

Introduction
In the future, when looking back to the latter half of the 20th century, the appearance of targeted therapies may be seen as the beginning of the end for some diseases. Such therapeutic innovations have strongly impacted certain types of diseases - those that have a pathological vector which demonstrates moderate to low variability and is clearly distinguished from normal healthy tissue. However, neoplastic and viral diseases have presented a demonstrably greater challenge than other types, such as infective or parasitic conditions, due partly to inherent adaptivity and to lack of disease-specific markers. The challenge to uncover target specificity in these and other diseases has been met by technological advances in molecular analysis and methodologies with particular emphasis recently on high throughput, data-heavy gene and protein screening protocols.
The B-cell lymphoma-2 (Bcl-2) family of proteins was first identified as the eponymous Bcl-2 protein in the early 1980's using recently developed DNA restriction analysis techniques (Erikson et al. 1984; Tsujimoto et al. 1984). The genetic translocation causing aberrant activity of the bcl-2 gene was found in the lymphocytes of patients with B-cell neoplasms and was subsequently observed to be broadly related to incidence of leukemia and lymphoma. Since then, the number and function of the Bcl-2 family of proteins has grown and continues to expand. An intricate mechanism mediating apoptosis was revealed for this family of pleotropic yet structurally related and directly interacting protein molecules. As a clear checkpoint in apoptosis related to cancer cell survival, the Bcl-2 family has raised a great deal of interest in the drug development community. This review introduces Bcl-2 function, then deals in more detail with the functional suppression of Bcl-2 by small molecule drugs, touches on the employment of Bcl-2 inhibition in tumor related angiogenesis and finally examines potential links between the choice of molecular models for drug design and eventual drug activity. Due to the continual appearance of new Bcl-2 inhibitory compounds, this review does not supply an exhaustive list of all the new experimental compounds. This review does, however, discuss in some detail the drugs under clinical trial and the experimental compounds with substantial in vitro and in vivo non-clinical data, highlighting rather unexpected findings of effects of Bcl-2 inhibition on tumor angiogenesis.
The Bcl-2 family
It is beyond the scope of the current review to fully discuss Bcl-2 family structure and function and a number of excellent reviews cover that area (Petros et al. 2004; Youle and Strasser. 2008). However an outline of Bcl-2 family structure and co-operative interactions will help understanding of Bcl-2 inhibitory drug effects discussed later. Bcl-2 was discovered after observation of frequent (14;18) gene translocation in follicular lymphoma and was shown to powerfully promote survival separate from proliferation in hematopoietic cell lines (Erikson et al. 1984; Tsujimoto et al. 1984; Vaux et al. 1988). It conferred a resistance to apoptosis that aided malignant transformation and that cancer cells exploited for survival (Croce et al. 1983; Hockenbery et al. 1991). Study of Bcl-2 expression and function revealed a larger family of structurally related proteins in control of mitochondrial directed apoptosis.
Bcl-2 itself is a 26 kDa protein consisting of five domains, four Bcl-2 homology (BH) domains and a transmembrane domain (Brunelle and Letai. 2009). These domains are shared to varying extent by the other Bcl-2 family members displaying different levels of homology to Bcl-2 and define the functional subgroup within the Bcl-2 apoptotic control mechanism. Anti-apoptotic proteins are represented primarily by Bcl-2, Bcl-xL, Mcl-1 and Bfl-1/A1 which generally bear all four BH domains, although the BH4 domain is not always present on Mcl-1 and Bfl-1 (Brunelle and Letai. 2009). Pro-apoptotic family members are divided into multi-domain ‘effectors’ such as Bax, Bak and Bok, which bear up to 3 BH domains and are pore forming proteins associated with mitochondria. Also pro-apoptotic are the BH3-only ‘facilitators’, such as Bid, Bad, Bim, Bik, NOXA and PUMA which variously inhibit the function of the anti-apoptotic members or promote that of the multi-domain pro-apoptotic members (Letai. 2008; Skommer et al. 2007; Youle and Strasser. 2008).
