

Abstract
Sonic hedgehog (SHH), a key regulator of embryonic neurogenesis, signals directly to neural stem cells (NSCs) in the subventricular zone (SVZ) and to astrocytes in the adult mouse forebrain. The specific mechanism by which the GLI2 and GLI3 transcriptional activators (GLI2A and GLI3A) and repressors (GLI2R and GLI3R) carry out SHH signaling has not been addressed. We found that the majority of slow-cycling NSCs express Gli2 and Gli3, whereas Gli1 is restricted ventrally and all three genes are downregulated when NSCs transition into proliferating progenitors. Surprisingly, whereas conditional ablation of Smo in postnatal glial fibrillary acidic protein-expressing cells results in cell-autonomous loss of NSCs and a progressive reduction in SVZ proliferation, without an increase in glial cell production, removal of Gli2 or Gli3 does not alter adult SVZ neurogenesis. Significantly, removing Gli3 in Smo conditional mutants largely rescues neurogenesis and, conversely, expression of a constitutive GLI3R in the absence of normal Gli2 and Gli3 abrogates neurogenesis. Thus unattenuated GLI3R is a primary inhibitor of adult SVZ NSC function. Ablation of Gli2 and Gli3 revealed a minor role for GLI2R and little requirement for GLIA function in stimulating SVZ neurogenesis. Moreover, we found that similar rules of GLI activity apply to SHH signaling in regulating SVZ-derived olfactory bulb interneurons and maintaining cortical astrocyte function. Namely, fewer superficial olfactory bulb interneurons are generated in the absence of Gli2 and Gli3, whereas astrocyte partial gliosis results from an increase in GLI3R. Thus precise titration of GLIR levels by SHH is critical to multiple functions of adult NSCs and astrocytes.
Introduction
The subventricular zone (SVZ) lining the lateral ventricles of the murine telencephalon is one of two sites where neurogenesis continues to occur in the adult (Alvarez-Buylla et al., 2008). Along with a variety of supporting niche cells and vasculature, the adult SVZ harbors three progenitor cell types: (1) slow-cycling glial-like neural stem cells (NSCs) and their progeny, (2) transit-amplifying cells (TACs), and (3) the more differentiated neuroblasts (NBs) that migrate anteriorly to supplement the interneuron and periglomerular cell populations of the olfactory bulb (OB; Ihrie and Alvarez-Buylla, 2011). A considerable body of evidence has established an essential role for Sonic hedgehog (SHH) in SVZ neurogenesis (Machold et al., 2003; Balordi and Fishell, 2007a, b; Han et al., 2008; Ihrie et al., 2011). In vertebrates, the zinc-finger transcription factors GLI1, GLI2, and GLI3, together with a receptor complex that includes smoothened (SMO), propagate SHH signaling (Hui and Angers, 2011). In the absence of ligand, GLI2 and GLI3 undergo proteolysis with GLI3 being preferentially cleaved into an N-terminal repressor form (GLI3R). In contrast, high-level SHH signaling preferentially converts GLI2 into an activator form (GLI2A) and downregulates GLI3R. Notably, Gli1 is a direct transcriptional target of GLI2/3A, and thus its expression serves as a readout of GLIA-mediated SHH signaling (Bai et al., 2002; 2004). Whether activation of GLI2A, the primary effector of SHH signaling in the spinal cord (Bai et al., 2004), or loss of GLI3R, dominant in the developing forebrain (Rallu et al., 2002), is central to maintaining adult SVZ neurogenesis remains to be determined.
Gli1 fate-mapping and expression studies in the postnatal forebrain have demonstrated that SHH signals to glial fibrillary acidic protein (GFAP)-expressing NSCs in the ventral SVZ (Ahn and Joyner, 2005; Ihrie et al., 2011), as well as to discrete astrocyte populations (Garcia et al., 2010). Mosaic removal of Smo in SVZ NSCs at postnatal day (P) 60 results in reduced proliferation and deficient regeneration of the SVZ (Balordi and Fishell, 2007b), whereas stimulation of SHH signaling in the mature brain augments both Gli1 transcription and SVZ proliferation (Machold et al., 2003). High-level SHH signaling also can alter the proportion of deep versus superficial OB interneurons produced (Ihrie et al., 2011), indicating two roles for SHH in SVZ neurogenesis. In addition, postnatal ablation of Smo causes cortical astrocytes to transition into a partial reactive astrogliosis-like state (Garcia et al., 2010), suggesting a third function of SHH in the adult brain is in maintaining astrocyte function.
Here we demonstrate that SHH signaling in the adult SVZ is restricted to NSCs, and required to cell-autonomously maintain label-retaining cells, progenitor proliferation, and OB neuron generation. We further uncovered that, in the absence of Smo, unattenuated GLI3R and, to a lesser extent, GLI2R cause the SVZ phenotypes as well as defects in cortical astrocyte function. In contrast, loss of GLIA function has little effect on either cell type. Thus SVZ NSC maintenance and production of OB neurons, as well as the function of cortical astrocytes, depend on the precise titration of GLIR levels by SHH.
Materials and Methods
Animals.
The following mouse lines were used and genotyped as previously described: Gli1nlacZ (Bai et al., 2002), Gli2nlacZ (Bai and Joyner, 2001), Gli3nlacZ (Garcia et al., 2010), mGfap-Cre (Garcia et al., 2004), Nestin-FlpoER and Rosa26MASTR(frt-STOP-frt-GFPcre) (Lao et al., 2012), Rosa26lox-STOP-lox-lacZ (Soriano, 1999), Rosa26lox-STOP-lox-tdTomato (Madisen et al., 2010), Smolox (Long et al., 2001), Gli2lox (Corrales et al., 2006), Gli2zfd (null allele of Gli2, referred to as Gli2−; Mo et al., 1997), Gli3lox (Blaess et al., 2008), Gli3Δ699 (Böse et al., 2002). Unexpectedly, we also found that the mGfap-Cre transgene recombines in the germline as when both the Cre and lox alleles were paternally inherited, a deleted allele was generated (referred to as “−”). All lines were maintained on an outbred Swiss Webster background. Mice of either sex were used for the analysis. Animals were housed on a 12 h light/dark cycle and were given access to food and water ad libitum. Experiments were performed in accordance with protocols approved by the Memorial Sloan-Kettering Cancer Center's Institutional Animal Care and Use Committee.
Bromodeoxyuridine administration.
Bromodeoxyuridine (BrdU; Sigma-Aldrich B9285) was administered via intraperitoneal injection. To label slow-cycling NSCs in the SVZ or newly generated OB granule cell interneurons (which incorporate the BrdU label during their last cell division) in the adult, 2–3-month-old mice received 50 mg/kg BrdU once a day for 5 d and were killed 3 weeks later. To label slow-cycling NSCs at P15, pups received two 100 mg/kg BrdU once a day for 2 d and were killed 3 weeks later (at ∼P40). A 3 week chase was chosen to study adult OB granule neuron generation based on previous evidence that all BrdU-labeled progenitors reach their final deep-to-superficial position in the OB by 3 weeks (Lemasson et al., 2005).
Tamoxifen administration.
Tamoxifen (Sigma-Aldrich, T5648-1G) was dissolved in corn oil (Sigma-Aldrich, C8267-500ML) at 20 mg/ml concentration, and then administered via oral gavage or intraperitoneal injection. For Gli1CreER/+;Rosa26YFP/+ fate-mapping, adult (2–3-month-old) mice received three 250 μg/g doses of tamoxifen over the course of 3 d and were killed for analysis 30 d later. For conditional mosaic ablation of Smo, P21 Nestin-FlpoER/+; Rosa26MASTR/+; Smolox/− mice or Nestin-FlpoER/+; Rosa26MASTR/+; Smolox/+ controls were administered tamoxifen via intraperitoneal injection once a day, every other day, for a total of three injections. Tamoxifen-induced animals were killed for analysis 5 weeks after the last injection.
Tissue processing.
For immunohistochemistry and immunofluorescence stainings, animals were anesthetized and transcardially perfused with PBS followed by chilled 4% paraformaldehyde (Fisher Scientific, T353-500). Brains were postfixed for 2–3 h and cryoprotected in 30% sucrose before freezing in Cryo-OCT (VWR, 25608-930). Frozen brains were sectioned on a cryostat and 40 μm sections were collected in 0.05% sodium azide in PBS and stored at 4°C until further processing. Brains from Smo mosaic mutant and control animals were cryosectioned at 20 μm and sections were stored at −20°C until further processing.
Immunohistochemistry.
Immunohistochemistry (IHC) was performed on free-floating sections using the following primary antibodies: rabbit anti-βGAL (ICN Biomedicals, 55976, and eBioscience, 14-6773-63), mouse anti-βGAL (Promega, Z3793), chicken anti-βGAL (Abcam, ab9361, and Aves, BGL-1040), rabbit anti-GFP (Invitrogen, A-11122), rat anti-GFP (Nacalai Tesque, 04404-84), rabbit anti-GFAP (DAKO, Z0334), mouse anti-GFAP (Millipore, MAB3402), chicken anti-GFAP (Fisher Scientific, AB5541), rabbit anti-S100β (DAKO, Z0311), mouse anti-S100β (Abcam, ab66028), sheep anti-BrdU (Biodesign International, M20107S, and Abcam, ab1893), rat anti-BrdU (Accurate Chemical and Scientific, OBT0030), rabbit anti-KI67 (Abcam, ab15580), goat anti-DCX (Santa Cruz Biotechnology, sc-8066), mouse anti-polysialated neuronal cell adhesion molecule (anti-PSA-NCAM; Millipore, MAB5324), rabbit anti-OLIG (Abcam, 33427), mouse anti-NeuN (Millipore Bioscience Research Reagents, MAB377), rabbit anti-calbindin (Swant, CB38). For BrdU IHC staining, sections were incubated in 2N HCl for 30 min before blocking in 10% serum and incubation in primary antibody; alternatively, antigen retrieval in 10 mm citrate buffer with 0.05% Tween 20 was performed at 95°C. For brightfield IHC staining, species-specific, biotinylated secondary antibodies (Vector) were used in combination with avidin-biotin complex (Vectastatin Elite ABC Kit, Vector, PK-6100). Staining was developed using diaminobenzadine (DAB; DAB Substrate Kit, Vector, SK-4100). For double and triple immunofluorescence labeling, species-specific AlexaFluor-tagged secondary antibodies were used (Invitrogen), followed by counterstaining with DAPI (Invitrogen, D3571).
XGAL staining.
Brains were lightly fixed using immersion fix (for P0 mGfap-Cre;R26lacZ/+ and adult Gli2nlacZ/+, Gli3nlacZ/+ and mutant Gli1nlacZ/+) or perfusion (for all other stages of mGfap-Cre;R26lacZ/+ mice). Brain sections were rinsed with XGAL washing buffer (2 mm MgCl2, 0.1% Igepal Ca-30, 0.05% deoxycholate in PBS), then incubated in XGAL reaction buffer (0.17 mm potassium ferrocyanide, 0.17 mm potassium ferricyanide, 1 mg/ml X-gal substrate) at 37°C. Sections were occasionally counterstained with nuclear fast red.
Imaging.
Stained sections were imaged on an inverted Zeiss Axio Observer.A1 microscope using Axiovision software. Single-cell analysis for antigen colocalization was performed using a 20× or 40× objective and Zeiss Apotome. Z-stacks were collected at approximately equal positions of the dorsal–ventral axis of the SVZ, as well as in the rostral migratory stream (RMS) or in the OB. Brightfield images were taken using 10× or 20× objectives.
Quantification and statistical analysis.
