

Case Summary
A female presented in infancy with hypotonia, undetectable serum glucose, lactic acidosis, and triglycerides > 5,000 mg/dl. The diagnosis of type 1A glycogen storage disease (GSD) was made by liver biopsy that showed increased glycogen and absent glucose-6-phosphatase enzyme activity. She was treated with dextrose feeding, which was replaced by frequent cornstarch feeding, with improvement of her metabolic parameters. At age 18 years she had marked hypertriglyceridemia (3,860 mg/dl) and eruptive xanthomas, and was treated with fenofibrate, atorvastatin, and fish oil. At age 29 years she was noted to have multiple liver adenomas, severe anemia, and hyperuricemia. Aggressive cornstarch therapy was commenced with a goal of maintaining her blood glucose levels > 75 mg/dl and lactate levels < 2 mmol/L. After 15 months on this regimen, her lipids levels (measured in mg/dl) off all medications were: total cholesterol 222, triglycerides 179, high density lipoprotein cholesterol 32, and calculated low density lipoprotein cholesterol 154. Her weight was stable with a body mass index of 24.8 kg/m2. Her liver adenomas had decreased in size, and her anemia and hyperuricemia had improved. She was homozygous for the R83C missense mutation in G6PC. Our data indicate that optimized metabolic control to maintain blood glucose levels > 75 mg/dl is critical in the management of this disease.
Key Words and Abbreviations: Glycogen storage disease type Ia, high density lipoproteins (HDL), low density lipoproteins (LDL), small dense LDL (sdLDL)
Case Report
A 31-year old woman had type Ia glycogen storage disease (GSD). She was the product of a normal gestation and weighed 6 pounds, 8 ounces at birth. She initially thrived, but required multiple formula changes due to feeding intolerance. At six months of age she was noted to have hypotonia and developmental delay. At age 7 months she was hospitalized for lethargy during a gastrointestinal illness. At 9 months of age she was documented to have an undetectable serum glucose level, lactic acidosis, plasma cholesterol levels > 500 mg/dl and plasma triglyceride levels > 5,000 mg/dl at a medical center in Washington DC. The patient had no glycemic response to a glucagon stimulation test. Glycogen storage disease type Ia was confirmed by a liver biopsy that revealed increased liver glycogen at 13% of wet weight and absent liver glucose-6-phosphatase enzyme activity.
She was placed on a treatment regimen with dextrose feeding every 3 hours during the day and a 10% dextrose nasogastric continuous infusion overnight. Despite significant clinical improvement, she continued to have lactic acidosis and suboptimal metabolic control, with documented blood glucose levels < 50 mg/dl. At age 3 years she was transitioned to cornstarch therapy every 3 hours during the day. At age 6 years nocturnal cornstarch administered every 4 hours was added. Over the ensuing years she has struggled with intermittent hypoglycemia, lactic acidosis, and significant hyperlipidemia. At age 18 years she was found to have a triglyceride level of 3,860 mg/dl and eruptive xanthomas, and was placed on fenofibrate therapy, to which atorvastatin was added to further improve her lipid control. She continued with cornstarch therapy during the day and night.
At age 26 years she moved to Boston and was referred to our clinic. At the time of her first visit, after an overnight fast, her blood glucose was 21 mg/dl, and at the time of blood drawing was completely alert, and had plasma triglyceride levels of 712 mg/dl. Her regimen was modified to include atorvastatin 20 mg/day, fenofibrate 145 mg/day, and 4 fish oil capsules/day. On this regimen her triglyceride levels ranged between 181 mg/dl and 442 mg/dl, with low density lipoprotein (LDL) cholesterol levels that remained below her target level of 160 mg/dl (124-158 mg/dl), and high density lipoprotein (HDL) cholesterol levels that ranged between 29 mg/dl and 41 mg/dl.