Despite great variation in protein sequence the Bcl-2 family multi-domain proteins, both pro- and anti-apoptotic, display a preserved secondary structure composed of a central hydrophobic helix surrounded by five or six amphipathic helices (Lama and Sankararamakrishnan. 2010). The hydrophobic groove acts as a binding site for the BH3 domains of other BH3 domain bearing family members (Kim et al. 2006; Lama and Sankararamakrishnan. 2010; Skommer et al. 2007). Despite three-dimensional structural homology of the BH domains within the Bcl-2 family it is remarkable that the hydrophobic groove has paradoxically conserved structural variation dividing pro-apoptotic and anti-apoptotic Bcl-2-like proteins. Specifically, it generally appears to be wider in the pro-apoptotic proteins than in the anti-apoptotic Bcl-2 family members (Lama and Sankararamakrishnan. 2010). With the exception of Bid, the BH3-only proteins are far more structurally diverse than the multi-BH domain Bcl-2-like proteins (Youle and Strasser. 2008). Bid alone resembles the multi-BH domain molecules and bears a hydrophobic groove resembling those of Bax and Bak in size, it is able to form multi-mer structures and also pores (Lama and Sankararamakrishnan. 2010; Youle and Strasser. 2008). This BH/BH-binding domain interaction is the primary control event for the Bcl-2 family mediated apoptotic balance and one that is exploited as a target by the small molecule inhibitors of Bcl-2 (Zeitlin et al. 2008). So despite strong familial similarity in the BH domains the prevalence of variation in the BH binding site within the Bcl-2 family may be expected to require equivalent variations in molecules designed to prevent the BH-BH-binding domain interaction, i.e. potential inhibitor drugs. Later we will discuss the concept that the final potency and specificity of a drug, modulating a protein-protein interaction, being dependent upon the initial choice of target protein and partners used for screening, binding assays, and structure-based design.
Apoptotic Bcl-2 control points
The mitochondrial apoptosis pathway is initiated by developmental signals or physiological stress (Dewson and Kluck. 2009; Reed. 2000; Youle and Strasser. 2008). These signals lead to activation of the Bcl-2 system and ultimately to homo- or hetero-oligomerization of Bax and Bak which form a pore within the mitochondrial outer membrane (Dewson and Kluck. 2009). This mitochondrial outer membrane pore formation results in damage to the mitochondrial membrane and release of apoptogenic mediators such as cytochrome C and Smac/Diablo which in turn activate caspase 3 and caspase 9 (Dewson and Kluck. 2009). Recent innovations in large scale protein mapping demonstrate that this caspase activation results in the cleavage and destruction of a wide variety of intracellular proteins, ultimately resulting in cell death (Dix et al. 2008).
Anti-apoptotic Bcl-2 and Bcl-xL are primarily associated with mitochondria but are also active in the endoplasmic reticulum where they play a key role in control of calcium release (Krajewski et al. 1993; Szegezdi et al. 2009; Youle and Strasser. 2008). Pro-apoptotic Bax and Bak are also primarily associated with mitochondria yet associate with the endoplasmic reticulum and may act as direct inhibitors of Bcl-2 and Bcl-xL at that site (Szegezdi et al. 2009; Youle and Strasser. 2008). Indeed calcium release from the endoplasmic reticulum is considered a key event during initiation of apoptosis which may involve Bcl-2 family regulation separate from the mitochondrial pathway (Szegezdi et al. 2009). Both Bcl-2 and Bcl-xL are also present in the nucleus performing roles related to both apoptotic function and cell cycle progression (Krajewski et al. 1993; Schmitt et al. 2007). Although Bid is able to insert into lipid membranes the other BH3-only proteins are generally located in the cytoplasm.