DAB-stained brain sections were used for quantitative analysis of BrdU label-retaining NSCs and Ki67+ progenitors in the SVZ, as well as for the analysis of BrdU-labeled newly generated OB interneurons. Cell counting and measurements of the brain areas of interest were performed on slides using an upright Zeiss Imager.M1 microscope with motorized stage using Stereoinvestigator image analysis software (MBF Bioscience). Analysis of proliferating cells in the SVZ was performed on 2–3 sections per animal, and included the SVZ of both ventricles, if they were intact, and both the septal and striatal ventricular sides of each SVZ, and was limited to the region 40 μm adjacent to the ventricle (which excludes a large portion of the migrating NBs in the dorsal striatal SVZ). The distribution of stem and progenitor cells along the dorsal–ventral axis of the SVZ was analyzed by measuring the length of the striatal and septal wall of each ventricle to find the wall midsection, and then establishing what percentage of the counted cells localize to the dorsal or ventral subregion. The distribution of newly born (BrdU+) interneurons in the OB was done by subdividing the area between the outer boundary of the granule cell layer and the core (RMS) into three regions, each of which represents one-third of the total radius. A minimum of two OB sections were analyzed for each animal. For the analysis of molecular markers expressed by slow-cycling cells (NSCs) in the SVZ of mutants, where Smo was ablated using mGfap-Cre, images of BrdU/GFAP/S100β triple stainings were acquired from 4–5 sections per animal on a Zeiss Axio Observer.A1 microscope at 40×. Cell counting was performed on the acquired images using Stereoinvestigator image analysis software (MBF Bioscience). A similar approach was taken for the analysis of GFP/GFAP/S100β triple stainings of mutants where Smo was ablated using Nestin-FlpoER; MASTR. All quantitative data analysis was performed using Prism software (GraphPad).
Results
The GLI transcription factors are expressed primarily in adult NSCs in the SVZ
The expression profile of Gli2 and Gli3, with respect to each of the adult neural progenitor cell types, has not been characterized. RNA in situ hybridization and qRT-PCR studies for Gli2 and Gli3 have demonstrated expression throughout the dorsal–ventral extent of the SVZ, whereas Gli1 transcription is primarily detected in the ventral half of the SVZ (Ahn and Joyner, 2005; Palma et al., 2005; Ihrie et al., 2011). The three Gli genes also are expressed in a subset of mature astrocytes throughout the adult forebrain (Garcia et al., 2010). Using LacZ knock-in reporter strains for all three Gli genes (Gli1nlacZ, Gli2nlacZ, Gli3nlacZ), we characterized the Gli2-expressing and Gli3-expressing cell populations in the adult SVZ compared with those expressing Gli1, to determine whether SHH signaling is preferentially restricted to a specific progenitor cell type based on expression of the two SHH effectors.
Analysis of βGAL protein expression in the SVZ of 2–3-month-old Gli-reporter mice revealed that, unlike the ventrally restricted Gli1 expression, Gli2-expressing and Gli3-expressing cells are distributed more evenly throughout the full dorsal–ventral extent of the SVZ (Fig. 1A–C). We examined βGAL expression in Gli2nlacZ/+ and Gli3nlacZ/+ mice in greater detail using timed XGAL enzymatic staining and observed that following 1.5 h of enzyme activity, βGAL expression in Gli3nlacZ/+ mice was detectable only in the dorsal SVZ, whereas Gli2 was expressed equally and more strongly in the dorsal and ventral SVZ (Fig. 1D,E, arrowheads). In addition, XGAL activity in Gli3nlacZ/+ brains did not become apparent in the ventral SVZ until 3 h of staining (Fig. 1D',D”,E',E”), indicating that Gli3 might be preferentially expressed at higher levels in the dorsal SVZ.
Figure 1.
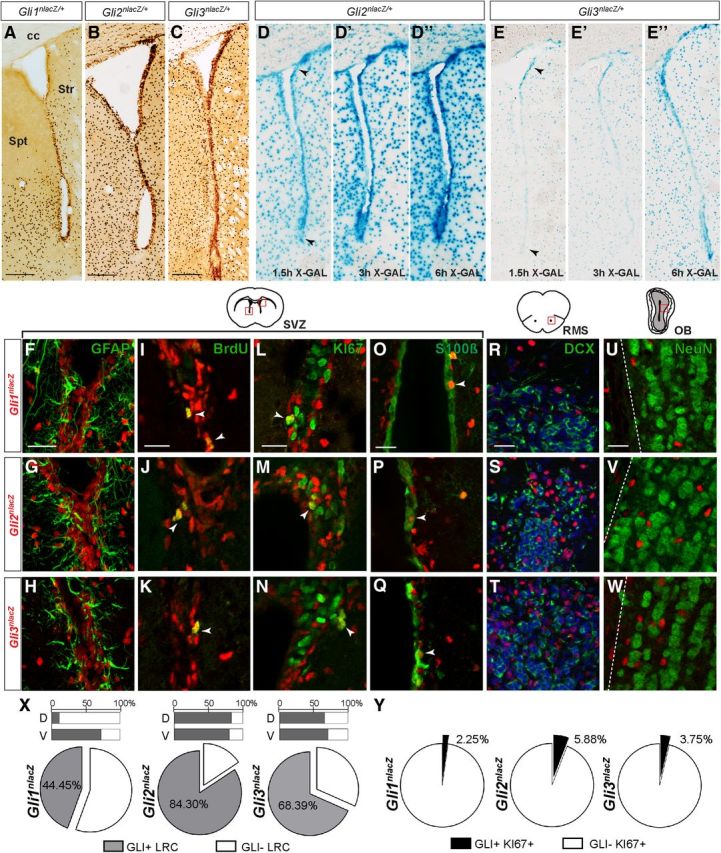
SHH signaling effectors GLI1, GLI2, and GLI3 are differentially expressed in the adult SVZ. A–C, Immunohistochemistry for βGAL (dark brown) activity from LacZ knock-in reporter alleles for Gli1, Gli2, and Gli3 shows expression of all three in the adult SVZ, with Gli1 expression primarily in the ventral portion of the SVZ. Scale bar, 200 μm. D–E”, XGAL enzymatic staining of coronal sections from adult Gli2nlacZ/+ (D–D”) and Gli3nlacZ/+ (E–E”) mice are shown after 1.5, 3, or 6 h of enzyme activity, and reveal that LacZ expression is strongest dorsally in Gli3nlacZ/+ SVZ and is more uniform in Gli2nlacZ/+ SVZ (arrowheads). F–H, GFAP is detected in many βGAL+ NSCs and astrocytes in the adult SVZ in the three reporter lines. I–Q, X, Y, The majority of BrdU label-retaining NSCs are GLI+ (I–K, X), whereas only a very small subset of dividing KI67+ progenitors (L–N, Y) or small proportion of S100β+ ependymal cells (O–Q) coexpresses βGAL. Red boxes in the schematic of the SVZ indicate the location of the images of the SVZ shown below (M, N, dorsal; ventral in others). R–T, In the RMS, DCX+ migratory NBs do not express any of the GLIs. U–W, βGAL expression is not detected in mature NeuN+ OB interneurons. Scale bars: (in F, I, L, O, R, U) F–W, 20 μm. Str, Striatum; spt, septum; cc, corpus callosum. X, Quantitative analysis of Gli expression in BrdU+ LRCs in the SVZ of the three reporter lines reveals that the vast majority of SVZ LRCs can respond to SHH signaling, as LRCs both in the dorsal (D-bar graph) and ventral (V-bar graph) SVZ are GLI2+ and GLI3+. However, only a subset of LRCs in the ventral SVZ is exposed to high levels of ligand (GLI1+).
Extensive analysis of individual double-labeled cells in the SVZ of adult Gli-reporter mice revealed that Gli2 and Gli3, similar to Gli1, are expressed in many GFAP+ cells, which includes NSCs and mature astrocytes (Fig. 1F–H). Strikingly, we observed Gli expression in the majority of BrdU label-retaining stem cells (LRCs) in the SVZ (Fig. 1I–K,X). We found that 84.30 ± 10.3% of SVZ LRCs expressed Gli2 and 68.39 ± 2.9% expressed Gli3 (207 LRCs total counted in n = 3 Gli2nlacZ/+ animals and 206 LRCs total counted in n = 3 Gli3nlacZ/+ mice). In contrast, 44.5 ± 3.8% of SVZ LRCs were GLI1+ (205 LRCs total counted in n = 6 animals). As expected, the majority of LRCs that expressed Gli1 were localized in the ventral half of the SVZ (72.08 ± 5.8% ventrally compared with 11.09 ± 3.7% dorsally), whereas GLI2+ and GLI3+ LRCs were uniformly distributed along the dorsal–ventral SVZ axis (Fig. 1X). This result is consistent with previous reports of SHH pathway activity in both mature astrocytes (Garcia et al., 2010) and long-lived adult NSCs (Ahn and Joyner, 2005; Balordi and Fishell, 2007a). Interestingly, quantitative analysis of expression of the three Gli genes within the actively cycling KI67+ progenitor population in the SVZ, consisting primarily of TACs and some NBs (Tavazoie et al., 2008; Ponti et al., 2013), revealed a very limited overlap with all of the Gli-expressing populations (Fig. 1L–N,Y). Only 2.25 ± 0.8% of all KI67+ cells expressed Gli1 (4752 KI67+ cells counted, n = 3), and 5.88 ± 2.4% (2051 KI67+ cells counted, n = 2) and 3.75 ± 0.7% (4005 KI67+ cells counted, n = 3) were found to be positive for GLI2 and GLI3, respectively. Similarly, only a very small subset of the βGAL+ cells within the SVZ expressed KI67 (Gli1nlacZ: 2.92 ± 0.3% of 3841 βGAL+ cells; Gli2nlacZ: 3.83 ± 1.3% of 4702 βGAL+ cells; Gli3nlacZ: 3.25 ± 0.4% of 4628 βGAL+ cells). In addition, we found Gli1, Gli2, and Gli3 to be transcribed in sparse S100β+ ependymal cells (analyzed n = 3 animals of each genotype; Fig. 1O–Q). Strikingly, no Gli1, Gli2, or Gli3 transcription was detected in doublecortin (DCX+) or PSA-NCAM-expressing committed migratory NBs in the SVZ or within the RMS leading to the OB (n = 3–5 for each genotype; Fig. 1R–T; data not shown). NeuN+ OB interneurons also did not express any of the three Gli genes (Fig. 1U–W). In summary, our results show that within the neural stem and progenitor pool in the SVZ, more mature neural progenitors cannot respond to canonical SHH signaling, as both Gli2 and Gli3 are downregulated at the transcriptional level already at the TAC and NB stage.
