE-201 and 24(S)-HC transformed a normally subthreshold stimulus into a stimulus capable of inducing LTP (Fig. 8A,B). At P120, a single, high-frequency tetanus produces only STP that fades to baseline >15–20 min (Fig. 8). In the presence of 24(S)-HC or SGE-201, the same single tetanus resulted in robust synaptic enhancement that persisted for over 1 h. The effects of 24(S)-HC were concentration-dependent with marginal effects at 0.1 μm and robust LTP at 1 μm (Fig. 8B). Similar to results in cultured hippocampal neurons (Fig. 2), SGE-201 was ∼10-fold more potent than 24(S)-HC (Fig. 8).
Figure 8.

Augmentation of LTP by 24(S)-HC. A, P120 hippocampal slices were challenged with a brief HFS that produces STP, but not LTP (open symbols). However, in the presence of SGE-201 the same stimulus effectively induced LTP (closed symbols). Responses in control slices were 91.9 ± 5.6% of baseline EPSP slope at 50% of maximum on the I/O curve, 60 min following HFS. With SGE-201 present, EPSP slopes were increased to 156 ± 10.8% of baseline (p < 0.001, N = 5 slices each). B, Similar experiment with 24(S)-HC. Black symbols represent effect of 1 μm 24(S)-HC (166.1 ± 19.4% of baseline; p < 0.001), whereas 0.1 μm 24(S)-HC did not significantly enhance LTP induction (103.9 ± 9.8% of baseline, p = n.s., N = 5 slices each). Insets for A and B show representative traces. Dashed lines are baseline and solid lines represent traces in the indicated oxysterol 60 min following HFS. Calibration bars: 1 mV, 5 ms.
In addition to enhancing LTP induction by weak tetanic stimulation in the adult hippocampus, both 24(S)-HC and SGE-201 reversed LTP deficits induced by pretreatment with the NMDAR channel blocker ketamine in hippocampal slices from juvenile (P30) rats (Fig. 9). In these experiments, pretreatment of slices with 1 μm ketamine for 30 min results in LTP inhibition that is observed subsequent to ketamine washout and that persists for >2 h. In the presence of 24(S)-HC or SGE-201, tetanic stimulation produced LTP that was sustained for over 1 h in ketamine-pretreated slices. Similar results were obtained with SGE-301 (data not shown).
Figure 9.

SGE-201 and SGE-301 reverse synaptic plasticity deficits following NMDA receptor blockade. A, B, Reversal of ketamine suppression of long-term potentiation (LTP) by 24(S)-HC (A) and SGE-201 (B) in P30 hippocampal slices. Open symbols are baseline response following ketamine administration (1 μm, 30 min preincubation) to 100 Hz × 1 s HFS (vertical arrow). The change in baseline EPSP slope was 93.1 ± 2.3% 60 min following HFS in ketamine-treated control slices. Solid symbols represent the same condition except 0.5 μm 24(S)-HC (A; 131.3 ± 6.7% of baseline, p < 0.001, N = 5) or SGE-201 (B; 129.1 ± 9.2% of baseline, p < 0.001, N = 5) was present; N = 5 each, p = 0.008. Calibration bars: 1 mV, 5 ms.
SGE-201 and SGE-301 reverse behavioral deficits induced by NMDAR channel blockers
Given the ability of SGE-201 and SGE-301 to enhance LTP induction in vitro, we determined whether systemic administration of these agents could reverse deficits in rodent behavioral models. In contrast to 24(S)-HC, systemic administration of SGE-201 or SGE-301 results in good systemic and brain concentrations (Fig. 10A). The brain concentration of SGE-301 is improved when compared with SGE-201 (Fig. 10A, inset), as are its potency and intrinsic efficacy in potentiating NMDA receptor function (e.g., Fig. 6B).
Figure 10.