Bcl-2 family interactions at the mitochondria
There continues to be some debate as to the exact nature of the interactions between members of the Bcl-2 family at the mitochondrial outer membrane. Although the anti-apoptotic Bcl-2, Bcl-xL, Mcl-1 and others may inhibit Bax and Bak mediated mitochondrial permeabilization directly via BH3 domain/hydrophobic groove interactions, there continues to be discussion over the importance of this interaction during actual apoptotic events. One model, the ‘neutralization’ model, dictates that inhibition of Bax activity by binding of Bcl-xL for example is reversed by intervention of the BH3-only proteins inhibiting Bcl-xL and thus indirectly allowing Bax/Bak pore formation and apoptosis (Chipuk and Green. 2008; Letai. 2008). Indeed apoptosis has been demonstrated in the absence of the BH3-only proteins Bid and Bim, the primary proposed activators of Bax and Bak pore formation (Willis et al. 2007). A second model, the ‘sensitizer’ model ascribes more complex control to the BH3-only proteins such that Bim and Bid are direct activators of Bax and Bak, inducing permissive conformational changes, whilst BH3-only proteins such as Bad, Bik, Noxa, Hrk and Bmf inhibit the anti-apoptotic proteins thus inducing sensitivity of the Bax/Bak pore forming mechanism to activation by Bim and Bid (Chipuk and Green. 2008; Letai. 2008; Youle and Strasser. 2008).
Notably, the complexity of Bcl-2 family interactions is such that a specific protocol for comparison has been developed for analysis of anti-Bcl-2 family proteins, BH3-profiling, in which the efficacy of BH3-only proteins is assessed for inhibition of binding to a range of Bcl-like proteins (Certo et al. 2006; Goldsmith et al. 2010). The authors show predictive capacity for this method for determining cancer cell sensitivity to small molecule inhibitors of Bcl-2. Interestingly, in a similar approach, a cell-free comparative screen was also developed for small molecule inhibitor binding to Bcl-2 anti-apoptotic family ligands in competition with a labeled Bid BH3 peptide fragment (Zhai et al. 2006). These fine intricacies illustrate the problems associated with development of any new inhibitor of Bcl-2-family interactions or even the interpretation of results obtained from existing ones.
Bcl-2 and tumor angiogenesis
Bcl-2 inhibition may act directly on tumor cells but may also target the nutrient supply to the tumor by disrupting the blood vessels that form around, and within, the tumor (Fig. 1). The effect of Bcl-2 on endothelial cells and angiogenic events is one that is becoming more recognized in recent years. Angiogenesis is the growth of new blood vessels from pre-existing vasculature and is an absolute requirement for tumor survival and expansion (Folkman. 1972). Over ten years ago Bcl-2 was shown to be upregulated in endothelial cells exposed to VEGF and that upregulation of Bcl-2 in these cells was sufficient to enhance tumor angiogenesis and tumor growth (Nor et al. 1999; Nor et al. 2001). More recently, it was demonstrated that Bcl-2 functions as a pro-angiogenic signaling molecule in endothelial cells through a pathway that involves activation of the canonical NF-kB pathways resulting in upregulation of the angiogenic chemokines CXCL1 and CXCL8 (Karl et al. 2005). Notably, Bcl-2 also activates the STAT3 signaling pathway in endothelial cells resulting in upregulation of VEGF secretion and induction of Bcl-2 expression in tumor cells via VEGFR1 (Kaneko et al. 2007). Of the Bcl-2 family, Bfl-1/A1 has also demonstrated importance as a mediator for endothelial protection. However, this appears to be transient and related to inflammatory response rather than tumor angiogenesis (Gerber et al. 1998; Karsan et al. 1996).
Figure 1.