At age 29 years she presented to an emergency room in Boston with severe pain and swelling in her left 3rd toe, and was treated initially with antibiotics and anti-inflammatory agents. She was subsequently diagnosed as having gout with an elevated uric acid of 11.3 mg/dl, and was treated with analgesic and anti-inflammatory medications and allopurinol; her symptoms subsided. At that time she was also noted to have marked anemia with a hemoglobin level of 5.7 g/dl, a hematocrit of 21.1%, and an elevated blood lactate level of 10.6 mmol/L. Upper gastrointestinal endoscopy and colonoscopy were normal. A magnetic resonance imaging (MRI) study of her abdomen showed multiple liver adenomas with 5 dominant lesions. The sizes and locations of the liver adenomas were: 1) 10 × 10 cm lesion with cystic changes and central scarring in the left lobe; 2) 12 × 10 cm multilobular and heterogenous lesion in the anterior segment of the right lobe; 3) 8.8 × 4.6 cm focal lesion in the posterior segment of the right lobe, 4) 4.3 × 3.0 cm lesion in the inferior aspect of the right lobe, and 5) 9.3 × 7.6 cm well encapsulated nodule in the inferior, medial aspect of the liver. Her serum alpha-fetoprotein level was normal. The patient had been on oral contraception for birth control and irregular menses; however this medication was discontinued since this type of therapy has been reported to stimulate the growth of liver adenomas. The patient's other medical problems include osteoporosis with a leg fracture at age 3 years, and several finger fractures during childhood and adolescence. A scan of her bones revealed significantly decreased bone density (> 2 standard deviations below normal in her spine and femurs). Oral calcium and vitamin D supplementation was started.
Due to the severity of her disease, the patient was referred to Dr. David Weinstein at the University of Florida, Gainesville, FL. The patient had been taking an empiric regimen of 2 teaspoons of cornstarch at 6AM, 10AM, and 2PM, together with 4 teaspoons at bedtime. After careful study on a metabolic ward, it was determined that these doses of cornstarch were inadequate. Her therapy was changed to a more intensive regimen of cornstarch: 40 grams at 8AM, 12:30PM, and 6PM, and 45 grams at 12:00AM and 3AM. The patient's estimated glucose requirement was about 10 grams/hour. On the above regimen, assuming complete digestion and absorption, she effectively received approximately 7.5 grams/hour during the day and 11.2 grams/hour overnight. In addition, she was placed on a diet with limited sucrose, galactose, and fructose. The patient was instructed to maintain her blood glucose levels > 75 mg/dl and began monitoring her blood glucose on a daily basis. She was able to minimize her episodes of hypoglycemia.
On this new regimen her prior intermittent headaches disappeared, and her vision became more stable using contact lenses. She had no other symptoms and her menstrual cycles became regular for the first time off oral contraceptive therapy. In addition, approximately 15 months after being on this regimen a repeat MRI of her liver showed a decrease in the size of her adenomas: 1. left lobe 8.8 × 5.4 cm versus 9.8 cm × 6.2 cm on prior exam (21.8% decrease in size), 2) hepatic segment 10.8 × 8.1 cm versus 10.8 × 9.8 cm previously (21.2% decrease in size), and inferior right lobe of the liver 7.9 × 6.4 cm versus 7.6 × 7.6 cm on prior exam (12.5% decrease in size).
There is no known family history of glycogen storage disease. Multiple relatives have hyperlipidemia and her mother is on cholesterol lowering therapy. She has a 33-year old brother who is in good health.
A physical examination most recently revealed: height 148.9 cm, weight 55 kg, with a body mass index of 24.8 kg/m2. Her weight had been quite stable for a number of years. Her blood pressure was 120/80 mmHg. Her head and neck examination was normal, as was her respiratory and cardiac examination. All her pulses were normal and there were no bruits. She had hepatomegaly with a liver edge 7 cm below the right costal margin. She did not have splenomegaly or focal tenderness or guarding in the abdomen. She had a well-healed scar below her liver. Her extremities were well perfused with no tenderness or edema. She had a tophus on the 3rd toe of her left foot with surrounding erythema. She had a normal neurologic examination.
Laboratory Analysis
At age 30 years routine clinical chemistry laboratory analysis performed in the non-fasting state on the optimized cornstarch regimen showed: plasma glucose 116 mg/dl, liver alanine (ALT) and aspartate transaminases (AST) 37 and 37 U/L, respectively (normal), alkaline phosphatase 465 U/L (elevated, normal < 126 U/L), blood urea nitrogen 9 mg/dl (normal), creatinine 0.4 mg/dl (normal), uric acid 4.4 mg/dl (normal). Her lipid profile was: total cholesterol: 255 mg/dl (elevated, normal < 200 mg/dl), triglyceride 229 (elevated, normal < 150 mg/dl), high density lipoprotein (HDL) cholesterol 28 (low, normal > 50 mg/dl), and calculated low density lipoprotein (LDL) 182 mg/dl, normal < 160 mg/dl). Her hemoglobin had increased to 10.7 g/dl (from 5.7 g/dl), hematocrit 35.2%, ferritin level 74 ng/ml (normal), and serum iron 25 ug/dl (low, normal 35-143 ug/dl).
