

Abstract
Pectus excavatum, the most frequent congenital chest wall deformity, may be rarely observed as a sole deformity or as a sign of an underlying connective tissue disorder. To date, only few studies have described correlations between this deformity and heritable connective tissue disorders such as Marfan, Ehlers-Danlos, Poland, MASS (Mitral valve prolapse, not progressive Aortic enlargement, Skeletal and Skin alterations) phenotype among others. When concurring with connective tissue disorder, cardiopulmonary and vascular involvement may be associated to the thoracic defect. Ruling out the concomitance of pectus excavatum and connective tissue disorders, therefore, may have a direct implication both on surgical outcome and long term prognosis. In this review we focused on biological bases of connective tissue disorders which may be relevant to the pathogenesis of pectus excavatum, portraying surgical and clinical implication of their concurrence.
Key words: pectus excavatum, Marfan syndrome, MASS, fibrillin, fibrillinopathies
Introduction
Pectus Excavatum (PE), the most frequent congenital chest wall deformity, is characterized by anomalous growth of several ribs and sternum, which makes the chest hollow (Figure 1). PE occurs in 1 in 300-1000 live births with a male/female ratio of approximately 4:1 with a tendency to become more evident during adolescence.1-4 The causes of PE are still unclear; although a familial predisposition has been observed and nearly half of the patients have relatives with various skeletal alterations, a direct genetic link has not yet been recognized.5,6 Patients with a non-syndromic familial isolated deformity have been described as a rare occurrence.7 Progression may be particularly evident during adolescence, an information unfamiliar to a lot of pediatricians who erroneously inform younger patients that this condition will resolve spontaneously with growth. At time of puberty, when patients grow rapidly, such deformity often abruptly accelerates, and a mild defect may quickly turn into severe. Usually this situation alarms parents and induces them to seek surgical consultation despite their pediatrician’s reassurance.8 Surgical repair of PE in most of the cases has a cosmetic indication. Since most patients with the deformity do not have other associated symptoms, treatment may not be strictly needed and will be dependent upon the development of symptoms or lack of self-acceptance. Although still unclear, physiotherapy in young patients may play a beneficial role slowing the development of the chest wall deformity and may possibly correct milder forms. The primary goal of surgical repair is to correct the chest deformity which improves first of all self-acceptance and may ameliorate patient’s breathing, posture and cardiac function. A variety of surgical procedures were developed in order to correct the defect by forward repositioning of the sternum. In 1949, Ravitch described a repair technique requiring subperichondrial resection of all deformed costal cartilages, xiphoid excision and transverse sternal osteotomy displacing the sternum anteriorly9,10 (Figure 2A). Some decades later, in 1998, a minimally invasive technique was developed by Nuss. This procedure involves placement under thoracoscopic view of a curved substernal bar which is placed and flipped into the chest acquiring concavity facing posteriorly in order to push sternum outward (Figure 2B). The bar is then removed after about three years, a period after which the chest is supposed to have acquired a stable structure.9-11 Nuss technique promptly gained a widespread approval, especially because in a different way from Ravitch procedure, it does not produce any visible scar in the anterior aspect of the chest. To date, several comparative studies in order to outline the real benefits of either techniques were published with inconsistent results;10,12-19 a systematic review and meta-analysis failed to provide overwhelming support to either approach, concluding that the use of validated pain scales or satisfaction surveys and the quantification of total narcotics would be helpful.20
Figure 1.
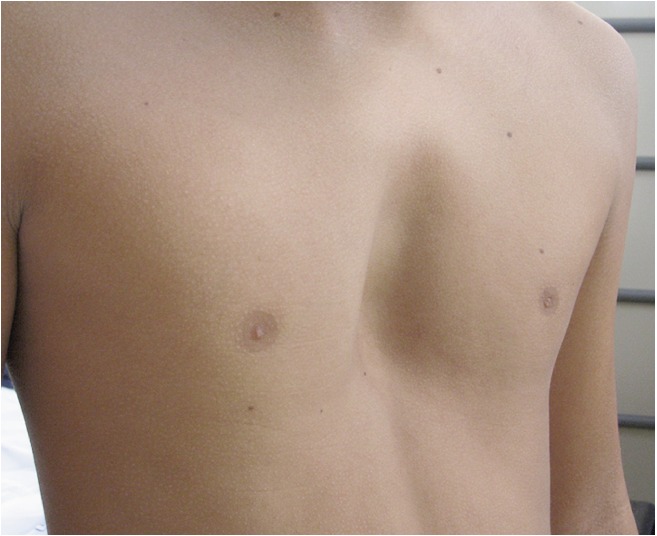
Chest picture of an adolescent affected by a moderate/severe form of pectus excavatum. Written consent was obtained from the patient and the patient’s parents for publication of this image.
Figure 2.