A mGfap-Cre transgene allows conditional gene ablation in SVZ NSCs postnatally and bypasses the embryonic requirement for SHH signaling
The constitutive Cre lines previously used for conditional inactivation of Smo in SVZ NSCs initiate expression in the embryo. For instance, Smo ablation at midgestation using the Nestin-Cre or hGfap-Cre transgenic lines results in neurodevelopmental defects, such as microcephaly and enlargement of the ventricles, as well as increased SVZ apoptosis at P8 and lethality by P30 (Machold et al., 2003; Balordi and Fishell, 2007a; Han et al., 2008), indicating a requirement for SHH signaling during embryonic brain development. Approaches to inactivate Smo or Shh after birth have relied on tamoxifen-inducible CRE (Balordi and Fishell, 2007b; Ihrie et al., 2011), and while they have been informative, such studies have been compromised by the inefficient mosaic recombination that is achieved with CreER lines. For the ablation of Smo and the Gli genes, we therefore chose a transgenic line that uses regulatory sequences from the mouse Gfap gene to drive CRE expression (mGfap-Cre) as it has previously been shown to result in efficient recombination in NSCs of the adult SVZ (Imura et al., 2003; Garcia et al., 2004). To define the precise timing of CRE activity in the SVZ after birth, we crossed the mGfap-Cre line to a R26lacZ reporter (Soriano, 1999) and analyzed βGAL activity at several time points in the postnatal brain. At P0, the SVZ was largely βGAL-negative, whereas at P3 a large portion of the SVZ was reporter-positive (Fig. 2A,B). mGfap-Cre recombination in the SVZ appeared complete by P7 (Fig. 2C–E). Use of a tdTomato reporter (Madisen et al., 2010) and double labeling revealed that at P7 recombination had occurred in most GFAP-expressing cells within the neurogenic niche (Fig. 2F). Consistent with βGAL activity in the SVZ, no cells from the CRE-recombined lineage were present in the OB at P0 and began to accumulate at P3 such that by P14 it appeared that βGAL+ cells constituted a large portion of the OB granule cell layer (GCL) population (Fig. 2A'–E',I). Analysis of tdTomato-labeled cells in the ependymal layer (identified as containing S100β+ cells, a marker of a subset of the multiciliated ependymal cells lining the ventricular wall) revealed sporadic CRE activity in ependymal cells at P7 and more extensive labeling at P16 (Fig. 2G,H). In summary, the mGfap-Cre transgene can be used to achieve highly efficient ablation of conditional alleles in NSCs postnatally and without disrupting embryonic neurogenesis and the establishment of the stem and progenitor cell milieu.
Figure 2.

mGfap-Cre transgene induces recombination in NSCs postnatally. A–E', Based on XGAL staining in mGfap-Cre; Rosa26lacZ/+ mice, little CRE activity was detected in the SVZ at birth (A), with increasing recombination at P3 (B) and P5 (C), and recombination in the SVZ appeared complete by P7 (D). Labeled cells in the OB granule cell layer originating from the mGfap-Cre lineage (A'–E') began to appear at P3 (B') and by P14 the majority of cells in the OB GCL expressed LacZ (E'). F, Analysis of reporter-positive cells in the SVZ of mGfap-Cre; Rosa26TdTom/+ mice with cell-specific markers revealed that most GFAP+ cells in the SVZ had undergone recombination by P7. G, H, mGfap-Cre was found to induce recombination in both S100β+ and S100β− ependymal layer cells, but only in a subset of cells at P7, whereas at P16 most cells expressed tdTomato. Arrowheads point to mGfap-Cre-expressing ependymal cells that have undergone recombination. Asterisks point to S100β+ cells that have not recombined. SVZ schematic indicates location of close-up images in F–H. I, By P60, reporter expression can be detected in the majority of NeuN+ cells as well as in non-neuronal cells in the OB. Scale bar: (in F) F–I, 20 μm.
Inactivation of Smo in mGfap-Cre-expressing cells leads to loss of LRCs and a gradual reduction in SVZ neurogenesis
Previous studies in which SHH signaling was inactivated by mosaic ablation of Smo in ∼70% of SVZ stem and progenitor cells, as well as astrocytes, at P60 (Nestin-CreER; Smolox/lox mutants) demonstrated that pathway activity is required in most cells to maintain the number of proliferating and neurosphere-forming cells in the SVZ (Balordi and Fishell, 2007b). Furthermore, after 6 d of antimitotic insult with cytarabine, only wild-type quiescent NSCs, which had escaped recombination, exhibited a moderate regenerative ability (Balordi and Fishell, 2007b). It was not tested, however, whether the reduction in SVZ neurogenesis in Smo conditional mutants is specifically due to loss of slow-cycling LRCs in the adult SVZ.
For comparison to conditional ablation of Gli2 and Gli3, we inactivated Smo postnatally using the mGfap-Cre transgene and examined multiple parameters of in vivo neurogenesis. As predicted, mGfap-Cre/+; Smolox/− [mGfap-Smo conditional knock-out (CKO)] mutants did not exhibit gross defects in brain morphology and survived into adulthood (Garcia et al., 2010), unlike Nestin-Cre; Smo CKOs (Machold et al., 2003; Balordi and Fishell, 2007a). Furthermore, a reduction in SVZ neurogenesis was observed in mGfap-Smo CKOs compared with controls that increased as the animals aged. At P15, only 1 week after mGfap-Cre-mediated deletion of Smo in SVZ NSCs, KI67+ progenitor proliferation in mGfap-Smo CKOs appeared slightly but not significantly reduced (p = 0.3) compared with Smolox/+ littermate controls (n = 3 control and mutants), and there was no obvious change in the cross-sectional area of the RMS (Fig. 3A,B,I). Analysis at P40 of the BrdU label-retaining NSC population present in the SVZ at P15 also revealed no significant change in the number of LRCs (p = 0.2; n = 3 Smolox/+ littermate controls and 4 mutants; Fig. 3I). Unlike mutants at P15, mGfap-Smo CKOs at 3 months had both a significant 62.6% decrease in the number of KI67+ progenitors compared with Smolox/lox littermate controls (p = 0.0001), and a 52.9% decrease in BrdU LRCs (p = 0.036; Fig. 3J; n = 3 littermate controls and 3–4 mutants). We also observed a corresponding large decrease in the cross-sectional area of the RMS in Smo CKOs compared with littermate controls (Fig. 3C,D). Moreover, 1-year-old mGfap-Smo CKO animals appeared to have little if any neuronal production based on cresyl violet and DCX staining of the RMS (Fig. 3E–H). However, some KI67+ cells could be found in the SVZ of aged mGfap-Smo CKOs. Quantitative analysis of progenitor proliferation revealed that, although neurogenesis is reduced in control animals at 1 year of age compared with 3 months, SVZ proliferation was significantly lower (p = 0.017) in mGfap-Smo CKO mutants compared with controls (n = 3 control and mutants; Fig. 3K) and lower than in mutants at 3 months. These results demonstrate a requirement for SHH signaling in the long-term maintenance of SVZ NSC function.
Figure 3.
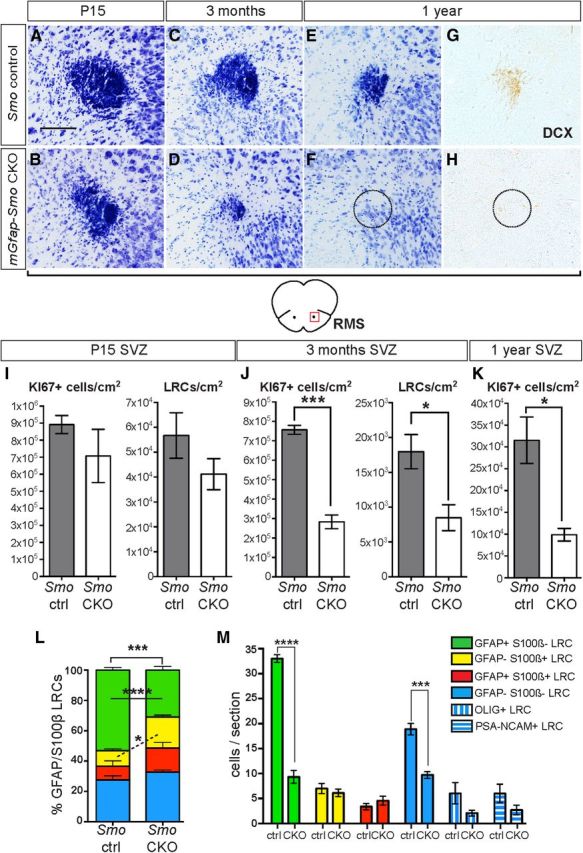
Inactivation of Smo in mGfap-Cre-expressing cells leads to a gradual reduction in neurogenesis. A, B, Cresyl violet staining of a cross section through the RMS of P15 mGfap-Smo CKOs shows no drastic change in the RMS size from that in control animals (Smolox/lox or Smolox/+). C–H, Cross-section of the RMS in adult mGfap-Smo CKOs and controls (Smolox/lox) reveals a progressive reduction of the RMS in controls but a substantial loss of DCX+ migrating progenitors by 1 year of age in mutant animals. Dashed line in G and H indicates expected location of RMS. Scale bar: (in A) A–H, 100 μm. I, J, Quantitative analysis of proliferating KI67+ progenitors and BrdU+ LRCs at P15 (I) and in 3-month-old mGfap-Smo CKOs and controls (Smo ctrl; J) demonstrates a dramatic depletion of the SVZ LRC pool and a concurrent reduction in KI67+ progenitor cell number at 3 months. K, By 1 year of age, the level of neurogenesis appears to decrease in control animals and SVZ proliferation in mGfap-Smo CKOs continues to be significantly lower than in controls and appears lower than at 3 months (*p < 0.05; ***p = 0.0001 unpaired t test; mean ± SEM). L, GFAP and S100β marker analysis of the LRCs in the SVZ of controls and mGfap-Smo CKOs reveals a significant decrease in GFAP+ S100β− LRCs (green) and an apparent complementary increase in the proportion of GFAP− S100β+ cells (yellow; *p < 0.05; ***p = 0.0001; ****p < 0.0001, 2-way ANOVA with Sidak's post hoc test; mean ± SEM). M, Normalizing the number of each subpopulation of LRCs to the number of cells per section reveals a specific loss of GFAP+ S100β− NSCs (green) but no increase in the number of GFAP− S100β+ glial/ependymal cells (yellow) or GFAP+ S100β+ astrocytes (red). As expected for cells that originate from NSCs, the number of GFAP− S100β− cells is significantly decreased (blue), and is reflected by a decrease in the number of OLIG+ LRCs (blue vertical stripes) and PSA-NCAM+ LRCs (blue horizontal stripes) in the SVZ of mGfap-Smo CKOs compared with controls (****p = 0.0001 and ***p = 0.0007, 2-way ANOVA with Sidak's post hoc test; mean ± SEM).
Consistent with the observed decrease in SVZ neurogenesis at 3 months, there was a considerable 68.2% decline in the number of newly generated OB granule cell interneurons that retain BrdU after a 3 week chase in adult mGfap-Smo CKO mice compared with controls (p = 0.0004; 46,142 ± 3349 cells/cm2 for n = 6 Smolox/lox littermate controls, and 14,653 ± 4340 cells/cm2 for n = 4 mutants). In addition, there was a 14.8% reduction in the area of the OB GCL in mGfap-Smo CKOs (p = 0.05; 1,760,263 ± 73,508 μm2 for n = 4 controls compared with 1,500,295 ± 78,145 μm2 for n = 5 mutants). Curiously, in contrast to the significant decrease in granule cell production in mGfap-Smo CKOs, we found no significant change in the total number of newly generated BrdU+ periglomerular cells (p = 0.3; 10,855 ± 1321 cells/cm2 for n = 3 Smolox/lox littermate controls, and 8504 ± 1520 cells/cm2 for n = 4 mutants), which originate from SVZ NSCs as well. Much like in adult Nestin-CreER; Smo CKOs (Balordi and Fishell, 2007b), our analysis of cleaved caspase-3 staining in the SVZ of P15 and P45 mGfap-Smo CKOs revealed no obvious increase in cell death compared with control animals (data not shown), indicating that cell apoptosis does not account for the loss in neuronal production. Together, our results demonstrate a progressive loss of neurogenesis in the SVZ of adult mGfap-Smo CKOs that stems from a major decrease in LRC number, revealing an in vivo requirement for active SHH signaling in preserving the long-term ability of NSCs to self-renew and generate TACs and their OB neuron progeny.