At age 31 years she had additional studies performed at Boston Heart Diagnostics, Framingham, MA (an advanced lipid testing laboratory) in the non-fasting state, which showed: total cholesterol 266 mg/dl (elevated), triglycerides 433 mg/dl (elevated), direct LDL cholesterol 169 mg/dl (elevated), small dense LDL cholesterol > 45 mg/dl (elevated), HDL cholesterol 24 mg/dl (decreased), apolipoprotein (apo) A-I 110 mg/dl (decreased), apoB 157 mg/dl (elevated), and lipoprotein (a) 7 mg/dl (normal). The methodology for these assays is as previously described and within and between run coefficients of variation were all < 5% (1,2). HDL particle analysis by two dimensional gel electrophoresis followed by apoA-I antibody immunoblotting in mg/dl of apoA-I measured as previously described (3,4) revealed: precursor very small pre-beta-1 HDL 9.3 (normal), very small alpha-4 HDL 8.3 (normal), small alpha-3 HDL 27.9 (normal), medium alpha-2 HDL 44.1 (normal), and large HDL alpha-1 7.0 (very low, normal > 20). Plasma sterol analysis by gas liquid chromatography measured as previously described (5,6) revealed: lathosterol (marker of cholesterol synthesis) < 20 umol/mmol of cholesterol (very low and below detection), campesterol (marker of cholesterol absorption) 85 umol/mmol/cholesterol (low), and beta sitosterol (marker of cholesterol absorption) 66 umol/mmol of cholesterol (low). Within and between run coefficients of variation for these assays (sterols and HDL particles) were < 10%.
Other analyses performed in this laboratory revealed: high sensitivity C reactive protein (hsCRP) > 100 mg/L (markedly elevated), lipoprotein associated phospholipase A2 (LpPLA2) 164 ng/ml (normal), hemoglobin A1c 5.8% (normal), insulin 19.7 uU/ml (elevated), creatine kinase 79 U/L (normal), ALT 21 U/L (normal), AST 25 U/L (normal), alkaline phosphatase 457 U/L (elevated), creatinine 0.4 (normal), blood urea nitrogen 11.1 (normal), uric acid 5.2 (normal), thyroid stimulating hormone 3.75 uIU/ml (normal), and NT-pro brain natriuretic peptide < 20 pg/dl (normal). Genetic analysis revealed: apoE genotype 3/3 (normal), factor V Leiden -/- (normal), and prothrombin 20210A variant -/- (normal). Analysis of the glucose-6-phosphatase gene (G6PC) as performed at the University of Florida documented that the patient was homozygous for a C to T substitution in the G6PC gene resulting in the substitution of arginine with cysteine at position 83 of the amino acid sequence (a previously reported mutation).
Comment and Discussion
Glycogen storage disease type Ia is an autosomal recessive disorder caused by a deficiency of glucose-6-phosphatase activity (7). Mutations in the G6PC gene cause a defect in the catalytic subunit of the enzyme (8). The case reported here was homozygous for a C to T substitution in the G6PC gene resulting in the substitution of arginine with cysteine at position 83 of the amino acid sequence (a previously reported mutation) (8-10). This defect causes an inability to convert glucose-6-phosphate to glucose, resulting in abnormal flux through the glycolytic pathway leading to increased production of pyruvate, which is converted to either lactate or triglycerides via acetyl CoA and fatty acids (see figure 1 as modified and taken from reference 7) (7). Glucose-6-phosphate can also be converted to uric acid via the pentose phosphate pathway, as well as to glycogen via glucose-1-phosphate, UDP-glucose, and amylopectin (see figure 1) (7). Therefore, unless patients with type 1 GSD regularly receive adequate amounts of exogenous glucose, they rapidly develop severe hypoglycemia, marked hypertriglyceridemia, hyperuricemia, and elevated lactate levels. The marked hypertriglyceridemia in this disease is related to enhanced de novo lipogenesis due to increased conversion of glucose-6-phosphate to triglycerides as shown in figure 1 (11).