Two surgical techniques are described. The first one, popularized by Ravitch (A), entails an anterior approach through which costal cartilages are removed and the sternum detached. The latter, proposed by Nuss (B) consists in the insertion of one or two concave steel bars into the chest, underneath the sternum. The bar is then flipped to a convex position so as to push outward on the sternum, correcting the deformity.
Patient age at the time of surgery is important for both speed of recovery and long-term results.11 Unlike the more invasive procedures such as Ravitch, there is no interference with growth plates when using Nuss procedure.21,22 This evidence supports the fact that the latter procedure is feasible at any age.
Concerns have been raised in patients younger than 6 years, since there are many years of subsequent growth during which the pectus excavatum may recur.8 Some authors agree that the optimal age is 7 to 14 years, because, before puberty, the patients’ chests are still soft and malleable; they show quick recovery, a rapid return to normal activities, and have excellent results.11 After puberty, the flexibility of the chest wall is decreased, requiring the insertion of two bars, making the procedure more difficult. It also takes the patients longer to recover.8 Long-term results, however, are superior in grown-up patients, particularly for those who have completed their growth at the time of bar removal. Since regression or improvement of PE is not related to cartilage and ligament fixation, the ideal phase for optimal PE correction should be around adolescence, when the costal cartilages are still plastic and flexible.23 In addition to this, since it is not negligible to share the indication for surgery among both parents and patient, at our Institution the procedure is carried out mainly in adolescents which are able to demonstrate a complete awareness of such surgical procedure.
In regards to clinical implications of surgical correction, the release of cardiac compression caused by the sunken chest is one of the most significant clinical consequence of surgical repair of PE. Previous reports and meta-analyses, supported by the hypothesis that the release of cardiac compression in PE patients improves the hemodynamic function, have been carried out demonstrating that cardiac function improves after surgical correction of PE.24-26 On the other hand, lung function improvement after surgical repair of PE is not yet generally accepted. In particular, it is still uncertain whether the pathophysiologic deficits in PE are primarily ventilatory or cardiovascular. Surgical management may increase a patient’s exercise tolerance and reduce symptoms because a depressed sternum compresses the right ventricular outflow tract and this anatomical constraint may produce in turn diminished lung capacity, arising with frequent respiratory tract infection, dyspnea, and pulmonary insufficiency.27,28 The post-operative cardiovascular and respiratory improvements are particularly applicable to patients with severe deformities, because surgery may increase significantly chest volume, making space available for lung expansion.29 A recent meta-analysis including a total of 2476 PE patients operated with either techniques, showed that although both procedures for the correction of PE displayed the same improvements in pulmonary function within 1 year after the procedure, Nuss technique showed an additional enhancement in pulmonary function after bar removal.23 In any case, there is still a need for further large-scale prospective trials and meta-analyses to better assess the extent of the beneficial effects of PE correction on pulmonary function.
PE may be frequently associated with heritable disorders of the connective tissue, such as MFS, Ehlers-Danlos, Poland, Mitral valve prolapse or MASS syndromes.30-32 Roughly half of the patients with MFS have a concurring PE.30,33-35 However, apart from limited reports on MFS,30,33-35 Ehlers-Danlos,33 and MASS phenotype,2 to date only few studies have described correlations between PE and connective tissue disorders.
Aetiopathogenesis of Pectus Excavatum
Many hypotheses have been proposed to explain the pathogenesis of PE, such as intrauterine compression, rickets, pulmonary restriction, diaphragm alterations resulting in posterior traction on the sternum, and primary failure of osteogenesis or chondrogenesis.5 The prevailing hypothesis has long been overgrowth of costal cartilage, further intensified during growth spurts, which pushes the sternal body inward.36,37 This theory was firstly introduced by Sweet in 1944 and many surgeons supported this theory subsequently,37 as none reported about the length of costal cartilage until Nakaoka.38 Nakaoka et al. measured costal cartilage length in PE patients and healthy controls from reconstructed images of three-dimensional computed tomography, where costal cartilage in PE patients resulted to be not any longer than in healthy controls.38 Several studies have then focused on histology and biochemistry of costal cartilage, albeit the results to date remain still inconclusive. Collagen type II is a major structural component of rib cartilage, therefore its metabolism and composition may play an important role in the aetiology and pathogenesis of PE.36 Biochemical and immunohistochemical analyses have shown abnormalities in the structure such as altered distribution and arrangement of type-2 collagen in costal cartilage, abnormal high levels of zinc and low levels of magnesium and calcium plus disturbance of collagen synthesis.5,39 Electron microscopy studies have revealed degenerated chondrocytes with fat droplets and other osmiophilic inclusions not better characterized, as well as atypical fibrils including long-spacing collagen.39 Additional data indicate that PE patients also exhibit disorders in endochondral ossification and alterations in the metabolism of acid mucopolysaccharides.5,40 Vacuolisation, polymorphism and nuclear pyknosis, along with higher cell number density, more variable cellular distribution, and more frequent myxoid matrix degeneration with focal necrosis occur which may reflect increased growth and decreased structural integrity in the