SGE-201 and SGE-301 reverse behavioral deficits following NMDA receptor blockade. A, Pharmacokinetic profiles of SGE-201(10 mg/kg, N = 3) and SGE-301(20 mg/kg, N = 2) following acute intraperitoneal administration in the mouse and rat respectively. Inset, Brain concentrations measured at 60 min following compound administration. Note that SGE-301 treatment resulted in a disproportionately higher brain concentration than SGE-201. B, SGE-201 reverses MK-801-nduced deficits in spontaneous alternations in the Y-maze in mice (N = 17–19/group). Percentage alternation was significantly reduced by MK-801 compared with vehicle (#p < 0.0001). SGE-201 restores alternation after MK-801, with increased percentage alternation in the 3 and 10 mg/kg SGE-201 groups compared with MK-801 alone (***p < 0.0005 and *p < 0.05). C, SGE-301 rescues social interaction deficits in PCP-experienced rats (N = 12–15/group). Vehicle or PCP (5 mg/kg, bid, i.p.) was administered twice daily from days 1 to 7. On day 14, SGE-301 was administered 60 min before testing. Time spent in active, nonaggressive social behavior was assessed during a 10 min session. PCP-experienced rats had significantly reduced interaction time compared with vehicle controls (#p < 0.0001). SGE-301 (3 and 10 mg/kg, i.p.) restored social interaction in PCP-experienced rats with significantly increased interaction time (****p < 0.0001 and *p < 0.05) versus PCP + vehicle group. D, SGE-301 rescues novel object recognition in PCP-experienced rats (12–15/group). On day 21, 7 d after social interaction testing, rats were administered SGE-301 intraperitoneally 60 min before object training. After the 30 min retention interval, object recognition was assessed in a 3 min test session. Discrimination ratio (time spent exploring the novel object/time spent exploring both objects during the test session) was calculated, so that a ratio of 0.5 corresponds to equal object preference (chance performance). Vehicle-treated PCP-experienced rats did not exhibit novel object preference and had a significantly reduced discrimination ratio (#p < 0.0001) compared with vehicle-treated rats. SGE-301 (1 and 3 mg/kg) significantly increased the discrimination ratio in PCP-experienced (*p < 0.05 and **p < 0.01vs vehicle + PCP), demonstrating rescue of the object recognition deficit.
The ability of SGE-201 to reverse MK-801-induced impairment in spatial working memory was assessed in a Y-maze spontaneous alternation task in mice. In this test, animals must recall which arms of the Y-maze they have previously explored in a trial, with an alternation consisting of successive entry into each of the three arms of the maze without re-entry into a previously visited arm. Acute administration of the NMDAR channel blocker, MK-801, significantly reduces the performance of animals in this task, as measured by percentage of alternations during a fixed testing interval. Treatment with SGE-201 produced a significant reversal of MK-801-induced deficits at 3 and 10 mg/kg, intraperitoneally (p < 0.0005 and p < 0.05, respectively; Fig. 10B). SGE-201 did not produce a significant effect on the overall number of arm entries in the Y-maze task (p > 0.05).
In rats, subchronic (7 d) administration of the NMDAR channel blocker, PCP (5 mg/kg, bid, i.p.) results in several behavioral deficits that persist for up to 14 d following cessation of treatment (Grayson et al., 2007; Snigdha and Neill, 2008). In the current study, at 7 d following cessation of PCP treatment, the duration of time spent in active social interaction was significantly (p < 0.001) reduced in PCP-experienced rats (32 ± 3 s) compared with vehicle-treated rats (62 ± 4 s; Fig. 10C). Acute administration of SGE-301 (3 and 10 mg/kg i.p.) significantly (p < 0.001 and p < 0.05, respectively) increased time spent in active social interactions in PCP-experienced rats (57 ± 4 s and 48 ± 3 s, respectively) compared with vehicle-treated PCP-experienced rats (32 ± 3 s; Fig. 10C). Administration of risperidone (0.2 mg/kg, i.p.) also significantly (p < 0.05) increased time spent in active social interactions (51 ± 3 s) when compared with vehicle-treated PCP-experienced rats (data not shown).
Subchronic administration of PCP also significantly impaired performance in the NOR task when tested 14 d following cessation of PCP treatment (Fig. 10D). Due to their natural proclivity for novelty, rodents normally spend more time exploring the novel object in the test session, as demonstrated by the vehicle-treated group. Novel object preference was eliminated in PCP-experienced rats, an effect that was significantly reversed by acute administration of SGE-301 at 1 and 3 mg/kg intraperitoneally (p < 0.05 and p < 0.01, respectively). Risperidone also improved novel object preference in PCP-experienced rats (p < 0.001, data not shown). SGE-301 had no significant effect on locomotor activity in PCP-experienced rats during the NOR task (p > 0.05).