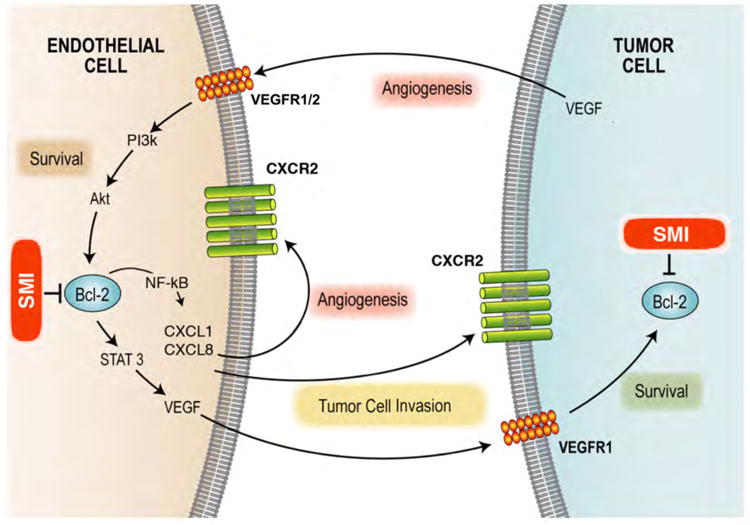
Non-Peptidic Small molecule Inhibitors of Bcl-2
Over the last decade there has been a great deal of interest in the therapeutic potential of modulating the Bcl-2 pathway towards cell death for specific pathological conditions. To this end a variety of routes have been investigated including antisense oligonucleotide drugs (Oblimersen, G3139, Genensense, Genta Inc. Berkeley Heights, NJ) and novel bioavailable peptide drugs (Gavathiotis et al. 2008; Liu et al. 2008). However, the small molecule inhibitors of Bcl-2 probably comprise the group of therapeutics with the biggest membership. Encouraged by molecular models such as the ‘sensitizer’ model of Bcl-2 control predicting that a removal of Bax/Bak inhibition may lead to apoptotic induction, drugs that might tip that balance continue to be developed.
Nature's bounty
Gossypol/AT-101
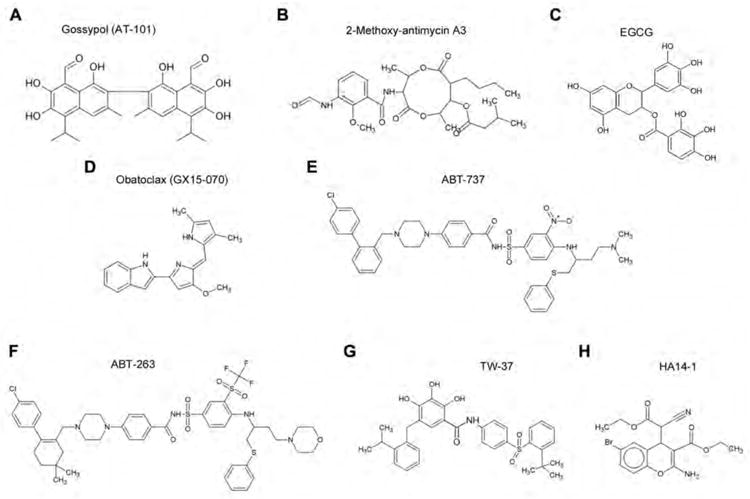
Gossypol is an orally available natural product of the cotton plant (Gossypium sp). Gossypol was previously investigated as a reversible male contraceptive, eventually undergoing trials in Brazil and China (Coutinho et al. 2000). Dose limiting toxicities were acceptable and also generally reversible. Around 20 years ago racemic gossypol was shown to inhibit growth of cancer cells and displayed limited clinical success. It was determined that the two enantiomers that composed racemic gossypol had different efficacies (Kitada et al. 2003). The observation was made that the negative enantiomer had 10-fold greater anti-proliferative capacity, and potentially a separate primary mechanism of action, compared to the positive enantiomer (Benz et al. 1990; Qiu et al. 2002). The negative enantiomer, ((-)-gossypol) has since been developed as an anti-cancer compound under the name AT-101 (Ascenta therapeutics Inc., Malvern, PA) (Figure 2A).
Figure 2.