presence of additional mechanical stress in PE patients and, to a certain extent, premature cartilage ageing.36 All the aforementioned alterations certainly indicate an abnormality in connective tissue biology which affects, among others, the ribs and sternum and which may contribute to the development of PE.41 Feng et al. have analysed the biomechanical properties of costal cartilage and showed that the biomechanical stability of costal cartilage in children with PE is impaired in response to tension, compression, and bending. Those authors have proposed that the abnormal costal cartilage in children with PE is unable to resist the stress of the respiratory gradient, leading to the formation of funnel chest. Furthermore, the movement of diaphragm, a main fetal breathing activity, might cause retraction of the weak costal cartilages and make them angled posteriorly so that PE would be present at birth. Unusual inspiratory efforts, as in case of crying spells, respiratory obstruction or hiccupping, might aggravate the effect of inspiratory retraction on the anterior sixth rib cartilage because of the strong spastic contraction of the diaphragm, and might induce or aggravate the deformity.41 This hypothesis is supported by the fact that the PE is more frequent in children with respiratory obstruction.41,42 Lastly, a recent study on 34 families provided evidence that PE can be an inherited disorder. Although some families demonstrated mendelian inheritance, most cases appeared to have multifactorial genetic background. It is reported that in families of individuals affected by PE there are relatives which have connective tissue disease traits, suggesting that PE may be due, in part, to some deficit in connective tissue disordersrelated genes such as FBN, collagen, and TGF.5
Heritable disorders of connective tissue
Heritable disorders of connective tissue (HDCTs) comprise a group of genetic disorders affecting proteins of connective tissue matrix, such as collagens, proteoglycans, elastins, fibrillins and laminins.43,44 HDCTs are described in Berlin International Nosology (1986) which defines several disorders such as MFS, MASS phenotype among others (Stickler syndrome, Ehlers-Danlos syndrome, Familial Articular Hypermobility syndrome, Skeletal Dysplasias with predominant joint laxity, Cutis Laxa, Pseudoxanthoma Elasticum, Epidermo lysis Bullosa, HDCTs secondary to metabolic defects, copper transport disorders, Osteogenesis Imperfecta, miscellaneous HDCTs).45 HDCTs generally result from single gene mutations and the patterns of inheritance may be autosomal dominant, autosomal recessive or X-linked.46 Abnormal folding and aggregation of the matrix macromolecules are caused by incorporation of one or more mutant polypeptides. Consequently, mutations which cause synthesis of structurally altered polypeptide chains usually produce more severe phenotypes than heterozygous null alleles.45 Fibrillin (FBN) proteins are the major components of extracellular microfibrils found in many connective tissues, elastic and non-elastic tissues.47 There are three human fibrillins, fibrillin-1, -2 and -3, which are encoded by different genes.48 FBN1 and FBN2 are well studied and mutations in these proteins cause a number of fibrillinopathies including MFS and congenital arachnodactily, respectively. FBN3 has been discovered more recently and is less well characterized. FBN1, the main component of the microfibrils which together with elastin form elastic fibres, is expressed throughout life, whereas FBN2 and FBN3 are thought to be primarily present during development.47 FBN1 serves two key physiological functions: it participates to the assembly of specialized, structurally relevant fibres within the extracellular matrix and provides context dependent modulation of TGF and bone morphogenetic protein (BMP) signalling, which in turn regulate matrix formation and remodeling.48 FBN1 is a 230-Kb gene containing 65 exons, situated on chromosome 15q21. More than 1750 mutations in FBN1 gene 1 are registered in the Universal Marfan Database (UMD)-FBN1 for MFS and its associated disorders. These include point mutations, large and small deletions, insertions, missense and splice mutations. Premature termination codon (PTC) and in-frame mutations are two major mutation categories in FBN1.48,49 FBN1 mutations have been associated with different clinical phenotypes: MFS, MASS phenotype, ectopia lentis syndrome, mitral valve prolapse syndrome, Shprintzen-Goldberg syndrome and Loeys-Dietz syndrome.50,51 MFS is a systemic autosomal dominant connective tissue disorder caused by several mutations in the gene encoding the glycoprotein FBN1. MFS has an incidence of 2-3 cases in 10,000,30,33 whilst PE is present in two thirds of patients with MFS.30,33-35 MFS clinical manifestations vary in range and severity and may involve several organs:51,52 cardiovascular diseases (aortic dilatation and dissection) are the major cause of morbidity and mortality.53,54 First in 1989, Glesby proposed the acronym MASS to define a group with concurrence of clinical features such as mitral valve prolapse,55 not progressive aortic enlargement, skeletal and skin alterations. MASS belongs to connective tissue disorders characterized by slight, generalized laxity. In patients with MASS phenotype, aortic root diameter may be at the upper limits of normal for body size, but there is no documented progression to aneurysm or predisposition to dissection and the finding is of limited clinical value;32,51,53,55,56 these patients do not suffer from ectopia lentis ( Table 1).32,51,56 MASS is described as an autosomal dominant disease and is caused by mutations in the FBN1 gene,32,57,58 which encodes the protein fibrillin 1 (FBN1) and appears to be commonly concurrent with PE.2 The prominent hypotheses on the pathogenesis of MFS, the most studied fibrillinopathy, focused both on the structural weakness of the tissues imposed by microfibrillar deficiency and upregulation of Transforming Growth Factor-β (TGFβ) signalling pathway.48,49 FBNs constitute the major backbone of multifunctional microfibrils in elastic and non-elastic extracellular matrices. FBN1 mutations have been shown to increase the susceptibility of