Figure 1. Overview of the Interrelationships of Glucose, Lactate, Triglyceride, Uric Acid, and Glygogen Metabolism in the Liver.
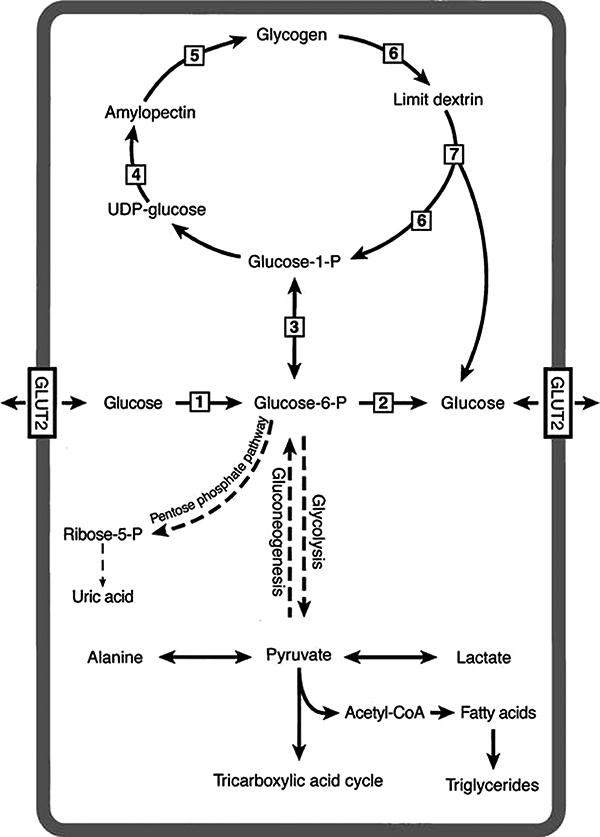
UDP-glucose is uridine diphosphoglucose. Steps in the process are catalyzed by: 1. hexokinase/glucokinase, 2. glucose-6-phosphatase (G6Pase), 3. phosphoglucomutase, 4. glycogen synthase, 5. branching enzyme, 6. glycogen phosphorylase, and 7. debranching enzyme (figure as modified from references 7 and 8). In glycogen storage disease type Ia there is a block in the conversion of glucose-6-phosphate to glucose due to a deficiency of glucose-6-phosphatase. This block results in markedly enhanced conversion of glucose-6-phosphate to: 1) to pyruvate and lactate; 2) to pyruvate and then to acetyl-CoA, fatty acids and finally to triglycerides; 3) to ribose-5-phosphate and then to uric acid; or to 4) to glucose-1-phosphate, then to UDP-glucose, then to amylopectin, and then to glycogen. The excess production of lactate, triglycerides, uric acid, and glycogen can be diminished by the administration of exogenous cornstarch (glucose polymer) on a regular and prescribed basis.
As an infant, the patient was documented to have serum triglycerides over 5,000 mg/dl, and at age 18 years she developed eruptive xanthomas with serum triglyceride levels of 3,860 mg/dl. At that time, she was receiving inadequate cornstarch therapy, and treatment with atorvastatin, fenofibrate, and fish oil maintained her triglycerides below 500 mg/dl. However, with better metabolic control that maintained her blood glucose levels > 75 mg/dl, off all medications she was able to reduce her triglyceride levels to 179 mg/dl. However, her latest value was 433 mg/dl, suggesting that her dietary regimen may not have been as optimal at the time of this latest sampling. Nevertheless, the data indicate that this form of therapy can be very effective in suppressing her de novo lipogenesis and hypertriglyceridemia, and is preferable to statin or fibrate therapy.
At the time of her latest sampling, we documented that she clearly had an elevated LDL cholesterol and small dense LDL cholesterol level. Our measurements of sterols indicated very striking suppression of cholesterol synthesis and some decrease in markers of cholesterol absorption, findings not previously reported in this disease. This suppression of cholesterol synthesis may be due to the increased shunting of fatty acids into the triglyceride pathway in this disease, and a resultant suppression of liver cholesterol production. In addition her elevated levels of apoB and LDL are probably due to delayed fractional clearance of LDL and down regulation of her hepatic LDL receptor activity. This latter effect may be due to excess liver glycogen and lipid storage, which may also account for her markedly elevated CRP level. As illustrated in this case adequate and careful supplementation with uncooked cornstarch (a slowly digested glucose polymer) results in marked improvement in triglyceride and uric acid levels, as well as improvement in liver adenomas over time.