Gossypol disrupts Bcl-2- and Bcl-xL-ligand interactions by blocking the BH3-binding site (Kitada et al. 2003). Racemic gossypol has also been shown to bind to a variety of specific targets including calcineurin, lactate dehydrogenase and glutathione-s-transferase (Baumgrass et al. 2001; Ford et al. 1991; Yu et al. 2001). Gossypol also modulates Bcl-2 family members at the genetic level in some instances, causing downregulation of anti-apoptotic proteins and upregulation of pro-apoptotic proteins (Huang et al. 2006; Meng et al. 2008; Wang et al. 2000b; Zhang et al. 2003). Indeed, AT-101 treatment induced dose dependent and time dependent increases in the Bax/Bcl-2 ratio corresponding with mitochondrial depolarization and increasing markers of apoptosis within multiple myeloma tumor cells (Kline et al. 2008). Notably however activation of apoptosis by gossypol appears to be via the direct modulation of the Bcl-2 pathway, primarily via relatively high affinity direct physical interactions and as such may be considered a true natural small molecule inhibitor of Bcl-2 (Kitada et al. 2003). Due to the wealth of information and review material regarding racemic gossypol the following section will examine the current efficacy data of AT-101, (-)-gossypol, specifically.
(-)-gossypol has been extensively examined in pre-clinical studies including prostate cancer (Huang et al. 2006; Huang et al. 2009; Meng et al. 2008; Xu et al. 2005), head and neck cancer (Oliver et al. 2004; Wolter et al. 2006), multiple myeloma (Kline et al. 2008), leukemia (Balakrishnan et al. 2009) and lymphoma models (Mohammad et al. 2005; Paoluzzi et al. 2008). (-)-gossypol displayed dose dependent anti-proliferative effects in 10 head and neck squamous cell carcinoma lines, some of which were cisplatin resistant (Oliver et al. 2004). At therapeutically relevant concentrations (i.e. 2.5-10 μmol/L) (-)-gossypol inhibited the cancer cell growth whilst human fibroblast lines were markedly less sensitive to growth inhibition (Oliver et al. 2004) (Oliver et al 2005). A follow-up study using two of these lines, UM-SCC-1 and UM-SCC-17B, in an in vivo murine model indicated that doses of 5 mg/kg and 15 mg/kg (-)-gossypol were sufficient to significantly inhibit tumor growth (Wolter et al. 2006). Interestingly the drug effects were persistent in several tumors after withdrawal of treatment with marked growth suppression lasting for two weeks before abrupt and virtually simultaneous relapse (Wolter et al. 2006). These pre-clinical observations are encouraging for potential clinical trials of AT-101 in head and neck cancer as high expression of Bcl-xL has been linked to poor prognosis in clinical disease (Kumar et al. 2008).