FBN1 to proteolysis in vitro leading to fragmentation of microfibrils.48 Fragmentation and disorganization of the elastic fibres in the aortic media layer is a histological marker of MFS, known as medial degeneration where the properties of microfibrils are often compromised because of their aberrant folding and assembly.48 Recent findings suggest that proteolysis due to increased metalloproteinase (MMP) activity, and consequent fragmentation of microfibrils in tissues of MFS patients may also contribute to the pathogenesis of this disorder. Therefore, FBN1 fragments may regulate MMP-1 expression, and MMPs dysregulation caused by fibrillin fragmentation may contribute to the development of MFS. Further findings also support an interaction between altered extracellular matrix and aberrant TGF signalling in MFS pathogenesis.48 An excessive TGF signalling characterizes the progression of aortic aneurysm in mouse models of MFS, possibly because FBN1 mutations promote abnormal Smad signalling by impairing the sequestration of latent TGF complexes into the extracellular matrix.48,59 Although molecular testing for FBN1 mutations is usually available at present, the diagnosis of MFS cannot rely only on the demonstration of a gene mutation,53,60 because of the large clinical variability and genetic heterogeneity. This implies that MFS still remains a clinical diagnosis and depends on a combination of major and minor signs as defined by Ghent criteria proposed in 1996 ( Table 2).32,53,61,62 Among these criteria, a severe PE which necessitates a surgical repair, for example, is a major one for MFS diagnosis.30,60 More recently, in 2010, Ghent criteria were revised and major changes were introduced.56 More importance was given to two cardinal features: aortic dilatation and ectopia lentis, which are now considered sufficient for diagnosis when concomitant. A more prominent role was assigned to molecular genetic testing of FBN1 and other relevant genes (e.g. TGFBR1, TGFBR2), whilst other less specific features (like recurrent or incisional hernia, apical blebs) were removed or considered less influential in the diagnostic evaluation. The revised criteria were simplified in order to facilitate a prompt diagnosis, providing specific recommendations for patients counseling and follow-up.56,63 Authors which applied the revised Ghent criteria to a large cohort of MFS patients, concluded that the definition of aortic root dilatation based on Z-score alone may underestimate aortic involvement, representing a pitfall in the revised criteria. Therefore, a re-evaluation of the aortic involvement criteria in adult patients with suspected MFS diagnosis may be warranted.63
Table 1.
Diagnostic criteria for the MASS (Mitral valve prolapse, not progressive Aortic enlargement, Skeletal and Skin alterations) phenotype.
| 1 | Mitral valve prolapse |
| 2 | Borderline and non-progressive aortic enlargement |
| 3 | Not specific skeletal alterations (pectus excavatum, long limbs) |
| 4 | Not specific skin alterations (striae atrophicae) |
| 5 | Myopia (non ectopia lentis) |
Table 2.
Diagnostic criteria for the Marfan Syndrome (Ghent Nosology).
| Skeletal system | Major criteria (at least 4 of the following constitutes a major criterion) |
Pectus carinatum Pectus excavatum, needing surgery Reduced upper to lower segment ratio or arm span to height ratio >1.05 Positive wrist and thumb signs Scoliosis >20° or spondylolisthesis Reduced extension of the elbows (>170°) Medial displacement of the medial malleolus causing pes planus Protrusio acetabuli of any degree, ascertained on x-ray |
| Minor criteria | Pectus excavatum of moderate severity Joint hypermobility Facial appearance (dolicocephaly, malar hypoplasia, enophthalmos, retrognathia, down-slanting palpebral fissures) |
|
| Highly arched palate with dental crowding | ||
| Ocular system | Major criteria | Ectopia lentis |
| Minor criteria | Abnormally flat cornea (as measured by keratometry) Increased axial length of globe (as measured by ultrasound) |
|
| Cardiovascular system | Major criteria | Dilatation of the ascending aorta with or without aortic regurgitation and involving at least the sinuses of Valsalva Dissection of the ascending aorta |
| Minor criteria | Mitral valve prolapse with or without mitral valve regurgitation Dilatation of main pulmonary artery, in the absence of valvular or peripheral pulmonary stenosis or any other obvious cause, under the age of 40 years Calcification of the mitral annulus below the age of 40 years Dilatation or dissection of the descending thoracic or abdominal aorta below the age of 50 years |
|
| Pulmonary system | Major criterion | None |
| Minor criterion | Spontaneous pneumothorax Apical blebs (ascertained by chest radiography) |
|
| Skin and integument | Major criteria | Lumbosacral dural ectasia ascertained by CT or MRI |
| Minor criteria | Striae atrophicae (stretch marks) not associated with marked weight change, pregnancy or repetitive stress Recurrent or incisional hernia |
|
| Family/genetic history | Major criteria | Having a parent, child, or sibling who meets the diagnostic criteria listed below independently Presence of a mutation in FBN1 known to cause the Marfan syndrome Presence of a haplotype around FBN1, inherited by descent, known to be associated with unequivocally diagnosed Marfan syndrome in the family |
| Minor criteria | None | |
| Requirement for diagnosis of Marfan syndrome | Index case | Major criteria in at least two different organ systems and involvement of a third organ system |
| Family member | Presence of a major criterion in the family history and one major criterion in an organ system and involvement of a second organ system |
To assess involvement of the skeletal system, at least two major criteria, or one major criterion plus two minor criteria must be present. To assess involvement of the ocular system, the major criterion or at least two minor criteria must be present. To assess involvement of the cardiovascular system, one major or minor criterion must be present. To assess involvement of the pulmonary system, one of the minor criteria must be present. To assess involvement of the skin and integument, the major criterion must be present.