Slow-cycling LRCs do not differentiate into glia when Smo is ablated
Since an increase in cell death does not appear to account for the decrease in slow-cycling NSCs (LRCs) in adult mGfap-Smo CKOs, we considered whether respecification (i.e., cell differentiation) of the slow-cycling NSCs could account for the phenotype. To determine whether the remaining LRCs in the SVZ of mGfap-Smo CKOs had assumed a different molecular identity, we used a combination of two glial markers, GFAP (expressed in NSCs and mature astrocytes) and S100β (present in some ependymal cells and in mature astrocytes but not in NSCs; Raponi et al., 2007). Quantitative analysis of the four subpopulations of LRCs revealed a significant drop (p = 0.0001) in the proportion of GFAP+ S100β− LRCs (slow-cycling NSCs) in the mutants compared with controls (n = 3 mutants and 3 littermate controls; Fig. 3L). No significant change was observed in the proportions of LRCs that were GFAP− S100β− (mostly OLIG+ and PSA-NCAM+ cells) or GFAP+ S100β+ (astrocytes) in mGfap-Smo CKOs, but there was an increase (p = 0.039) in the proportion that were GFAP− S100β+ (astrocytes and ependymal cells; Fig. 3L). However, when the number of each LRC subpopulation was calculated per section for each animal, we found no difference in the number of GFAP− S100β+ LRCs between mutant and control animals (Fig. 3M). In contrast, the number of GFAP+ S100β− LRCs (slow-cycling NSCs) remained significantly decreased (p = 0.0001; Fig. 3M). In addition, the number of GFAP− S100β− LRCs, which are progeny of NSCs, was significantly decreased (p = 0.0005; Fig. 3M). Quantification of OLIG+ and PSA-NCAM+ LRCs per section supported a decrease in both populations. Hence it appears that the decrease in slow-cycling LRCs in mGfap-Smo CKOs is due to a reduction in the number of NSCs incorporating BrdU (i.e., undergoing cell division) rather than an increase in the rate of terminal differentiation (respecification of NSCs into mature glia).
Reduced neurogenesis in mGfap-Smo CKOs results from a cell-autonomous requirement for Smo in adult NSCs
In mGfap-Smo CKOs, the SHH receptor SMO is ablated in mature GFAP-expressing astrocytes, in some ependymal cells, and in GFAP+ NSCs. Since astrocytes could be an essential component of the SVZ stem cell niche, the reduced SVZ neurogenesis observed in mGfap-Smo CKOs might be partially due to a defect in the microenvironment. To test whether SHH signaling plays a cell-autonomous role in NSCs to maintain normal levels of neurogenesis, we used a new mosaic mutant approach termed “MASTR” (mosaic analysis with spatial and temporal control of recombination; Lao et al., 2012) to ablate Smo in scattered Nestin-expressing cells in 3-week-old animals. A tamoxifen-inducible Nestin-FlpoER transgene and a Rosa26MASTR(lox-STOP-lox-GFPcre) allele were combined with the Smo conditional allele to simultaneously ablate Smo (Smo−/lox) and mark mutant cells with GFP (Fig. 4A). Nestin-FlpoER/+; R26MASTR/+ animals with one conditional allele for Smo (Smo+/lox) were used as controls and Nestin-FlpoER/+; R26MASTR/+; Smo−/lox mice as mosaic mutants (Nestin-MASTR Smo ctrl and mosaics, respectively). The mice were administered three doses of tamoxifen starting at P21 and analyzed 5 weeks later. Marker expression analysis in controls confirmed that FLP (site-specific recombinase) is active in many S100β+ ependymal cells and in scattered GFAP+ S100β+ astrocytes and in GFAP+ S100β− NSCs and their KI67+ progeny (Fig. 4B,C). Quantitative analysis of the proportion of GFP+ cells that expressed GFAP and/or S100β showed a significant decrease in GFAP+ S100β− NSCs (p = 0.042) in Nestin-MASTR Smo mosaics compared with controls. As with LRCs in mGfap-Smo CKOs, the proportion of S100β+ GFAP− cells was correspondingly increased (p = 0.046; Fig. 4D). Furthermore, analysis of GFP+ cells that were KI67+ revealed a significant decrease in progenitors of NSCs (p = 0.045; n = 3 mutants and littermate controls; Fig. 4E). Thus the phenotype of Nestin-MASTR Smo mosaic mutant cells is similar to that in mGfap-Smo CKOs. Since most astrocytes in the SVZ microenvironment of Nestin-MASTR Smo mosaics remain wild type and there is little SHH signaling in the ependyma (see above), these results demonstrate a cell-autonomous requirement for Smo in maintaining NSCs.
Figure 4.

Mosaic removal of Smo postnatally reveals a cell-autonomous requirement for SHH signaling in SVZ NSCs. A, Schematic illustrating the genetic approach taken to mosaicly ablate Smo only in a subpopulation of Nestin-expressing cells. Nestin-FlpoER/+, R26MASTR/+; Smolox/− mosaic mutant (Nestin-MASTR Smo mosaics) or Nestin-FlpoER/+; R26MASTR/+; Smolox/+ control (Nestin-MASTR Smo ctrl) animals were given tamoxifen at P21–P26, which induces FLP to translocate into the nucleus and delete the STOP cassette in the R26MASTR allele, allowing expression of GFPcre, which induces recombination of the loxP sites in Smo, producing a null allele. Importantly, all Smo mutant cells and their progeny are permanently marked with GFP. B, Five weeks after tamoxifen administration, control animals express GFP in many SVZ ependymal cells (S100β+ GFAP−, arrowhead), some astrocytes (S100β+ GFAP+, asterisk) and NSCs (S100β− GFAP+, arrow). C, Some GFP+ NSCs have also given rise to KI67+ GFP+ progenitors. Scale bar: (in B) B, C, 50 μm. D, Quantitative analysis of the proportion of GFAP-expressing and/or S100β-expressing cells reveals that the proportion of GFP+ cells that are GFAP+ S100β− NSCs (green) is greatly reduced in Nestin-MASTR Smo mosaics compared with controls, whereas the proportion of GFP+ cells that are GFAP− S100β+ astrocytes and ependymal cells (yellow) is increased. No significant change was observed in the proportion of GFP+ cells that are GFAP+ S100β+ astrocytes (red) or GFAP− S100β− cells (blue; a mixture of S100β− ependymal cells and NSC progenitors; *p < 0.05 2-way ANOVA with Sidak's post hoc test; mean ± SEM). E, Analysis of the proportion of GFP+ cells that are KI67+ progenitors reveals a significant reduction in production of TACs in Nestin-MASTR Smo CKOs (*p < 0.05 unpaired t test; mean ± SEM).
Either GLI2 or GLI3 activity is sufficient to maintain adult SVZ neurogenesis
Both Gli2 and Gli3, unlike Gli1, are indispensible for normal development and their ablation results in embryonic lethality with major brain defects (Fuccillo et al., 2006). The contribution of each of the GLIs to SHH-regulated processes in neurodevelopment, however, varies along the rostral–caudal axis of the CNS. Specifically, GLI2A function is absolutely necessary in the spinal cord for the establishment of the floor plate and a subset of ventral interneuron types (Matise et al., 1998; Bai et al., 2002; Hui and Angers, 2011), whereas GLI3R plays only a minor role in patterning the intermediate neural tube (Persson et al., 2002; Bai et al., 2004). GLI2R activity is not required for CNS development (Bai and Joyner, 2001). In contrast, attenuation of GLI3R levels is crucial for telencephalon patterning during development, whereas GLI2A is largely expendable (Park et al., 2000; Rallu et al., 2002; Fuccillo et al., 2004). Unlike during early development, the specific roles of GLI2 and GLI3 as transcriptional activators/repressors in adult neurogenesis have not been addressed.
To determine whether GLI2 or GLI3 is the primary mediator of SHH signaling in GFAP+ NSCs postnatally, we analyzed the state of neuron production in adult mice (2–4-month-old) lacking either Gli2 or Gli3 in the SVZ. Similar to mGfap-Smo CKOs, neither mGfap-Cre/+; Gli2lox/− (mGfap-Gli2 CKO), nor mGfap-Cre/+; Gli3lox/− (mGfap-Gli3 CKO) mutants exhibited gross defects in brain morphology. In contrast to mGfap-Smo CKO mutants, however, quantitative analysis of the number of KI67+ progenitors in the SVZ revealed no significant change in proliferation when Gli2 (p = 0.6) or Gli3 (p = 0.7) was inactivated using mGfap-Cre (n = 4 Gli2lox/lox littermate controls and 4 mGfap-Gli2 CKOs; n = 3 Gli3lox/lox littermate controls and 3 mGfap-Gli3 CKOs; Fig. 5A,B). Consistent with these results, quantitative analysis of newly generated BrdU-labeled interneurons in the OB of mGfap-Gli2 or mGfap-Gli3 CKOs revealed no significant deviation from littermate controls (57,261 ± 7567 cells/cm2 for n = 3 Gli2 controls compared with 48,988 ± 2886 BrdU+ cells/cm2 for n = 3 mGfap-Gli2 CKOs, p = 0.4; 45,365 ± 5261 cells/cm2 for n = 3 Gli3 controls compared with 49,882 ± 12,819 BrdU+ cells/cm2 for n = 3 mGfap-Gli3 CKOs, p = 0.7). The lack of a clear phenotype with respect to TAC and NB progenitor proliferation and formation of OB interneurons after removal of Gli2 or Gli3 in SVZ NSCs, compared with the defects seen in mGfap-Smo CKOs, could indicate an overlap of function between GLI2 and GLI3 in adult SVZ neurogenesis. In support of the notion of partial redundancy between GLI2 and GLI3 in transducing high levels of SHH, we found that Gli1 expression in the ventral SVZ was obviously reduced in mGfap-Gli2 CKOs and only slightly reduced in mGfap-Gli3 CKOs, whereas Gli1 expression in the SVZ of mGfap-Smo CKOs was completely absent (Fig. 5C–F).
Figure 5.
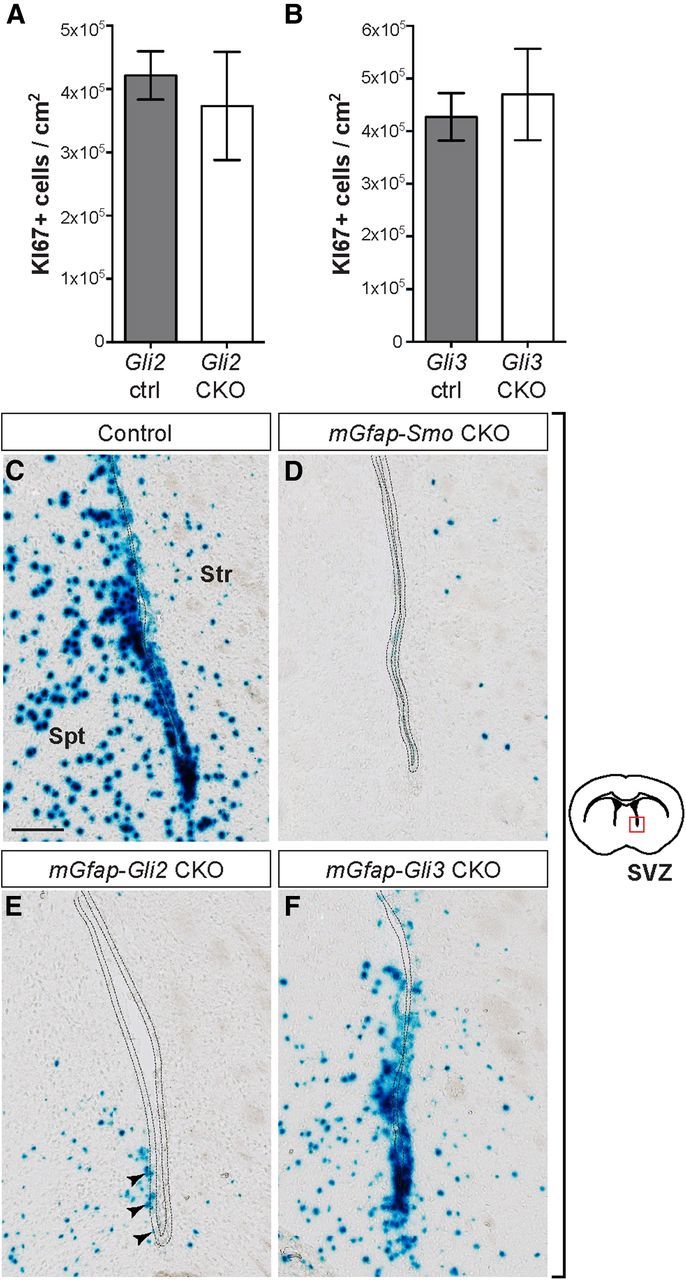
Ablation of Gli2 or Gli3 in NSCs has no significant effect on neurogenesis and does not abolish Gli1 expression in the adult SVZ. A, B, Quantitative analysis of progenitor proliferation in the adult SVZ of mGfap-Gli2 CKOs (A) and mGfap-Gli3 CKOs (B), compared with controls (Gli2 ctrl and Gli3 ctrl, respectively), indicates no significant change in neurogenesis (unpaired t test, mean ± SEM). C–F, XGAL staining for βGAL activity in the SVZ of adult mGfap-Smo CKOs (mGfap-Cre; Smolox/−; Gli1nlacZ/+) shows complete loss of Gli1 expression in the mutant compared with a Smolox/+; Gli1nlacZ/+ control animal (C, D). SVZ cells (dotted line) in mGfap-Gli2 and mGfap-Gli3 CKOs, which carry a Gli1nlacZ/+ reporter allele (mGfap-Cre; Gli2lox/−; Gli1nlacZ/+ and mGfap-Cre; Gli3lox/−; Gli1nlacZ/+, respectively), express a low level of Gli1 in the ventral SVZ (E, F). Scale bar: (in C) C–F, 100 μm. Str, Striatum; spt, septum.