On the basis that Bcl-2 and Bcl-xL are overexpressed in, variously, 80% to 100% of hormone refractory prostate cancers and that this overexpression appears related to poor prognosis (-)-gossypol was tested in combination with radiation treatment in a human prostate cancer line, PC-3 (Xu et al. 2005). (-)-gossypol was an effective radiosensitizer at all doses tested in a colony formation assay and displayed synergic enhancement of the radiation induced colony growth inhibition at doses of 1-5 μmol/L (Xu et al. 2005). (-)-gossypol also inhibited the in vitro invasion and migration of a metastatic prostate cancer cell line isolated from rat lungs (Huang et al. 2006; Huang et al. 2009). Clinically, 30 mg/p.o./q.d. AT-101 for 21 days has been well tolerated in treatment of castrate-resistant prostate cancer with modest clinical response (Liu et al. 2009). Including this study there are four phase I/II trials of AT-101 including combination studies with prednisone and docetaxel or with the anti-androgen bicalutamide (ClinicalTrials.gov). Three phase I and phase II clinical trials for AT-101 in B-cell malignancies have also been constructed with two completed (ClinicalTrials.gov). These cancers have been comprehensively studied for Bcl-2 inhibitory chemotherapeutics due to a general dependence on Bcl-2 for functional survival of these tumor cells. Early in the development of the drug, (-)-gossypol was shown to inhibit growth of a diffuse large-cell lymphoma (DLBCL) line in vitro and to attenuate tumor growth in vivo in combination with a standard CHOP (cyclophosphamide-Adriamycin-vincristine-prednisolone) regime (Mohammad et al. 2005). In combination with carfilzomib (proteosome inhibitor), etoposide (topoisomerase inhibitor), doxorubicin (DNA intercalating antibiotic) or the DNA alkylating agent 4-hydroperoxyxcyclophosphamide (4-HC), AT-101 caused variously synergic growth inhibition or significantly increased apoptosis of DLBCL and mantle cell lymphoma cell lines (Paoluzzi et al. 2008). Notably, antagonism was observed for AT-101 with bortezomib, a second proteosome inhibitor, but also with 4-HC when given simultaneously with AT-101 instead of after AT-101 pre-treatment (Paoluzzi et al. 2008). The effect of drug administration schedule on the response, which can range from simple antagonism to additivity or to synergy, is a common theme throughout the functional class of small molecule inhibitors of Bcl-2.
Antimycin A
The case for other natural small molecule inhibitors of Bcl-2 is less clear. Antimycin A was first isolated from the bacteria streptomyces and is noted as a potent inhibitor of mitochondrial respiration. Specifically antimycin A targets and disrupts function of complex III of the respiratory chain (Guidarelli et al. 1997). Resulting from an observation of apoptotic sensitivity to antimycin in cells over expressing Bcl-xL, almost incidentally, one group discovered that antimycin bound directly to Bcl-xL and also Bcl-2 (Kim et al. 2001; Tzung et al. 2001). Indeed mutation of the hydrophobic BH3-binding site on Bcl-xL attenuated the cytotoxic efficacy of antimycin A on TAMH murine hepatocytes thus reinforcing the role of antimycin as a pro-apoptotic agent acting directly through Bcl-2 protein binding (Manion et al. 2004). The early serendipitous discovery of antimycin A selective binding to Bcl-2-like proteins led to development of antimycin analogues lacking respiratory chain toxicity but maintaining Bcl-2 targeted pro-apoptotic potential. Within the cancer therapeutic literature, the primary derivative of antimycin A is 2-methoxy-antimycin and there is currently limited information on this compounds activity (Wang et al. 2005).
Currently in pre-clinical development, the 2-methoxy-antimycin A3 analogue (Figure 2B) of 2-methoxy-antimycin was effective in killing mesothelioma cancer cells preferentially over non-cancer cells at concentrations of 10 – 50 μg/ml (Cao et al. 2007). Indeed 2-methoxy-antimycin A3, administered intraperitoneally (2 mg/kg, i.p.) showed promising efficacy in vivo in combination with cisplatin (2 mg/kg) without displaying overt toxicities (Cao et al. 2007). Notably, and in contrast to gossypol, the same study showed that 2-methoxy antimycin A3 did not alter cellular expression levels of Bcl-2, Bcl-xL, Mcl-1, Bax or Bak. Interestingly, 2-methoxy antimycin A3 sensitized otherwise resistant prostate cancer cells to apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (Huerta-Yepez et al. 2004). This is clinically relevant as TRAIL is a relatively specific target for killing tumor cells in vivo, as are most small molecule inhibitors of Bcl-2, and it will be interesting to see if in vitro efficacies translate to in vivo co-operative effects. Importantly, TRAIL or drugs activating the TRAIL pathway via the death receptor are currently in clinical Phase I/II trials where they are being reasonably well tolerated and are showing some response as both single agent and in combination (Wiezorek et al. 2010).