Discussion
The synthesis of 24(S)-HC, the major cholesterol metabolite in brain, is critical to brain cholesterol metabolism and turnover (Lund et al., 2003; Russell et al., 2009). Unlike cholesterol, 24(S)-HC is membrane-permeable and readily enters the peripheral circulation, where it is subsequently metabolized and excreted (Lütjohann et al., 1996; Lund et al., 2003). 24(S)-HC has been measured at very high concentrations in brain (10–20 μg/g tissue or ∼25 μm) in a variety of mammalian species (Smith et al., 1972; Meljon et al., 2012). Russell et al. (2009) have identified the highly conserved cytochrome P450 enzyme (cholesterol 24-hydroxylase, CYP46A1) responsible for synthesis of 24(S)-HC from cholesterol (Lund et al., 1999). This enzyme is expressed predominantly in the endoplasmic reticulum of neuronal cell bodies and dendrites, but not axons (Ramirez et al., 2008). Importantly, cholesterol 24-hydroxylase deficiency disrupts hippocampal LTP and memory acquisition in mice (Kotti et al., 2006) and this disruption of hippocampal LTP is rescued by geranylgeraniol diphosphate, a downstream nonsteroidal isoprenoid metabolite of mevalonate (Kotti et al., 2008). Our data suggest that 24(S)-HC may also directly impact synaptic plasticity by modulating postsynaptic NMDARs in an autocrine manner near sites of 24(S)-HC synthesis (Ramirez et al., 2008). Interestingly, the potency of 24(S)-HC at NMDARs is almost 10-fold greater than at LXRs, the other major target of 24(S)-HC (Janowski et al., 1996; Kalaany and Mangelsdorf, 2006), and the potency of 24(S)-HC at NMDARs in our study may represent an underestimate given the slow onset time of oxysterol action. Other potent LXR agonist oxysterols, such as 22(R)-HC and 20(S)-HC, are not active NMDAR modulators (Fig. 1B), and neither SGE-201 nor SGE-301 are LXR agonists (data not shown). Combined with the excised membrane patch data in Figure 5, these observations indicate that LXRs do not mediate the modulatory effects of 24(S)-HC on NMDARs.
Oxysterols are now well recognized as important signaling molecules that interact with various soluble intracellular as well as membrane-bound receptors. In addition to serving as ligands for LXRs in a variety of tissues (Janowski et al., 1996; Kalaany and Mangelsdorf, 2006), oxysterols bind with relatively high affinity to membrane proteins including Insig proteins (Radhakrishnan et al., 2007) and oxysterol-binding proteins (Banerji et al., 2010), which act as oxysterol/cholesterol sensors to regulate cholesterol synthesis and lipid metabolism. Oxysterols also activate the Hedgehog (Hh) signaling pathway (Corcoran and Scott, 2006) by binding to and allosterically activating the seven trans-membrane protein Smoothened which mediates signal transduction by Hh ligands (Nachtergaele et al., 2012). Thus, like steroids, oxysterols can subserve cell signaling functions by binding to either soluble or membrane-bound receptors and via both transcriptional and post-translational mechanisms.
Our data on 24(S)-HC and NMDAR-mediated synaptic transmission are reminiscent of previous work on neuroactive steroids (Paul and Purdy, 1992), which directly enhance or inhibit synaptic transmission at GABA-ARs (Majewska et al., 1986; Paul and Purdy, 1992) or NMDARs (Wu et al., 1991). PREGS, one purported neurosteroid (Vallee et al., 1997), potentiates NMDARs (Wu et al., 1991) and inhibits GABA-ARs (Akk et al., 2001). Moreover, direct administration of PREGS to the CNS of rodents has been reported to improve learning and memory and to reverse the amnestic effects of NMDAR blockers (Flood et al., 1992; Mathis et al., 1994). Nonetheless, PREGS seems unlikely to play a significant role in the physiological regulation of NMDARs as it is a relatively weak NMDAR PAM (EC50 ≥ 50 μm) (Wu et al., 1991) and its presence in rodent brain is controversial (Liere et al., 2009). Given its potency and high brain concentrations, 24(S)-HC is a more compelling candidate to modulate NMDARs under physiological or pathophysiologic conditions. Further work however will be required to determine whether 24(S)-HC modulates NMDARs under physiological conditions. The availability of potent CYP46A1 inhibitors (Shafaati et al., 2010) and CYP46A1-deficient mice (Lund et al., 2003) should facilitate these studies. Nonetheless, regardless of a physiologic role for 24(S)-HC in regulating NMDAR function, it appears that the novel oxysterol NMDAR modulatory site described here represents a potential target for drug discovery. The potencies of 24(S)-HC and its closely related synthetic analogs SGE-201 (Madau et al., 2009) and SGE-301 attest to the striking selectivity of 24-hydroxylated oxysterols for NMDARs. Figure 1A shows that these compounds bear significant structural similarity, particularly with respect to the 24-hydroxyl group of the sterol side chain (at C-17) and the steroid backbone. The lack of any measurable activity of other endogenous oxysterols at NMDARs (Fig. 1B) as well as additional compounds synthesized in an attempt to optimize the drug-like properties of this series (data not shown), further supports the specificity of these 24(S)-hydroxylated oxysterols for NMDARs. Importantly, synthetic oxysterol derivatives, like SGE-201 and SGE-301, have desirable in vivo drug-like properties, including excellent brain concentrations after parenteral dosing (Fig. 10A). The reversal of NMDAR blocker-induced impairment of cognitive and social behavior following treatment with SGE-201 or SGE-301 suggests that this novel oxysterol NMDAR modulatory site could serve as a target for designing drugs to treat a variety of neuropsychiatric disorders where augmenting NMDAR function may be of therapeutic benefit.