Discussion
As previously reported, biochemical and histopathological studies in PE patients have shown abnormalities in the costal cartilage,5,36,39 and this presumably implies that nearly the totality of patients affected by PE displays various degrees of connective tissue laxity. Although PE is seen in individuals with primary connective tissue defects, it is likely that the outcome of these patients, when surgically treated, differs from those who have only chest deformity without signs of heritable connective tissue disorder.
A few studies have addressed the surgical outcome of patients upon operation for the correction of PE with or without associated connective tissue disorder. In a comparison between seven children affected by PE and MFS and 38 ones affected by sole deformity, the authors claimed that they had to use a substernal strut to ensure a satisfactory correction of the defect especially in MFS patients.35 These results have been confirmed also by other authors.30,34 In a report of 28 patients with MFS and 30 patients with a supposed sole deformity it was concluded that PE may be a part of a heritable disorder of connective tissue such as MFS and that in patients with MFS or other connective tissue diseases surgical repair should be deferred, whether possible, until skeletal maturity is nearly established; moreover, for the cases subjected to surgical correction the use of an internal stabilization is strongly recommended.64 A study on a large series of patients was then published in 2002, regarding 303 patients affected by PE who underwent minimally invasive surgical technique according to Nuss.11 The series contained 16 individuals with MFS and 49 with a diagnosis of nonspecific connective tissue disease.65 Among the reported complications, late failure of the procedure occurred in four patients, three of which had MFS. Furthermore, four other patients as a consequence of the surgical procedure developed overcorrection, three had concurrent MFS and one had Ehlers-Danlos syndrome. Nonetheless, in the majority of MFS patients, the long-term result was reputed excellent.65 In another similar study, the authors found that the patients with sole PE did not differ from those affected by connective tissue diseases (MFS, Ehlers-Danlos syndrome and nonspecific connective tissue disease) neither in operation time nor in estimated mean blood loss nor in the number of bars needed to achieve correction. Moreover there was no statistical difference between the rates of postoperative complications in the two groups.33 Lastly, in 2010, Redlinger et al.30 studied 3 groups of patients who underwent surgical repair of PE and compared the operative and postoperative results among patients with MFS, those with marfanoid features, and all other patients and concluded that there was no difference in terms of surgical outcome.30 To date there is no report focusing on the surgical outcome of patients with MASS phenotype undergoing PE surgical correction.
In regards to aortic aneurysm and dissection, which are considered notorious causes of morbidity and mortality in MFS patients,50,58 there are scarce findings on the evolution of aortic diameter over time in patients with features of connective tissue disorder without full blown MFS. Mortensen et al. performed an observational study to assess the evolution of aortic disease in 78 patients in which MFS was ruled out, measuring the augmentation index at a heart rate of 75 beats per minute and central pulse pressure (CPP) by applanation tonometry (APT). During twenty months after APT, they observed progression of aortic diameters in 15 patients, and aortic surgery or aortic dissection in 3 individuals.57 Rybczynski et al.60 evaluated prospectively 279 patients with suspected MFS. They confirmed MFS in 138 individuals and another hereditary disorder of connective tissue in 69 individuals, in 30 of which the cardiovascular system was involved.
They concluded that many different, inherited collagen disorders have a poorly defined cardiovascular risk, therefore future studies are required to establish long-term cardiovascular prognosis. Therefore it is likely that most patients with an inherited connective tissue disorder do not progress towards aortic aneurysm or dissection, but it still remains unclear how to identify those at increased risk, especially for patients who do not have the stigmata of MFS. To sort out this issue, larger prospective cohorts of patients with detailed clinical and echocardiographic follow-up over years are warranted.