Unattenuated GLIR suppresses adult NSC function
Since NSCs in adult mGfap-Smo CKOs lack all GLIA function and have an increase in GLIR, it is possible that either the increase in GLIR or complete loss of GLIA function causes the deficit in SVZ neurogenesis. We first tested whether the decrease in neuron production in the absence of SMO is specifically due to unattenuated levels of GLI3R by analyzing adult (2–4-month-old) mGfap-Cre/+; Gli3lox/−; Smolox/− (mGfap-Gli3;Smo CKO) double mutants. Strikingly, removal of Gli3 in mGfap-Smo CKOs resulted in only an ∼24.7% decrease in the number of KI67+ progenitor cells in the SVZ of mGfap-Gli3;Smo CKOs, which was not significant (p = 0.25; n = 3 Gli3lox/+;Smolox/+ littermate controls and 4 mutants), compared with the significant 62.6% decline observed in mGfap-Smo CKOs. Furthermore, there was no significant decrease in the number of slow-cycling NSCs (LRCs; p = 0.1; n = 3 Gli3lox/+;Smolox/+ littermate controls and 3 mutants; Fig. 6A,B) or newly generated OB interneurons (p = 0.86, 64,572 BrdU+ cells/cm2 in controls vs 62,456 cells/cm2 in mutants) and periglomerular cells (p = 0.37, 11,624 BrdU+ cells/cm2 in controls vs 13,734 cells/cm2 in mutants). Consistent with this, we also observed a clear phenotypic rescue at the level of the RMS in mGfap-Gli3;Smo CKOs (Fig. 6C–K). These results demonstrate that removing GLI3R in mGfap-Gli3;Smo CKOs results in a substantial rescue of neurogenesis.
Figure 6.

Unattenuated GLI2R and GLI3R suppress adult SVZ neurogenesis. A, B, Quantitative analysis of the number of proliferating KI67+ progenitors, as well as LRCs in the SVZ of mGfap-Gli3;Smo CKOs show neurogenesis is no longer significantly reduced compared with controls (Gli3;Smo ctrl), indicating a rescue compared with mGfap-Smo CKOs (Fig. 3H). C–K, Cross sections of the RMS in control, mGfap-Smo CKOs, and mGfap-Gli3;Smo CKOs showing the specific increase in the cresyl violet-labeled, KI67+ and DCX+ migrating NB progenitors after Gli3 ablation in mGfap-Smo CKOs. Scale bar: (in C) C–K, 100 μm. L, No significant effect was observed on SVZ progenitor proliferation after ablation of all GLIA and GLIR function in mGfap-Gli2;Gli3 CKOs. M, Expression of a constitutive GLI3R isoform in the absence of GLIA in mGfap-Gli2;Gli3Δ699 CKOs causes a mild decrease in the number of SVZ progenitors compared with littermate controls (unpaired t test; mean ± SEM).
To determine whether the small reduction in SVZ proliferation in mGfap-Gli3;Smo CKOs is due to the remaining GLI2R in the mutants and thus can be rescued by removing Gli2, we analyzed neuron production in adult mGfap-Cre/+; Gli2lox/−; Gli3lox/− (mGfap-Gli2;Gli3 CKO) double mutants that lack all GLIR in addition to GLIA activity. Interestingly, SVZ progenitor proliferation in mGfap-Gli2;Gli3 CKOs (n = 3 controls and 3 mutants) was reduced by only ∼12.9% (Fig. 6L). We observed no significant decrease in the number of newly generated OB interneurons (p = 0.098, 87,695 BrdU+ cells/cm2 in controls vs 73,712 cells/cm2 in mutants) or periglomerular cells (p = 0.61, 24,269 cells/cm2 in controls vs 22,943 cells/cm2 in mutants) in mGfap-Gli2;Gli3 CKOs. Our double-mutant results show that the primary function of SHH signaling is to attenuate GLIR levels (GLI2R and GLI3R) in adult NSCs, allowing other transcription factors to induce neurogenesis, and that augmenting GLIA plays only a minor role in boosting SVZ NSC function.
To further examine the specific contribution of increased GLI3R levels to the deterioration of neurogenesis in the SVZ of adult mGfap-Smo CKOs, we generated mGfap-Cre/+; Gli2lox/−; Gli3lox/Δ699 (mGfap-Gli2;Gli3Δ699 CKO) mutants, which carry no functional alleles of Gli2 but have one copy of Gli3 expressing a low level of a constitutive GLI3R protein in GFAP+ cells (Böse et al., 2002). Quantitative analysis of the KI67+ progenitor population in the SVZ showed a 37.5% decrease in KI67+ progenitors when compared with Gli2lox/+; Gli3lox/+ littermate controls (n = 4 controls and mutants), although it was not statistically significant (p = 0.27; Fig. 6M). The less prominent neurogenesis phenotype in mGfap-Gli2;Gli3Δ699 CKOs compared with mGfap-Smo CKOs is likely due to a lower level of GLI3R produced by the single Gli3Δ699 allele and no GLI2R.
GLI3R upregulation compromises both dorsal and ventral SVZ proliferation
High-level SHH signaling (GLI1+ cells) is restricted to the ventral portion of the SVZ. Even so, since GLI3R rather than GLIA is critical to SHH signaling and Gli3 is expressed throughout the SVZ, we investigated whether SVZ neurogenesis is equally compromised in the dorsal and ventral SVZ when SHH signaling is obliterated. We first determined the extent of decrease in proliferating KI67+ progenitors in the dorsal and ventral halves of the SVZ in mGfap-Smo CKOs and littermate controls and observed a dramatic decrease in both the dorsal (p = 0.0001) and ventral (p=0.0001) regions of the mutants (Fig. 7A,B). Surprisingly, in the anterior SVZ of mGfap-Smo CKOs, progenitor proliferation was more strongly reduced dorsally (by 73.9%) than in the ventral SVZ (by 50.4%), whereas in control animals the distribution of KI67+ progenitors was fairly even between the dorsal and ventral SVZ (two-way ANOVA interaction p = 0.035; n = 3 Smolox/lox littermate controls and 4 mGfap-Smo CKOs; Fig. 7B). The greater defect in cell proliferation in mGfap-Smo CKOs dorsally could be a result of the higher level of Gli3 transcription dorsally (as seen from the X-gal staining in Gli3nlacZ in Fig. 1E–E”) resulting in more GLI3R in the dorsal SVZ, where GLI3R inhibits TAC production to a greater extent than in ventral NSCs.
Figure 7.
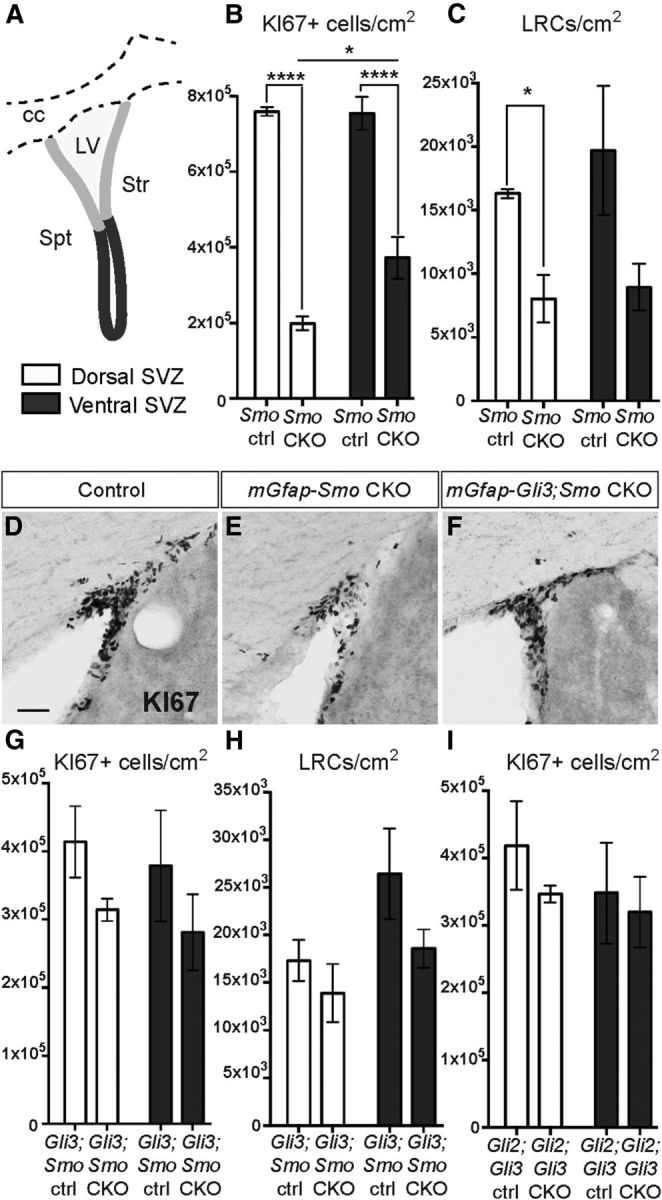
GLIR upregulation compromises proliferation throughout the SVZ. A, Schematic diagram showing the dorsal (light gray) and ventral (dark gray) SVZ regions that were included in the analysis. Str, Striatum; Spt, septum; cc, corpus callosum; LV, lateral ventricle. B, C, Quantitative analysis of the number of KI67+ progenitors and LRCs per square centimeter in the dorsal (white bars) or ventral (gray bars) SVZ shows a significant decrease in the density of KI67+ progenitors in both regions of mGfap-Smo CKOs compared with Smolox/lox controls (Smo ctrl) that is more pronounced dorsally (B; n = 3 controls and 4 mutants; *p = 0.016, ****p < 0.0001; 2-way ANOVA with Sidak's post hoc test; mean ± SEM). A decrease in the density of the LRCs also was observed in the dorsal and ventral SVZ of mGfap-Smo CKOs compared with controls with no obvious change in the distribution between dorsal and ventral (C; n = 3 controls and 3 mutants). D–F, Dorsolateral SVZ sections stained for KI67 show a decrease in the number of KI67+ cells in the dorsal SVZ of mGfap-Smo CKOs when compared with controls (D, E), and a rescue of proliferation in the dorsal SVZ when Gli3 is ablated in mGfap-Smo CKOs (F). Scale bar: (in D) D–F, 50 μm. G, H, Quantitative analysis shows the recovery in the density of KI67+ progenitors and LRCs in mGfap-Gli3;Smo CKOs compared with controls (Gli3;Smo ctrl); as well as a similar distribution of KI67+ cells in the dorsal and ventral SVZ of mGfap-Gli2;Gli3 CKOs compared with controls (I) (Gli2;Gli3 ctrl; 2-way ANOVA with Sidak's post hoc test; mean ± SEM).