Footnotes
This work was supported by Sage Therapeutics and National Institutes of Health Grants MH078823, MH077791, GM47969, and AA017413, and the Bantly Foundation. We thank J.C. Dodart, Frank Salituro, Gabriel Botella, Kiran Reddy, Carlos Loya, Ann Benz, and Amanda Taylor for their technical support and suggestions.
S.M.P. is a founder and shareholder of Sage Therapeutics. J.J.D., A.J.R., G.M.B., B.Y.C., and R.S.H are either employees and (or) shareholders of Sage Therapeutics. C.F.Z. is a member of the Sage Therapeutics Scientific Advisory Board. The remaining authors declare no competing financial interests.
References
- Abdrachmanova G, Chodounska H, Vyklický L., Jr Effects of steroids on NMDA receptors and excitatory synaptic transmission in neonatal motoneurons in rat spinal cord slices. Eur J Neurosci. 2001;14:495–502. doi: 10.1046/j.0953-816x.2001.01663.x. [DOI] [PubMed] [Google Scholar]
- Akk G, Bracamontes J, Steinbach JH. Pregnenolone sulfate block of GABAA receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol. 2001;532:673–684. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–11613. doi: 10.1523/JNEUROSCI.4173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztely F, Wigström H, Gustafsson B. The relative contribution of NMDA receptor channels in the expression of long-term potentiation in the hippocampal CA1 region. Eur J Neurosci. 1992;4:681–690. doi: 10.1111/j.1460-9568.1992.tb00177.x. [DOI] [PubMed] [Google Scholar]
- Banerji S, Ngo M, Lane CF, Robinson CA, Minogue S, Ridgway ND. Oxysterol binding protein-dependent activation of sphingomyelin synthesis in the Golgi apparatus requires phosphatidylinositol 4-kinase IIalpha. Mol Biol Cell. 2010;21:4141–4150. doi: 10.1091/mbc.E10-05-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991;349:156–158. doi: 10.1038/349156a0. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Bowlby MR. Pregnenolone sulfate potentiation of N-methyl-d-aspartate receptor channels in hippocampal neurons. Mol Pharmacol. 1993;43:813–819. [PubMed] [Google Scholar]
- Connick JH, Cadzow L, Madau P, Jeggo R, Erdemli G, Thomson F. Org (1) potentiates NMDA-receptor mediated responses in vivo. Soc Neurosci Abstr. 2009;35:613.1. [Google Scholar]
- Corcoran RB, Scott MP. Oxysterols stimulate sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci U S A. 2006;103:8408–8413. doi: 10.1073/pnas.0602852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Wang H, Tan Y, Zaia KA, Zhang S, Tsien JZ. Inducible and reversible NR1 knockout reveals crucial role of the NMDA receptor in preserving remote memories in the brain. Neuron. 2004;41:781–793. doi: 10.1016/S0896-6273(04)00072-8. [DOI] [PubMed] [Google Scholar]
- Flood JF, Morley JE, Roberts E. Memory-enhancing effects in male mice of pregnenolone and steroids metabolically derived from it. Proc Natl Acad Sci U S A. 1992;89:1567–1571. doi: 10.1073/pnas.89.5.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson B, Idris NF, Neill JC. Atypical antipsychotics attenuate a sub-chronic PCP-induced cognitive deficit in the novel object recognition task in the rat. Behav Brain Res. 2007;184:31–38. doi: 10.1016/j.bbr.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Harrison NL, Vicini S, Barker JL. A steroid anesthetic prolongs inhibitory postsynaptic currents in cultured rat hippocampal neurons. J Neurosci. 1987;7:604–609. doi: 10.1523/JNEUROSCI.07-02-00604.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefco V, Yamada K, Hefco A, Hritcu L, Tiron A, Nabeshima T. Role of the mesotelencephalic dopamine system in learning and memory processes in the rat. Eur J Pharmacol. 2003;475:55–60. doi: 10.1016/S0014-2999(03)02115-0. [DOI] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Petrovic M, Chodounska H, Vyklicky L., Jr Molecular mechanism of pregnenolone sulfate action at NR1/NR2B receptors. J Neurosci. 