It seems clear that several patients affected by PE display a typical association with connective tissue disorders, which may span from mild form like simple laxity without morbidity associated, to more severe forms such as MFS and Ehlers-Danlos syndrome. Yet at present, for most of them there is no information on the risk of complications or about the long-term prognosis, therefore strategies for clinical management remain still indefinite, especially for those patients which undergo a surgical operation to correct the deformity. Anyway, when a PE is concurrent with a connective tissue disorder of whatsoever gravity, we reckon that it is advisable a thorough clinical and echocardiographic follow-up over time, first of all because of the possible worsening of mitral valve prolapse which is a feature present in many of these patients and secondly to possibly avert the onset of notorious cardiovascular complications such as aortic dissection, a situation commonly seen in the most severe connectivopathies.
References
- 1.Williams AM, Crabbe DC.Pectus deformities of the anterior chest wall. Paediatr Respir Rev 2003;4:237-42 [DOI] [PubMed] [Google Scholar]
- 2.Tocchioni F, Ghionzoli M, Pepe G, Messineo A.Pectus Excavatum and MASS phenotype: an unknown association. J Laparoendosc Adv Surg Tech A 2012;22:508-13 [DOI] [PubMed] [Google Scholar]
- 3.Cartoski MJ, Nuss D, Goretsky MJ, et al. Classification of the dysmorphology of pectus excavatum. J Pediatr Surg 2006;41:1573-81 [DOI] [PubMed] [Google Scholar]
- 4.Rattan AS, Laor T, Ryckman FC, Brody AS.Pectus excavatum imaging: enough but not too much. Pediatr Radiol 2012;40:168-72 [DOI] [PubMed] [Google Scholar]
- 5.Creswick HA, Stacey MW, Kelly RE, Jr., et al. Family study of the inheritance of pectus excavatum. J Pediatr Surg 2006;41:1699-703 [DOI] [PubMed] [Google Scholar]
- 6.Jaroszewski D, Notrica D, McMahon L, et al. Current management of pectus excavatum: a review and update of therapy and treatment recommendations. J Am Board Fam Med 2010;23:230-9 [DOI] [PubMed] [Google Scholar]
- 7.Kotzot D, Schwabegger AH.Etiology of chest wall deformities-a genetic review for the treating physician. J Pediatr Surg 2009;44:2004-11 [DOI] [PubMed] [Google Scholar]
- 8.Goretsky MJ, Kelly RE, Jr., Croitoru D, Nuss D.Chest wall anomalies: pectus excavatum and pectus carinatum. Adolesc Med Clin 2004;15:455-71 [DOI] [PubMed] [Google Scholar]
- 9.Nasr A, Fecteau A, Wales PW.Comparison of the Nuss and the Ravitch procedure for pectus excavatum repair: a meta-analysis. J Pediatr Surg 2010;45:880-6 [DOI] [PubMed] [Google Scholar]
- 10.Antonoff MB, Erickson AE, Hess DJ, et al. When patients choose: comparison of Nuss, Ravitch, and Leonard procedures for primary repair of pectus excavatum. J Pediatr Surg 2009;44:1113-9 [DOI] [PubMed] [Google Scholar]
- 11.Nuss D, Kelly RE, Jr., Croitoru DP, Katz ME.A 10-year review of a minimally invasive technique for the correction of pectus excavatum. J Pediatr Surg 1998;33:545-52 [DOI] [PubMed] [Google Scholar]
- 12.Jo WM, Choi YH, Sohn YS, et al. Surgical treatment for pectus excavatum. J Korean Med Sci 2003;18:360-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller KA, Woods RK, Sharp RJ, et al. Minimally invasive repair of pectus excavatum: a single institution’s experience. Surgery 2001;130:652-9 [DOI] [PubMed] [Google Scholar]
- 14.Fonkalsrud EW, Beanes S, Hebra A, et al. Comparison of minimally invasive and modified Ravitch pectus excavatum repair. J Pediatr Surg 2002;37:413-7 [DOI] [PubMed] [Google Scholar]
- 15.Boehm RA, Muensterer OJ, Till H.Comparing minimally invasive funnel chest repair versus the conventional technique: an outcome analysis in children. Plast Reconstr Surg 2004;114:668-75 [DOI] [PubMed] [Google Scholar]
- 16.Inge TH, Owings E, Blewett CJ, et al. Reduced hospitalization cost for patients with pectus excavatum treated using minimally invasive surgery. Surg Endosc 2003;17:1609-13 [DOI] [PubMed] [Google Scholar]
- 17.Lam MW, Klassen AF, Montgomery CJ, et al. Quality-of-life outcomes after surgical correction of pectus excavatum: a comparison of the Ravitch and Nuss procedures. J Pediatr Surg 2008;43:819-25 [DOI] [PubMed] [Google Scholar]