Tea polyphenols
Both green and Black tea polyphenols have been touted as virtual ‘cure-alls’ and amongst the proven functions of these molecules is the capacity to bind to and inhibit Bcl-2 function (Leone et al. 2003; Zhai et al. 2006). Catechins, and particularly the epigallocatechin-3-gallate constituent (Figure 2C), are the main components of the tea polyphenol family displaying anti-cancer activity in vitro and in vivo. However they bind a large variety of other apoptosis, cell cycle and cell survival related molecules with affinity similar to catechin affinity for Bcl-2 (Ahmad et al. 2002; Patra et al. 2008; Peng et al. 2006; Pianetti et al. 2002; Sakata et al. 2004; Tachibana et al. 2004). Thus, despite a wealth of data linking catechins and Bcl-2 and despite the clear potential and utility of green tea polyphenols as anticancer therapeutics implicated by a large volume of literature, this class of compounds will not be considered further in the review as true specific small molecule inhibitors of Bcl-2.
Products of the Laboratory
Obatoclax
Nature often informs science and provides a foothold for drug development. A candidate for earliest patented small molecule inhibitor of Bcl-2 is Obatoclax (GX15-070) (Figure 2D) developed by Gemin X Biotechnologies Inc. from derivatives of the bacterially derived streptorubin B (Daïri et al. 2007; Shore and Viallet. 2005). Despite undergoing several clinical trials there has been limited academic literature available on this compound other than in abstracted form until the last two to three years. This most recent period has seen a rapid burst of literature about this drug. Obatoclax first demonstrated pro-apoptotic capacity in the literature in isolated clinical samples from patients with chronic lymphocytic leukemia with activity comparable to HA14-1, an established small molecule inhibitor of Bcl-2, however no mention of mechanism was made in this study (Campas et al. 2006). Obatoclax is described as a pan-Bcl-2 inhibitor with inhibitory activity against all the pro-survival Bcl-2-family proteins (Zhai et al. 2006). Perhaps in light of results obtained from ABT-737, a well documented small molecule Bcl-2 inhibitor discussed below, several studies specifically focused on obatoclax inhibition of Mcl-1 function. Obatoclax disrupted Mcl-1 inhibition of Bak in cell free systems and induced activation of Bax in isolated mitochondria (Nguyen et al. 2007; Smoot et al. 2010). Additionally, the drug induced cytotoxicity and attenuated Bcl-2, Mcl-1 and Bcl-xL sequestration of Bak, Bax and Bim under a variety of conditions in mantle cell lymphoma, chronic lymphocytic leukemia, acute myeloid leukemia, non-small cell lung cancer and cholangiocarcinoma cell lines (Konopleva et al. 2008; Li et al. 2008; Perez-Galan et al. 2007; Perez-Galan et al. 2008; Smoot et al. 2010). Notably, in a comparative analysis of several mantle cell lymphoma cell lines, sensitivity to obatoclax mediated cytotoxicity seems related to Bcl-2 expression level (Perez-Galan et al. 2007). A later study by this group indicated even more specifically that drug activity was closely related to Bcl-2 phosphorylation state. Whilst activity of obatoclax is within the mid nanomolar to mid micromolar range for these cells, and in fact most cancer cells investigated, resistance to the drug due to overexpression of Bcl-2 may be a potential problem in clinical therapy of similar cancers. The authors of the previous study suggested that co-administration of inhibitors of the ERK kinase pathway, which reduced Bcl-2 phosphorylation and concomitantly increased obatoclax efficacy, may be one solution to this problem (Perez-Galan et al. 2008).