One possible explanation for a greater reduction in proliferating cells in the dorsal SVZ of mutants is that the progenitor pool in the dorsal region includes migrating NBs derived from more posterior and ventral regions of the SVZ. We therefore determined whether there is a preferential decrease in LRCs in the dorsal SVZ of mGfap-Smo CKOs. However, no significant difference was observed between the number of LRCs in the dorsal and ventral SVZ of mutants or controls, although the total number of LRCs was lower in the mutants (two-way ANOVA interaction p = 0.68; n = 3 Smolox/lox littermate controls and 3 mGfap-Smo CKOs; Fig. 7C). Interestingly, removal of Gli3 in GFAP-expressing cells mutant for Smo restored the normal dorsoventral distribution of KI67+ progenitors (two-way ANOVA interaction p = 0.97; n = 4 Gli3lox/+; Smolox/+ littermate controls and 4 mGfap-Gli3;Smo CKOs; Fig. 7D–G), and had no effect on the LRC distribution (two-way ANOVA interaction p = 0.53; n = 4 Gli3lox/+; Smolox/+ littermate controls and 5 mutants; Fig. 7H). In addition, we found no change in the dorsal–ventral distribution of proliferating cells in mGfap-Gli2;Gli3 CKOs (two-way ANOVA interaction p = 0.70; n = 3 littermate controls and 4 CKOs). These results demonstrate that SHH signaling through GLI3R is required for neurogenesis in both the ventral and the dorsal SVZ.
GLIR and not GLIA is required to maintain the normal proportion of deep and superficial OB interneurons
We were next interested in whether GLIR and/or GLIA play a role in the production of OB interneurons. Fate-mapping studies in adult mice have shown that Gli1-expressing NSCs preferentially generate OB interneurons that localize to the deep granule cell layer of the OB adjacent to the RMS (Merkle et al., 2007; Ihrie et al., 2011). Indeed, using genetic inducible fate mapping (GIFM), we found that 3 weeks after administering tamoxifen to Gli1CreER/+; R26yfp/yfp adult mice (Gli1CreER GIFM), 49 ± 1.73% of fate-mapped NeuN+ interneurons localized to the deep OB layer, whereas only 34 ± 1.0% and 17 ± 2.0% occupied the intermediate and superficial layers, respectively (Fig. 8A–C). In contrast, the proportions of OB neurons labeled with BrdU (after a 3 week chase) in the deep, intermediate, and superficial layer were ∼37, 38, and 25% respectively (Fig. 8D–G). Thus, Gli1-expressing cells preferentially, but not exclusively, give rise to deep OB neurons. In addition, it was previously shown that ectopic upregulation of SHH signaling in GFAP+ cells in the adult dorsal SVZ results in a preferential increase in deep-layer OB interneuron generation, whereas conditional ablation of Shh at P60 leads to a decrease in the proportion of deep-layer interneurons (Ihrie et al., 2011).
Figure 8.
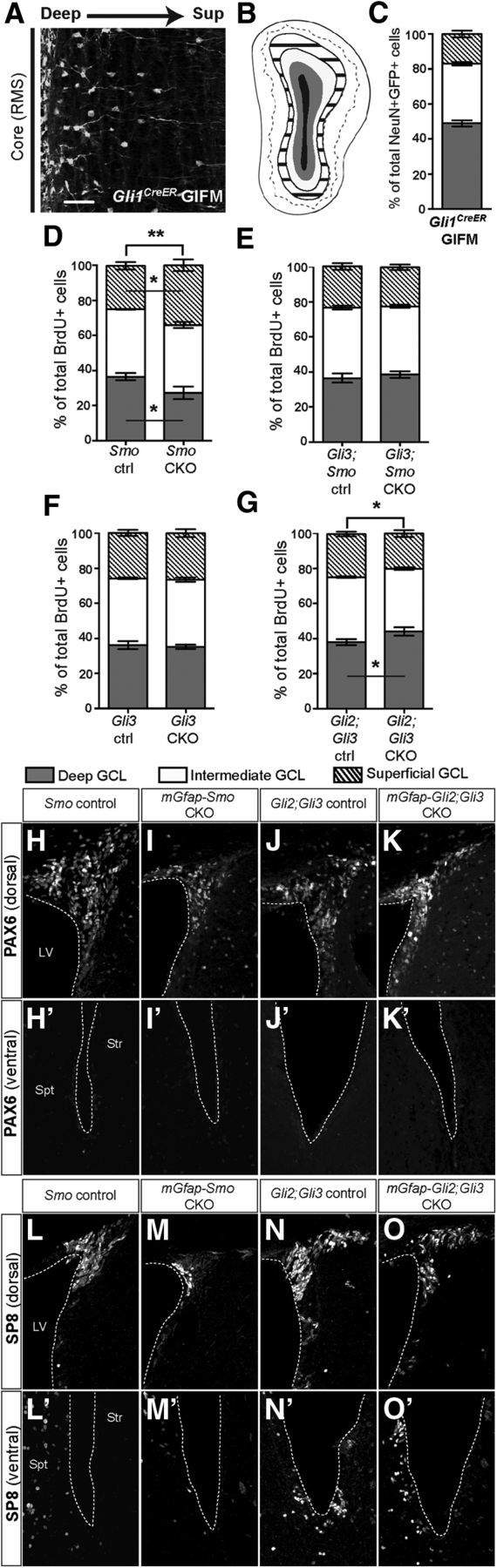
SHH is required postnatally to regulate the normal proportions of interneurons that settle in the deep to superficial axis of the OB granule layer. A, Coronal section through the OB of a Gli1CreER/+; R26yfp/yfp mouse 30 d after tamoxifen treatment (Gli1CreER GIFM) at 3 months shows that fate-mapped interneuron progeny localize preferentially to the region closest to the RMS (core). Scale bar, 50 μm. B, Schematic of an OB granule cell layer subdivided into deep (gray area), intermediate (white area), and superficial (striped area) domains based on lines drawn to demarcate the borders between domains 1/3 and 2/3 of the radius between the outline of the RMS (core; in black) and the outer rim of the GCL. C, Quantitative representation of the distribution of interneurons derived from Gli1-expressing NSCs shows that the majority of Gli1CreER fate-mapped mature (NeuN+) interneurons localize to the deep and intermediate GCL domains. D–G, Quantitation of the distribution of newly generated (BrdU+) interneurons within the GCL shows that in mGfap-Smo CKOs (D) there is a shift toward generating more superficial layer interneurons and a reduction in deep OB interneurons (2-way ANOVA with Sidak's post hoc test; ANOVA interaction p value **p = 0.0053; mean ± SEM), whereas in mGfap-Gli3;Smo CKOs (E) the distribution is not altered compared with controls (2-way ANOVA with Sidak's post hoc test; mean ± SEM). Loss of Gli3 alone (F) is not sufficient to induce a change in the OB interneuron distribution compared with controls (2-way ANOVA with Sidak's post hoc test; mean ± SEM). In contrast, conditional ablation of both Gli2 and Gli3 in mGfap-Gli2;Gli3 CKOs (G) results in a slight increase in the frequency of generation of deep-layer OB interneurons compared with controls (ANOVA interaction p value *p = 0.02; mean ± SEM). H–K', Staining for the dorsal molecular marker PAX6 in mGfap-Smo CKOs (H, H', I, I') and mGfap-Gli2;Gli3 CKOs (J, J', K, K') revealed no change in PAX6 expression and hence in progenitor respecification along the dorsal–ventral axis of the SVZ. Analysis for the putative repatterning of the SVZ in the same two mutants using the dorsally expressed marker SP8 yielded similar results (L–O'). A minimum of n = 3 littermate controls and 3 mutants were used for the analysis of each experimental group.
We first tested whether Smo ablation using mGfap-Cre results in a similar preferential loss of deep-layer OB interneurons. Indeed, in addition to the substantial overall reduction in the total number of newly generated interneurons in mGfap-Smo CKO mutants, the proportion of cells labeled with BrdU (after a 3 week chase) in each of the three layers was altered (two-way ANOVA interaction p = 0.0053), such that significantly more cells occupied the superficial OB layer (p = 0.046) and fewer cells occupied the deep layer (p = 0.046; 36.50% deep, 38.50% intermediate, and 24.75% superficial interneurons in n = 4 littermate controls, compared with 27.25% deep, 38.75% intermediate, and 34.75% superficial interneurons in n = 4 mGfap-Smo CKOs; Fig. 8D). Notably, attenuating GLI3R levels in mGfap-Gli3;Smo double mutants largely restored the balance in deep versus superficial OB interneuron production (p = 0.56; n = 4 Gli3lox/+;Smolox/+ littermate control animals and 4 mGfap-Gli3;Smo CKOs; Fig. 8E). In addition, removal of Gli3 alone did not lead to a preferential increase in the number of deep-layer OB interneurons in mGfap-Gli3 CKOs (p = 0.89) compared with littermate Gli3lox/lox controls (Fig. 8F), demonstrating that downregulation of GLI3R alone is not sufficient to stimulate production of deep-layer OB interneurons in vivo. Finally, our analysis of the distribution of newly generated OB interneurons in mGfap-Gli2;Gli3 double CKOs lacking all GLIA and GLIR activity strikingly revealed a significant increase in the production of deep-layer interneurons (p = 0.02), seemingly at the expense of superficial ones (two-way ANOVA interaction p = 0.02; 37.97% deep, 37.13% intermediate, and 24.57% superficial interneurons in n = 3 littermate controls, compared with 44.00% deep, 35.88% intermediate, and 20.08% superficial interneurons in n = 4 mGfap-Gli2;Gli3 CKOs; Fig. 8G). This result points to a role for GLI2R in inhibiting deep-layer OB interneuron production in mGfap-Smo and mGfap-Gli3;Smo CKOs. In summary, our results reveal that proper regulation of both GLI2R and GLI3R levels, rather than GLIA activity in the SVZ or astrocytes in the RMS, is a mechanism by which SHH signaling might influence the proportions of deep and superficial OB interneurons produced.
Similarly, to determine whether there was a decrease in production of periglomerular cells expressing calbindin (CalB+), another OB cell type previously shown to preferentially derive from the ventral SVZ and depend on SHH signaling (Merkle et al., 2007; Ihrie et al., 2011), we compared the proportion of CalB+ BrdU-labeled newly generated cells within the glomeruli of mGfap-Smo CKOs and controls. Surprisingly, despite the fact that the overall number of newly generated periglomerular cells in the mutants was unaffected, we observed a significant reduction in newly generated CalB+ cells 3 weeks after BrdU labeling of progenitors (p = 0.039; 11.65 ± 1.76% for n = 3 controls compared with 5.14 ± 0.53% for n = 3 mutants). One possible explanation for this result is a shift toward the production of other periglomerular subtypes derived from the SVZ. We tested whether the complete lack of GLIR in mGfap-Gli2;Gli3 CKOs affected production of CalB+ periglomerular neurons in an opposite way to that of mGfap-Smo CKOs but found no significant change compared with controls (p = 0.76; 13.03 ± 0.24% for n = 3 controls compared with 13.84 ± 2.47% for n = 3 mutants). Thus, unlike granule cells, a complete lack of GLI function does not alter production of CalB+ periglomerular neurons, at least when assayed 3 weeks after BrdU labeling. It is nevertheless still possible that other populations of periglomerular neurons are sensitive to a reduction in GLIR levels.