- 18.Molik KA, Engum SA, Rescorla FJ, et al. Pectus excavatum repair: experience with standard and minimal invasive techniques. J Pediatr Surg 2001;36:324-8 [DOI] [PubMed] [Google Scholar]
- 19.Kelly RE, Jr., Cash TF, Shamberger RC, et al. Surgical repair of pectus excavatum markedly improves body image and perceived ability for physical activity: multicenter study. Pediatrics 2008;122:1218-22 [DOI] [PubMed] [Google Scholar]
- 20.Nasr A, Fecteau A, Wales PW.Comparison of the Nuss and the Ravitch procedure for pectus excavatum repair: a meta-analysis. J Pediatr Surg 2010;45:880-6 [DOI] [PubMed] [Google Scholar]
- 21.Haller JA, Jr., Colombani PM, Humphries CT, et al. Chest wall constriction after too extensive and too early operations for pectus excavatum. Ann Thorac Surg 1996;61:1618-25 [DOI] [PubMed] [Google Scholar]
- 22.Calik M, Aribas OK, Kanat F.The effect of costal cartilage resection on the chest wall development: a morphometric evaluation. Eur J Cardiothorac Surg 2007;32:756-60 [DOI] [PubMed] [Google Scholar]
- 23.Chen Z, Amos EB, Luo H, et al. Comparative pulmonary functional recovery after Nuss and Ravitch procedures for pectus excavatum repair: a meta-analysis. J Cardiothorac Surg 2012;7:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maagaard M, Tang M, Ringgaard S, et al. Normalized cardiopulmonary exercise function in patients with pectus excavatum three years after operation. Ann Thorac Surg 2013;96:272-8 [DOI] [PubMed] [Google Scholar]
- 25.Kinuya K, Ueno T, Kobayashi T, et al. Tc-99m MAA SPECT in pectus excavatum: assessment of perfusion volume changes after correction by the Nuss procedure. Clin Nucl Med 2005;30:779-82 [DOI] [PubMed] [Google Scholar]
- 26.Sigalet DL, Montgomery M, Harder J, et al. Long term cardiopulmonary effects of closed repair of pectus excavatum. Pediatr Surg Int 2007;23:493-7 [DOI] [PubMed] [Google Scholar]
- 27.Quigley PM, Haller JA, Jr., Jelus KL, et al. Cardiorespiratory function before and after corrective surgery in pectus excavatum. J Pediatr 1996;128:638-43 [DOI] [PubMed] [Google Scholar]
- 28.Malek MH, Berger DE, Housh TJ, et al. Cardiovascular function following surgical repair of pectus excavatum: a metaanalysis. Chest 2006;130:506-16 [DOI] [PubMed] [Google Scholar]
- 29.Morshuis WJ, Folgering HT, Barentsz JO, et al. Exercise cardiorespiratory function before and one year after operation for pectus excavatum. J Thorac Cardiovasc Surg 1994;107:1403-9 [PubMed] [Google Scholar]
- 30.Redlinger RE, Jr., Rushing GD, Moskowitz AD, et al. Minimally invasive repair of pectus excavatum in patients with Marfan syndrome and marfanoid features. J Pediatr Surg 2010;45:193-9 [DOI] [PubMed] [Google Scholar]
- 31.Ho NC, Tran JR, Bektas A.Marfan’s syndrome. Lancet 2005;366:1978-81 [DOI] [PubMed] [Google Scholar]
- 32.Le Parc JM, Molcard S, Tubach F, et al. Marfan syndrome and fibrillin disorders. Joint Bone Spine 2000;67:401-7 [PubMed] [Google Scholar]
- 33.Olbrecht VA, Nabaweesi R, Arnold MA, et al. Pectus bar repair of pectus excavatum in patients with connective tissue disease. J Pediatr Surg 2009;44:1812-6 [DOI] [PubMed] [Google Scholar]
- 34.Scherer LR, Arn PH, Dressel DA, et al. Surgical management of children and young adults with Marfan syndrome and pectus excavatum. J Pediatr Surg 1988;23:1169-72 [DOI] [PubMed] [Google Scholar]
- 35.Golladay ES, Char F, Mollitt DL.Children with Marfan’s syndrome and pectus excavatum. South Med J 1985;78:1319-23 [DOI] [PubMed] [Google Scholar]
- 36.Fokin AA, Steuerwald NM, Ahrens WA, Allen KE.Anatomical, histologic, and genetic characteristics of congenital chest wall deformities. Semin Thorac Cardiovasc Surg 2009;21:44-57 [DOI] [PubMed] [Google Scholar]
- 37.Sweet RH.Pectus excavatum: report of two cases successfully operated upon. Ann Surg 1944;119:922-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakaoka T, Uemura S, Yoshida T, et al. Overgrowth of costal cartilage is not the etiology of pectus excavatum. J Pediatr Surg 2010;45:2015-8 [DOI] [PubMed] [Google Scholar]
- 39.Rupprecht H, Hummer HP, Stoss H, Waldherr T.[Pathogenesis of chest wall abnormalities—electron microscopy studies and trace element analysis of rib cartilage]. Z Kinderchir 1987;42:228-9 [Article in German] [DOI] [PubMed] [Google Scholar]