Another clinically significant problem in these patients is anemia. The reason for the severe anemia in glycogen storage disease type Ia has now been clarified. Weinstein and colleagues have reported that patients with glycogen storage disease type Ia and large hepatic adenomas have a microcytic anemia associated with low iron levels, which is refractory to iron therapy. This anemia is linked to the presence of hepatic adenomas, and adenoma resection or liver transplantation has been associated with a return to normal hemoglobin levels (12-14). The anemia that develops in these patients appears to be an effect of aberrant expression of hepcidin by the adenomas. Hepcidin is a peptide produced in the liver that is detectable in serum and urine. Hepcidin binds and internalizes a protein known as MTP1 that facilitates iron absorption in the intestine, rendering it dysfunctional (15). A dramatic increase in hepcidin mRNA expression has been observed in glycogen storage disease patients with adenomas compared with other patients who had unaffected liver tissue. Our patient's severe anemia was probably related to her liver adenomas. More optimal metabolic control decreased her adenoma size, probably decreased hepcidin levels, and significantly decreased the severity of her anemia. Confirmation for this concept of the role of liver hepcidin comes from studies in mice in which hepcidin gene expression has been knocked out or where human hepcidin has been overexpressed using transgenic mice (15).
There are 14 types of glycogen storage diseases, which have been classified based on their enzyme deficiencies (see figure 1). Hyperlipidemia is seen in types 0, I, III, VI and IX, but is most prominent in type Ia. In glycogen storage disease type III, VI and IX, hyperlipidemia is thought to be mainly due hypoglycemia and activation of glucose counterregulation resulting in increased fatty acid flux from adipose tissue to the liver to provide an alternative source of fuel. In contrast, in glycogen storage disease type Ia, the severe hypertriglyceridemia is due to lack of conversion of glucose-6-phosphate to glucose (step 2, figure 1), resulting in excess glucose-6-phosphate being shunted towards pyruvate, lactate, triglyceride, uric acid, and glycogen production (7).
The novel features in this case, not previously reported, are the suppression of markers of cholesterol synthesis (lathosterol) and absorption (beta-sitosterol and campesterol), presumably because of increased endogenous triglyceride production. Other novel features previously unreported include documentation of increased levels of non-HDL cholesterol, apoB, LDL cholesterol, small dense LDL cholesterol, and decreased levels of HDL cholesterol, apoA-I, and apoA-I in large alpha-1 migrating HDL, all abnormalities observed in patients with hypertriglyceridemia (her levels were 433 mg/dl at the time of these evaluations). A third set of novel features reported here are the markedly elevated CRP levels, with normal levels of LpPLA2, indicating excess lipid storage in the liver, but not in peripheral macrophages.
What is clear is that in patients with glycogen storage disease type Ia it is very important to achieve metabolic control by maintaining serum glucose levels > 75 mg/dl throughout the day and night, which improves the hypertriglyceridemia, hyperuricemia, lactic acidosis, and anemia, and decreases the size of the liver adenomas (7, 14, 17). Referral to a specialized center is recommended to optimize therapy. This rare disorder clearly demonstrates the linkage between glucose, triglyceride, lactic acid, uric acid, and glycogen metabolism.