One reason for the bias toward CalB+ periglomerular and superficial OB interneuron production in mGfap-Smo CKOs could be a change in dorsal–ventral “patterning” of the SVZ due to increased GLIR levels. To test for a “dorsalization” (i.e., a gain of dorsally restricted gene expression in the ventral SVZ), we analyzed the expression of two different dorsal SVZ molecular markers, PAX6 and SP8, in mGfap-Smo CKOs and controls (Fig. 8H–I',L–M'). However, we found no significant change in the pattern of PAX6 or SP8 expression between mutant and control animals. Similarly, we saw no indication of “ventralization” (loss of dorsal markers dorsally) in the SVZ in mGfap-Gli2;Gli3 CKOs using PAX6 and SP8 (Fig. 8J–K',N–O'). These results argue against a direct respecification of the progenitors in the SVZ. Alternative explanations suggest that either (1) the dorsal genes we analyzed are not specification genes, or (2) GLIR levels in the ventral SVZ of mGfap-Smo CKOs are not high enough to induce a change in PAX6/SP8 expression.
GLI3R level modulation regulates mature astrocyte function
Given our discovery that an increase in GLIR levels rather than loss of GLIA is responsible for two distinct functions of SVZ NSCs regulated by SHH (maintenance of LRCs and OB interneuron production), we asked whether a similar rule applies to forebrain astrocyte function. We have previously shown that conditional ablation of Smo in postnatal astrocytes using mGfap-Cre results in a partial reactive astrogliosis phenotype in the adult cortex (Garcia et al., 2010). mGfap-Smo CKOs display a remarkable increase in the number of GFAP-expressing cortical astrocytes, which at the cellular level develop enlarged cell bodies and thicker GFAP-stained processes. To determine whether this phenotype in astrocytes is caused by an increase in GLI3R levels, we analyzed expression of GFAP in the adult cortex of mGfap-Gli3;Smo double mutants compared with mGfap-Smo CKOs. Indeed, in addition to a clear rescue of astrocyte hypertrophy, GFAP expression was also noticeably downregulated in the cortex of mGfap-Gli3;Smo CKOs (n = 3 mGfap-Smo CKOs compared with n = 5 mGfap-Gli3;Smo CKOs; Fig. 9A–C). Analysis of cortical astrocytes in mGfap-Gli2;Gli3 double mutants (n = 7; Fig. 9F) and mGfap-Gli3 (n = 4) or mGfap-Gli2 (n = 6) single CKOs (Fig. 9D; data not shown) also revealed no obvious change in the number or morphology of GFAP+ astrocytes. Moreover, expression of GLI3R in the absence of the normal GLI2 or GLI3 proteins in the cortex of mGfap-Gli2;Gli3Δ699 CKOs (n = 4) led to an increase in the number of GFAP-expressing astrocytes and partial astrocyte hypertrophy in mutant cortices (Fig. 9E), similar to that in mGfap-Smo CKOs. Together, our results reveal that as in SVZ NSCs, GLI3R plays a dominant role in the transduction of SHH signaling in mature astrocytes in the uninjured forebrain (Fig. 10).
Figure 9.

Unattenuated GLI3R levels alter astrocyte function. A–C, IHC staining of sections of adult cortex for GFAP shows that mGfap-Smo CKO astrocytes exhibit a partial reactive astrogliosis phenotype (A, B), whereas removal of Gli3 in mGfap-Smo CKOs rescues astrocyte morphology and drastically reduces the population of GFAP-overexpressing glia in the cortex (C). D–F, No obvious change in the morphology of GFAP-expressing astrocytes is observed in mGfap-Gli3 CKOs (D) or in mGfap-Gli2;Gli3 double CKOs (F), whereas expression of GLI3R in the absence of GLIA (mGfap-Gli2;Gli3Δ699 CKOs) recapitulates the astrocyte hypertrophy and GFAP overexpression observed in mGfap-Smo CKOs (E). A–F, Insets, Example of cortical astrocyte morphology in the various mutants and control. Scale bar: (in A) A–F, 50 μm.
Figure 10.

SVZ proliferation, OB interneuron specification, and astrocyte morphology require precise titration of GLIR levels by SHH. Summary of the three distinct SHH-dependent phenotypes observed in mouse mutants lacking Smo and/or Gli2 and Gli3 or expressing GLI3R (Gli3Δ699) in GFAP-expressing cells in the postnatal forebrain. Phenotypic changes that are statistically significant based on analyzing 3–4 animals are indicated by an asterisk. The GLI proteins that remain in the mutants are indicated above the phenotypes, demonstrating the degree to which regulation of the correct dose of GLI3R and GLI2R is necessary for each developmental process, and the very limited role of GLIA. Mice lacking Gli2 or Gli3 alone have no obvious defects in the adult forebrain (not indicated). In mice lacking only Smo, no GLI2A or GLI3A is produced and instead the levels of GLI3R and GLI2R are increased. This results in a loss of slow-cycling NSCs in the SVZ, which leads to a major decease in the percentage of proliferating cells in the SVZ (SVZ proliferation), a decrease in the number of newly born OB neurons (not indicated), an alteration in the proportion of deep versus superficial OB interneurons produced (OB %deep : %superficial; control percentage shown in brackets), and an increase in the number of GFAP-expressing astrocytes and morphological changes indicating partial gliosis (↑GFAP in astrocytes). SVZ neurogenesis is decreased to a lesser extent in mice lacking Gli2 and only expressing a low level of a GLI3R (mGfap-Gli2;Gli3Δ699 CKO), whereas the defect in astrocyte function is similar to that in mGfap-Smo CKOs. In mice lacking Smo and Gli3, and in which only GLI2R is expressed (mGfap-Smo;Gli3 CKO), there is only a mild reduction in neurogenesis, a slight increase in astrocyte gliosis, and OB interneuron production and the proportion of deep and superficial neurons is restored to normal levels. In contrast, in mice lacking Gli2 and Gli3 that produce no GLI 2/3A or GLI2/3R proteins (mGfap-Gli2;Gli3 CKO), neurogenesis is almost normal and astrocyte function appears normal, but the ratio of deep/superficial OB interneurons is changed in the opposite direction to that in Smo mutants.
Discussion
In this study, we addressed the specific roles of the zinc-finger transcription factors GLI2 and GLI3 in the postnatal SVZ and cortical astrocytes, and determined the extent to which the repressor or activator function of each GLI contributes to maintaining three processes. In terms of SVZ neurogenesis, our comparative analysis of mGfap-Gli2 and mGfap-Gli3 conditional mutants compared with mGfap-Smo CKOs revealed that either Gli2 or Gli3 alone is sufficient in GFAP-expressing cells to maintain a normal level of SVZ proliferation and OB neuron production. One possible explanation for these results was redundancy between GLI2 and GLI3 activators in transducing SHH signaling, as there is evidence for GLI3A having a role along with GLI2A in the embryo (Bai et al., 2004; Bowers et al., 2012). However, our double-mutant studies argue against the GLIA side of the pathway playing a major role in SVZ neurogenesis. First, we found only a slight decrease in neurogenesis in mGfap-Gli2;Gli3 CKO animals lacking both Gli genes. Second, we found that mGfap-Smo CKOs that also lack Gli3 have a major rescue of neurogenesis compared with mGfap-Smo CKOs, indicating that an increase in GLI3R contributes to the neurogenesis defect in mGfap-Smo CKOs. Third, mGfap-Gli2;Gli3 CKOs lacking all GLIR activity have a more normal level of SVZ proliferation than mGfap-Gli3;Smo CKOs in which GLI2R remains functional, indicating GLI2R also contributes to the mGfap-Smo CKO phenotype. A similar minor role for GLI2R has been observed in the developing limb (Bowers et al., 2012). Finally, expression of a constitutive repressor form of GLI3 in GFAP-expressing cells leads to a decrease in neurogenesis. Together, our results demonstrate only a minor requirement for GLIA-mediated SHH signaling in promoting SVZ neurogenesis, similar to the embryonic forebrain (Park et al., 2000; Rallu et al., 2002; Fuccillo et al., 2004), and have uncovered that GLI2R augments a major role of GLI3R in reducing neurogenesis in the postnatal SVZ.
We found that Gli2 and Gli3 expression in the adult SVZ, and with this the ability to activate canonical SHH signaling, is limited primarily to mature astrocytes and astrocyte-like slow-cycling or activated NSCs. Our analysis of BrdU label-retaining NSCs in the SVZ revealed that the majority of LRCs in both the dorsal and ventral SVZ express Gli2 and Gli3. Furthermore, the three Glis are expressed only in rare KI67+ progenitors, arguing against activation of SHH signaling in TACs and NBs. Since Ascl1 expression, traditionally viewed as a demarcation of TACs, has been observed in Gli1-expressing cells in the adult SVZ (Palma et al., 2005; Balordi and Fishell, 2007a), our results indicate that it is only the subset of long-lived ASCL1+ adult NSCs (Kim et al., 2011) that express Gli1. Our results of Gli2 and Gli3 expression are consistent with the previously reported lack of Gli1 expression in SVZ NBs (Palma et al., 2005; Balordi and Fishell, 2007a). The major loss of SVZ NSCs in mGfap-Smo CKOs and Nestin-MASTR Smo mosaics further demonstrates a primary role for SHH signaling postnatally specifically in slow-cycling NSCs. In contrast, a role for SHH in the regulation of adult NB migration toward the OB has been suggested (Angot et al., 2008), raising the possibility of an additional chemoattractive role for SHH. Although in vitro NB migration was shown to be SMO-dependent, as receptor antagonists were able to disrupt migration, it is highly likely that NB migration is regulated through a noncanonical mechanism that is GLI-independent (Yam et al., 2009). Taking into account that GLIA/R activity can influence proliferation in many tissues during embryonic development (Bai et al., 2004), and that GLI1 is able to modulate cell-cycle progression (Kenney and Rowitch, 2000), it is possible that SHH regulates the switch between NSC quiescence and proliferative activation, or the decision-making process between self-renewal and progenitor generation. Consistent with this, our marker analysis of LRCs in mGfap-Smo CKOs and GFP+ cells in Nestin-MASTR Smo mosaics indicates NSCs lacking Smo do not change fate into astrocytes but instead proliferate less often.