2004;24:10318–10325. doi: 10.1523/JNEUROSCI.2099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Chodounska H, Vyklicky L., Jr Subtype-dependence of N-methyl-d-aspartate receptor modulation by pregnenolone sulfate. Neuroscience. 2006;137:93–102. doi: 10.1016/j.neuroscience.2005.08.058. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Norepinephrine promotes long-term potentiation in the adult rat hippocampus in vitro. Synapse. 1999;31:196–202. doi: 10.1002/(SICI)1098-2396(19990301)31:3<196::AID-SYN4>3.0.CO%3B2-K. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- Kostakis E, Smith C, Jang MK, Martin SC, Richards KG, Russek SJ, Gibbs TT, Farb DH. The neuroactive steroid pregnenolone sulfate stimulates trafficking of functional NMDA receptors to the cell surface via a non-canonical G-protein and Ca++ dependent mechanism. Mol Pharmacol. 2013;84:261–274. doi: 10.1124/mol.113.085696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, Russell DW. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci U S A. 2006;103:3869–3874. doi: 10.1073/pnas.0600316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotti T, Head DD, McKenna CE, Russell DW. Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc Natl Acad Sci U S A. 2008;105:11394–11399. doi: 10.1073/pnas.0805556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Tsien JZ. Memory and the NMDA receptors. N Engl J Med. 2009;361:302–303. doi: 10.1056/NEJMcibr0902052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liere P, Pianos A, Eychenne B, Cambourg A, Bodin K, Griffiths W, Schumacher M, Baulieu EE, Sjövall J. Analysis of pregnenolone and dehydroepiandrosterone in rodent brain: cholesterol autoxidation is the key. J Lipid Res. 2009;50:2430–2444. doi: 10.1194/jlr.M900162-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug; sernyl. AMA Arch Neurol Psychiatry. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278:22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- Lütjohann D, Breuer O, Ahlborg G, Nennesmo I, Sidén A, Diczfalusy U, Björkhem I. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A. 1996;93:9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madau P, Clark AG, Neale SA, Smith L, Hamilton N, Thomson F, Connick JH, Belelli D, Lambert JJ. Org 1, Org 2 and Org 3 potentiate NMDA-receptor mediated responses in vitro. Program #613.2. Neuroscience Meeting Planner; Chicago, IL: Society for Neuroscience; 2009. 2009. Online. [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- Malayev A, Gibbs TT, Farb DH. Inhibition of the NMDA response by pregnenolone sulphate reveals subtype selective modulation of NMDA receptors by sulphated steroids. Br J Pharmacol. 2002;135:901–909. doi: 10.1038/sj.bjp.0704543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis C, Paul SM, Crawley JN. The neurosteroid pregnenolone sulfate blocks NMDA antagonist-induced deficits in a passive avoidance memory task. Psychopharmacology (Berl) 1994;116:201–206. doi: 10.1007/BF02245063. [DOI] [PubMed] [Google Scholar]
- Meljon A, Theofilopoulos S, Shackleton CH, Watson GL, Javitt NB, Knölker HJ, Saini R, Arenas E, Wang Y, Griffiths WJ. Analysis of bioactive oxysterols in newborn mouse brain by LC/MS. J Lipid Res. 2012;53:2469–2483. doi: 10.1194/jlr.D028233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- Mouriño A, Blair P, Wecksler W, Johnson RL, Norman AW, Okamura WH. Studies on vitamin D (calciferol) and its analogues: 15. 24-Nor-1alpha,25-dihydroxyvitamin D3 and 24-nor-25-hydroxy-5,6-trans-vitamin D3. J Med Chem. 1978;21:1025–1029. doi: 10.1021/jm00208a005. [DOI] [PubMed] [Google Scholar]
- Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, Rohatgi R. Oxysterols are allosteric activators of the oncoprotein smoothened. Nat Chem Biol. 2012;8:211–220. doi: 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan N, Quast C, Boxall AR, Shahid M, Rosenmund C. Mechanism and impact of allosteric AMPA receptor modulation by the ampakine CX546. Neuropharmacology. 2001;41:650–663. doi: 10.1016/S0028-3908(01)00133-2. [DOI] [PubMed] [Google Scholar]
- Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur J Biochem. 1989;186:17–22. doi: 10.1111/j.1432-1033.1989.tb15171.x. [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/S0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- Plattner A, Pataki J. Über steroide und sexualhormone. (86. Mitteilung). Über die hydrierungsprodukte der Δ5, 6; 20, 22–3b-Oxy-nor-choladiensaure. Helv Chim Acta. 1943;26:1241–1252. doi: 10.1002/hlca.19430260427. [DOI] [Google Scholar]
- Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Andersson S, Russell DW. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol. 2008;507:1676–1693. doi: 10.1002/cne.21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW, Halford RW, Ramirez DM, Shah R, Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem. 2009;78:1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafaati M, Mast N, Beck O, Nayef R, Heo GY, Björkhem-Bergman L, Lütjohann D, Björkhem I, Pikuleva IA. The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24S-hydroxylase in vitro and in vivo. J Lipid Res. 2010;51:318–323. doi: 10.1194/jlr.M900174-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwinski A, Monnet FP, Schumacher M, Morin-Surun MP. Pregnenolone sulfate enhances long-term potentiation in CA1 in rat hippocampus slices through the modulation of N-methyl-d-aspartate receptors. J Neurosci Res. 2004;78:691–701. doi: 10.1002/jnr.20332. [DOI] [PubMed] [Google Scholar]
- Smith LL, Ray DR, Moody JA, Wells JD, Van Lier JE. 24-hydroxycholesterol levels in human brain. J Neurochem. 1972;19:899–904. doi: 10.1111/j.1471-4159.1972.tb01406.x. [DOI] [PubMed] [Google Scholar]
- Snigdha S, Neill JC. Efficacy of antipsychotics to reverse phencyclidine-induced social interaction deficits in female rats-A preliminary investigation. Behav Brain Res. 2008;187:489–494. doi: 10.1016/j.bbr.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Tamminga CA. Schizophrenia and glutamatergic transmission. Crit Rev Neurobiol. 1998;12:21–36. doi: 10.1615/CritRevNeurobiol.v12.i1-2.20. [DOI] [PubMed] [Google Scholar]
- Tokuda K, O'Dell KA, Izumi Y, Zorumski CF. Midazolam inhibits hippocampal long-term potentiation and learning through dual central and peripheral benzodiazepine receptor activation and neurosteroidogenesis. J Neurosci. 2010;30:16788–16795. doi: 10.1523/JNEUROSCI.4101-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallée M, Mayo W, Darnaudéry M, Corpéchot C, Young J, Koehl M, Le Moal M, Baulieu EE, Robel P, Simon H. Neurosteroids: deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proc Natl Acad Sci U S A. 1997;94:14865–14870. doi: 10.1073/pnas.94.26.14865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, He Y, Eisenman LN, Fields C, Zeng CM, Mathews J, Benz A, Fu T, Zorumski E, Steinbach JH, Covey DF, Zorumski CF, Mennerick S. 3β-hydroxypregnane steroids are pregnenolone sulfate-like GABAA receptor antagonists. J Neurosci. 2002;22:3366–3375. doi: 10.1523/JNEUROSCI.22-09-03366.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. Biology of the NMDA Receptor, Chap 11. Boca Raton, FL: CRC; 2009. Extracellular modulation of NMDA receptors. [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH. Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-d-aspartate receptor. Mol Pharmacol. 1991;40:333–336. [PubMed] [Google Scholar]
- Zorumski CF, Izumi Y. NMDA receptors and metaplasticity: mechanisms and possible roles in neuropsychiatric disorders. Neurosci Biobehav Rev. 2012;36:989–1000. doi: 10.1016/j.neubiorev.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]