- 40.Serafin J, Swiatkowski J, Majkusiak R, Nowakowski P.40-year experience in surgical treatment of congenital chest deformations—ethiopathogenesis, operative techniques and clinical results. Acta Chir Orthop Traumatol Cech 2003;70:207-13 [PubMed] [Google Scholar]
- 41.Feng J, Hu T, Liu W, et al. The biomechanical, morphologic, and histochemical properties of the costal cartilages in children with pectus excavatum. J Pediatr Surg 2001;36:1770-6 [DOI] [PubMed] [Google Scholar]
- 42.van Klaveren RJ, Morshuis WJ, Lacquet LK, et al. Congenital bronchial atresia with regional emphysema associated with pectus excavatum. Thorax 1992;47:1082-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grahame R.Heritable disorders of connective tissue. Baillieres Best Pract Res Clin Rheumatol 2000;14:345-61 [DOI] [PubMed] [Google Scholar]
- 44.Beighton P, de Paepe A, Danks D, et al. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet 1988;29:581-94 [DOI] [PubMed] [Google Scholar]
- 45.Bruckner-Tuderman L, Bruckner P.Genetic diseases of the extracellular matrix: more than just connective tissue disorders. J Mol Med (Berl) 1998;76:226-37 [DOI] [PubMed] [Google Scholar]
- 46.Cole WG.Etiology and pathogenesis of heritable connective tissue diseases. J Pediatr Orthop 1993;13:392-403 [DOI] [PubMed] [Google Scholar]
- 47.Sabatier L, Miosge N, Hubmacher D, et al. Fibrillin-3 expression in human development. Matrix Biol 2011;30:43-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao LG, Luo F, Hui RT, Zhou XL.Recent molecular biological progress in Marfan syndrome and Marfan-associated disorders. Ageing Res Rev 2010;9:363-8 [DOI] [PubMed] [Google Scholar]
- 49.Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007;81:454-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brautbar A, LeMaire SA, Franco LM, et al. FBN1 mutations in patients with descending thoracic aortic dissections. Am J Med Genet A 2010;152A:413-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dietz HC.Marfan Syndrome. In: Pagon RA, Adam MP, Bird TD, et al.GeneReviews™ [Internet]. Seattle: University of Washington, Seattle; 1993-2013 [PubMed] [Google Scholar]
- 52.Nollen GJ, Mulder BJ.What is new in the Marfan syndrome? Int J Cardiol 2004;97:103-8 [DOI] [PubMed] [Google Scholar]
- 53.Judge DP, Dietz HC.Marfan’s syndrome. Lancet 2005;366:1965-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stuart AG, Williams A.Marfan’s syndrome and the heart. Arch Dis Child 2007;92:351-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glesby MJ, Pyeritz RE.Association of mitral valve prolapse and systemic abnormalities of connective tissue. A phenotypic continuum. JAMA 1989;262:523-8 [PubMed] [Google Scholar]
- 56.Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476-85 [DOI] [PubMed] [Google Scholar]
- 57.Mortensen K, Baulmann J, Rybczynski M, et al. Augmentation index and the evolution of aortic disease in marfan-like syndromes. Am J Hypertens 2010;23:716-24 [DOI] [PubMed] [Google Scholar]
- 58.Chaffins JA.Marfan syndrome. Radiol Technol 2007;78:222-36; quiz 237-9 [PubMed] [Google Scholar]
- 59.Carta L, Smaldone S, Zilberberg L, et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem 2009;284:5630-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rybczynski M, Bernhardt AM, Rehder U, et al. The spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndrome. Am J Med Genet A 2008;146A:3157-66 [DOI] [PubMed] [Google Scholar]
- 61.Loeys B, De Backer J, Van Acker P, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat 2004;24:140-6 [DOI] [PubMed] [Google Scholar]
- 62.Frydman M.The Marfan syndrome. Isr Med Assoc J 2008;10:175-8 [PubMed] [Google Scholar]
- 63.Radonic T, de Witte P, Groenink M, et al. Critical appraisal of the revised Ghent criteria for diagnosis of Marfan syndrome. Clin Genet 2011;80:346-53 [DOI] [PubMed] [Google Scholar]
- 64.Arn PH, Scherer LR, Haller JA, Jr., Pyeritz RE.Outcome of pectus excavatum in patients with Marfan syndrome and in the general population. J Pediatr 1989;115:954-8 [DOI] [PubMed] [Google Scholar]
- 65.Croitoru DP, Kelly RE, Jr., Goretsky MJ, et al. Experience and modification update for the minimally invasive Nuss technique for pectus excavatum repair in 303 patients. J Pediatr Surg 2002;37:437-45 [DOI] [PubMed] [Google Scholar]