Acknowledgments
This research was supported by P50 HL083813-01 project grant from the National Institutes of Health and contract 53-3K-06 from the United Department of Agriculture Research Service. Support for this project was also provided, in part, by the National Institutes of Health (NIH) and National Center for Research Resources (NCRR) CTSA grant k1UL1RR029890. Additional philanthropic support was provided from the Scott Miller Glycogen Storage Disease Program Fund at the University of Florida. Contributions: all authors evaluated the patient, and contributed to the concept, design, drafting and critiquing of the article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingelsson E, Schaefer EJ, Contois JH, McNamara JR, Sullivan L, Keyes MJ, Pencina MJ, Schoonmaker C, Wilson PW, D'Agostino RB, Vasan RS. Clinical utilty of different lipid measures for prediction of coronary heart disease in men and women. JAMA. 2007;298:776–785. doi: 10.1001/jama.298.7.776. [DOI] [PubMed] [Google Scholar]
- 2.Ai M, Otokozawa S, Asztalos BF, Ito Y, Nakajima K, White CC, Cupples LA, Wilson PW, Schaefer EJ. Small dense low density lipoprotein cholesterol and coronary heart disease: results from the Framingham Offspring Study. Clin Chem. 2010;56:967–76. doi: 10.1373/clinchem.2009.137489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asztalos BF, Cupples LA, Demissie S, Horvath KV, Cox CE, Batista MC, Schaefer EJ. High-density lipoprotein subpopulation profile and coronary heart disease prevalence in male participants in the Framingham Offspring Study. Arterioscler Thromb Vasc Biol. 2004;24:2181–2187. doi: 10.1161/01.ATV.0000146325.93749.a8. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol. 2010;21:289–97. doi: 10.1097/MOL.0b013e32833c1ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthan NB, Pencina M, Larocquw JM, Jacques PF, D'Agostino RB, Schaefer EJ, Lichtenstein AH. Alterations in cholesterol absorption and synthesis characterize Framingham Offspring study participants with coronary disease. J Lipid Res. 2009;50:1927–35. doi: 10.1194/jlr.P900039-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Himbergen TM, Matthan NR, Resteghini NA, Otokozawa S, Ai M, Stein EA, Jones PH, Schaefer EJ. Comparison of the effects of maximal dose atorvastatin and rosuvastatin therapy on cholesterol synthesis and absorption markers. J Lipid Res. 2009;50:730–739. doi: 10.1194/jlr.P800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Rev Endocr Metab Disord. 2003;1:95–102. doi: 10.1023/a:1021831621210. [DOI] [PubMed] [Google Scholar]
- 8.Lei KJ, Pan CJ, Liu JL, Shelly LL, Chou JY. Structure-function analysis of human glucose-6-phospahatase, the enzyme deficient in glycogen storage disease type Ia. J Bio Chem. 1995;270:11882–11886. doi: 10.1074/jbc.270.20.11882. [DOI] [PubMed] [Google Scholar]
- 9.Janecke AR, Mayatapek E, Utermann G. Molecular genetics of type 1 glycogen storage disease. Mol Genet Metab. 2001;73:117–25. doi: 10.1006/mgme.2001.3179. [DOI] [PubMed] [Google Scholar]
- 10.Mayatepek E, Hoffmann B, Meissner T. Inborn errors of carbohydrate metabolism. Best Practice & Research Clinical Gastroenterology. 2010;24:607–618. doi: 10.1016/j.bpg.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Bandsma RH, Prinsen BH, van Der Velden Mde S, Rake JP, Boer T, Smit GP, Reijngoud DJ, Kuipers F. Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr Res. 2008;63:702–7. doi: 10.1203/PDR.0b013e31816c9013. [DOI] [PubMed] [Google Scholar]
- 12.Weinstein DA, Roy CN, Fleming MD, Loda M, Wolfsdorf JI, Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood. 2002;100:3776–3781. doi: 10.1182/blood-2002-04-1260. [DOI] [PubMed] [Google Scholar]
- 13.Mean RT., Jr Hepcidin and anaemia. Blood reviews. 2004;18:219–225. doi: 10.1016/S0268-960X(03)00066-3. [DOI] [PubMed] [Google Scholar]
- 14.Haruni FI. Hepcidin and the anemia of chronic disease. Annals of Clinical & Laboratory Science. 2006;36:3–6. [PubMed] [Google Scholar]
- 15.Nicolas G, Bennoun M, Porteu A. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci U S A. 2002;99:4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernier AV, Sentner CP, Correia CE, Theriaque DW, Shuster JJ, Smit GP, Weinstein DA. Hyperlipidemia in glycogen storage disease type III: effect of age and metabolic control. J Inherit Metab Dis. 2008;6:729–32. doi: 10.1007/s10545-008-0919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang DQ, Fiske LM, Carreras CT, Weinstein DA. Natural history of hepatocellular adenoma formation in glycogen storage disease Type I. J Pediatr. 2011;159:442–6. doi: 10.1016/j.jpeds.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]