In addition to regulating neurogenesis in the SVZ, our results have important implications for the recent evidence that the SVZ is molecularly patterned along the dorsal–ventral axis and SHH signaling can influence the proportion of OB neurons that occupy the deep versus superficial layers (Merkle et al., 2007; Ihrie et al., 2011). We showed that constant negative regulation of GLI3R activity by SHH is required both in the dorsal GLI1− NSCs and GLI1+ ventral SVZ, as the number of LRCs and proliferating TACs is significantly decreased in both regions of the SVZ of mGfap-Smo CKOs. Interestingly, as was previously observed in mice lacking Shh in the adult forebrain (Ihrie et al., 2011), we found that in the few remaining OB neurons generated in adult mGfap-Smo CKOs, the proportion of deep-layer OB interneurons is preferentially reduced. Moreover, in mGfap-Gli2;Gli3 double CKOs that have little reduction in neurogenesis, we observed a significant increase in the proportion of deep-layer interneurons produced, whereas in mGfap-Gli3;Smo CKOs that maintain GLI2R, the distribution appeared normal. Our study of the requirement for GLI2/3 function in OB neuron generation in the postnatal brain indicates that a complete inhibition of all GLIR activity, and not GLIA function, pushes SVZ stem cells toward production of deep-layer OB neurons. In addition, a low level of GLIR is required to produce the normal proportion of superficial OB neurons. In light of our results, the likely explanation for the finding that ectopic expression of SmoM2 (an oncogenic activated form of SMO) in NSCs of the dorsal SVZ can bias dorsal NSCs toward producing deep-layer interneurons (Ihrie et al., 2011) is that activated SmoM2 removes all GLIR, producing a molecular environment similar to our mGfap-Gli2;Gli3 CKOs. Altogether, our results demonstrate that an antagonistic interaction between GLIR and SHH is crucial to the generation of the correct proportions of interneurons along the deep to superficial axis of the OB. Our analysis of two dorsally restricted genes in the SVZ revealed no spatial change in mGfap-Smo or mGfap-Gli2;Gli3 CKOs, revealing that SHH signaling is not required to regulate Pax6 and Sp8 expression.
An interesting aspect of SHH function outside the neurogenic niches of the adult forebrain is its role in maintaining astrocyte function. We have demonstrated that SHH signaling in mature astrocytes is not only mediated by the GLI transcription factors, but also that differentiated astrocytes in the adult cortex are exquisitely sensitive to an increase in GLIR. On one hand, ablation of Gli3 in mGfap-Smo CKOs largely rescues the reactive astrogliosis phenotype characteristic of cortical astrocytes mutant for Smo. On the other hand, expression of a constitutive GLI3R in the absence of normal GLI2 or GLI3 produces a great degree of reactive astrogliosis. As gliosis is normally a sign of injury or disease, our studies indicate that an increase of GLIR in astrocytes of mGfap-Smo CKOs leads to changes in gene expression that either directly compromises astrocytes or alters astrocyte–neuron reciprocal communication in specific brain regions, such as the cortex. In light of a recent paper showing that SHH controls production of GFAP+ forebrain cells capable of forming neurospheres in vitro (Sirko et al., 2013), we speculate that GLI3R is the main effector of this additional SHH-regulated adult forebrain process that could be critical to repair following injury. Despite the requirement for SHH signaling in astrocyte function, our mosaic mutant analysis demonstrates a cell-autonomous requirement for SHH to maintain neurogenesis and the population of GFAP+ S100β− NSCs in the SVZ.
Finally, we uncovered that in mGfap-Smo CKO mutants there is a gradual deterioration of SVZ neurogenesis, indicating that SHH activity preserves NSCs throughout life. Furthermore, our examination of cell death in P14 and P45 mGfap-Smo CKOs indicates that apoptosis is not a major cause of the loss of LRCs in the SVZ. However, if NSCs alone were to die over a protracted period of time in mGfap-Smo CKOs, cell death might not be readily detectable. On the other hand, lowering SHH signaling often leads to changes in cell-fate decisions during development, including in the telencephalon (Nery et al., 2001; Loulier et al., 2006), which raised the possibility that ablation of SHH signaling results in an alteration in the identity of adult SVZ NSCs. Hence one possibility, in addition to cell death or differentiation, is that loss of SHH signaling (increased GLI3R) pushes NSCs in mGfap-Smo CKOs to slip into permanent quiescence. Interestingly, we have shown that SVZ neurogenesis significantly decreases with age (Fig. 3), similar to the aging dentate gyrus (Bonaguidi et al., 2011; Lugert et al., 2012), where NSCs can transform into differentiated astrocytes (Encinas et al., 2011). Although our marker analysis indicates SVZ NSCs in mGfap-Smo CKOs do not become astrocytes, in light of this recent discovery and our finding of a role for SHH in maintaining LRCs and thus the proliferative ability of NSCs, as well as in governing astrocyte function, SHH stands out as a possible antiaging signal (Dashti et al., 2012) that could be responsible for prolonging the life of neurogenic niches in the mature brain.
Footnotes
This work was supported by a grant from the National Cancer Institute (CA128158). We are grateful to Lalanti Venkatasubramanian for technical support, Zhimin Lao for experimental animals, Alexandre Wojcinski and Celia Andreu-Agullo for thoughtful comments on the manuscript, and past and present members of the laboratory for helpful discussions.
The authors declare no competing financial interests.
References
- Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Kohwi M, Nguyen TM, Merkle FT. The heterogeneity of adult neural stem cells and the emerging complexity of their niche. Cold Spring Harb Symp Quant Biol. 2008;73:357–365. doi: 10.1101/sqb.2008.73.019. [DOI] [PubMed] [Google Scholar]
- Angot E, Loulier K, Nguyen-Ba-Charvet KT, Gadeau AP, Ruat M, Traiffort E. Chemoattractive activity of sonic hedgehog in the adult subventricular zone modulates the number of neural precursors reaching the olfactory bulb. Stem Cells. 2008;26:2311–2320. doi: 10.1634/stemcells.2008-0297. [DOI] [PubMed] [Google Scholar]
- Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development. 2001;128:5161–5172. doi: 10.1242/dev.128.24.5161. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/S1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Balordi F, Fishell G. Hedgehog signaling in the subventricular zone is required for both the maintenance of stem cells and the migration of newborn neurons. J Neurosci. 2007a;27:5936–5947. doi: 10.1523/JNEUROSCI.1040-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balordi F, Fishell G. Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J Neurosci. 2007b;27:14248–14259. doi: 10.1523/JNEUROSCI.4531-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaess S, Stephen D, Joyner AL. Gli3 coordinates three-dimensional patterning and growth of the tectum and cerebellum by integrating Shh and Fgf8 signaling. Development. 2008;135:2093–2103. doi: 10.1242/dev.015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ, Ming GL, Song H. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell. 2011;145:1142–1155. doi: 10.1016/j.cell.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böse J, Grotewold L, Rüther U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet. 2002;11:1129–1135. doi: 10.1093/hmg/11.9.1129. [DOI] [PubMed] [Google Scholar]
- Bowers M, Eng L, Lao Z, Turnbull RK, Bao X, Riedel E, Mackem S, Joyner AL. Limb anterior–posterior polarity integrates activator and repressor functions of GLI2 as well as GLI3. Dev Biol. 2012;370:110–124. doi: 10.1016/j.ydbio.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- Dashti M, Peppelenbosch MP, Rezaee F. Hedgehog signalling as an antagonist of ageing and its associated diseases. Bioessays. 2012;34:849–856. doi: 10.1002/bies.201200049. [DOI] [PubMed] [Google Scholar]
- Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA, Fishell G, Koulakov A, Enikolopov G. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell. 2011;8:566–579. doi: 10.1016/j.stem.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo M, Rallu M, McMahon AP, Fishell G. Temporal requirement for hedgehog signaling in ventral telencephalic patterning. Development. 2004;131:5031–5040. doi: 10.1242/dev.01349. [DOI] [PubMed] [Google Scholar]
- Fuccillo M, Joyner AL, Fishell G. Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:772–783. doi: 10.1038/nrn1990. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Petrova R, Eng L, Joyner AL. Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J Neurosci. 2010;30:13597–13608. doi: 10.1523/JNEUROSCI.0830-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008;11:277–284. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–537. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- Ihrie RA, Alvarez-Buylla A. Lake-front property: a unique germinal niche by the lateral ventricles of the adult brain. Neuron. 2011;70:674–686. doi: 10.1016/j.neuron.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrie RA, Shah JK, Harwell CC, Levine JH, Guinto CD, Lezameta M, Kriegstein AR, Alvarez-Buylla A. Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron. 2011;71:250–262. doi: 10.1016/j.neuron.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura T, Kornblum HI, Sofroniew MV. The predominant neural stem cell isolated from postnatal and adult forebrain but not early embryonic forebrain expresses GFAP. J Neurosci. 2003;23:2824–2832. doi: 10.1523/JNEUROSCI.23-07-02824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/MCB.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Ables JL, Dickel LK, Eisch AJ, Johnson JE. Ascl1 (Mash1) defines cells with long-term neurogenic potential in subgranular and subventricular zones in adult mouse brain. PLoS One. 2011;6:e18472. doi: 10.1371/journal.pone.0018472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao Z, Raju GP, Bai CB, Joyner AL. MASTR: a technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep. 2012;2:386–396. doi: 10.1016/j.celrep.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson M, Saghatelyan A, Olivo-Marin JC, Lledo PM. Neonatal and adult neurogenesis provide two distinct populations of newborn neurons to the mouse olfactory bulb. J Neurosci. 2005;25:6816–6825. doi: 10.1523/JNEUROSCI.1114-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Loulier K, Ruat M, Traiffort E. Increase of proliferating oligodendroglial progenitors in the adult mouse brain upon Sonic hedgehog delivery in the lateral ventricle. J Neurochem. 2006;98:530–542. doi: 10.1111/j.1471-4159.2006.03896.x. [DOI] [PubMed] [Google Scholar]
- Lugert S, Vogt M, Tchorz JS, Müller M, Giachino C, Taylor V. Homeostatic neurogenesis in the adult hippocampus does not involve amplification of Ascl1(high) intermediate progenitors. Nat Commun. 2012;3:670. doi: 10.1038/ncomms1670. [DOI] [PubMed] [Google Scholar]
- Machold R, Hayashi S, Rutlin M, Muzumdar MD, Nery S, Corbin JG, Gritli-Linde A, Dellovade T, Porter JA, Rubin LL, Dudek H, McMahon AP, Fishell G. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/S0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 1998;125:2759–2770. doi: 10.1242/dev.125.15.2759. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Mirzadeh Z, Alvarez-Buylla A. Mosaic organization of neural stem cells in the adult brain. Science. 2007;317:381–384. doi: 10.1126/science.1144914. [DOI] [PubMed] [Google Scholar]
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, Joyner AL, Hui C. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- Nery S, Wichterle H, Fishell G. Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Development. 2001;128:527–540. doi: 10.1242/dev.128.4.527. [DOI] [PubMed] [Google Scholar]
- Palma V, Lim DA, Dahmane N, Sánchez P, Brionne TC, Herzberg CD, Gitton Y, Carleton A, Alvarez-Buylla A, Ruiz i Altaba A. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132:335–344. doi: 10.1242/dev.01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Persson M, Stamataki D, te Welscher P, Andersson E, Böse J, Rüther U, Ericson J, Briscoe J. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002;16:2865–2878. doi: 10.1101/gad.243402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti G, Obernier K, Guinto C, Jose L, Bonfanti L, Alvarez-Buylla A. Cell cycle and lineage progression of neural progenitors in the ventricular-subventricular zones of adult mice. Proc Natl Acad Sci U S A. 2013;110:E1045–E1054. doi: 10.1073/pnas.1219563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallu M, Machold R, Gaiano N, Corbin JG, McMahon AP, Fishell G. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development. 2002;129:4963–4974. doi: 10.1242/dev.129.21.4963. [DOI] [PubMed] [Google Scholar]
- Raponi E, Agenes F, Delphin C, Assard N, Baudier J, Legraverend C, Deloulme JC. S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia. 2007;55:165–177. doi: 10.1002/glia.20445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M, Fischer IR, Plesnila N, Staufenbiel M, Haass C, Snapyan M, Saghatelyan A, Tsai LH, Fischer A, Grobe K, Dimou L, et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog glia. Cell Stem Cell. 2013;12:426–439. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, Doetsch F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–288. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam PT, Langlois SD, Morin S, Charron F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 2009;62:349–362. doi: 10.1016/j.neuron.2009.03.022. [DOI] [PubMed] [Google Scholar]