Obatoclax (1.5 mg/kg/d via arterial infusion) has shown single agent activity in vivo in a syngeneic rat orthotopic model of cholangiocarcinoma where it significantly increased survival times (Smoot et al. 2010). There does not appear to be a great deal more information regarding obatoclax in vivo in the available literature. In the clinic however, phase I and phase II trials are underway with some results published for obatoclax treatment of solid tumor malignancies, refractory leukemia and myelodysplasia, and also advanced chronic lymphocytic leukemia (O'Brien et al. 2009; Paik et al. 2010; Schimmer et al. 2008). In total however there are 16 clinical trials in the US with obatoclax directed at leukemia, lymphoma, myeloma and lung cancers (www.clinicaltrials.gov). As single agent, doses up to 40 mg/m3 were administered, although doses of up to 28 mg/m3 were well tolerated and recommended for therapeutic application (O'Brien et al. 2009; Schimmer et al. 2008). In conjunction with topoisomerase inhibitor topotecan administration obatoclax was tolerated up to 14 mg/m3 administered intravenously on days 1 and 3 weekly during a three week treatment (Paik et al. 2010). In all published cases, modest but encouraging clinical activity was observed for obatoclax.
ABT-737
ABT-737 (Figure 2E), mentioned above, is a small molecule inhibitor of Bcl-2 developed by Abbot Laboratories. It has specificity and nanomolar affinity for all the major pro-survival Bcl-2 family members except Mcl-1 and Bfl1/A1 (Oltersdorf et al. 2005; Wendt et al. 2006; Zhai et al. 2006). Basic research was performed on this compound however it was not found to be orally available. This led to development of a structurally similar yet orally available compound ABT-263 (Park et al. 2008; Tse et al. 2008). ABT-737 showed single agent regression of a solid tumor in xenograft models but due to low binding capacity for Mcl-1 or Bfl1/A1 it was poorly effective in treatment of tumor cells, or in vivo xenografts, over-expressing these molecules (Chen et al. 2007; Konopleva et al. 2006; Oltersdorf et al. 2005; van Delft et al. 2006). Indeed a number of studies have been published, with a view to potential therapeutic co-administration, showing potentiation of ABT-737 activity with downregulation of Mcl-1 expression, via RNA inhibition, or functional neutralization of the molecule with the cyclin dependent kinase inhibitor Seliciclib, the protein synthesis inhibitor cycloheximide, the DNA alkylating agent Melphalan or inhibitors of the MEK/ERK kinase pathway (Chen et al. 2007; Konopleva et al. 2006; Trudel et al. 2007; van Delft et al. 2006). However, it is important to note that at least one study indicated that efficacy of ABT-737 was Mcl-1 independent in newly isolated chronic lymphocytic leukemia cells grown under conditions mimicking the lymph node (Vogler et al. 2009). This report suggested that, whilst circulating cells may be sensitive to Bcl-2 inhibition, cells entering lymph nodes may escape death due to pro-survival signals from stromal cells in the lymph node microenvironment. In that study, survival signals resulted in rapid and massive upregulation of Bcl-2 and moderate upregulation of Bcl-xL in the cancer cells (Vogler et al. 2009). It should be noted that some argument exists over the nature of stimulation from stromal cells in in vitro or ex vivo models but even the possibility of drug resistance via this mechanism would have potentially important implications for clinical use of all pharmacological inhibitors of Bcl-2.
As seen with obatoclax, phosphorylation of Bcl-2 resulted in loss of efficacy of ABT-737 (Konopleva et al. 2006). Mechanism of action of ABT-737, and presumably potentially, also ABT-263 appears to involve displacement of Bcl-2 from Bax and Bak dependent activation of apoptosis with no requirement for Bim in this process (Konopleva et al. 2006). The study showed that although the drug indeed inhibited Bcl-2/Bim interactions, knockdown of Bim isoforms in HL-60 human leukemic cells did not affect the efficacy of ABT-737. Interestingly, a separate study looking at chronic lymphocytic leukemia cells indicated that displacement of Bim from Bcl-2 was essential to ABT-737 mediated apoptosis as it allowed Bim to then activate Bax resulting in mitochondrial release of pro-apoptotic signals (Del Gaizo Moore et al. 2007). As Bim has been postulated to act as a direct activator of Bak/Bax mediated mitochondrial depolarization and apoptosis induction these observations would perhaps suggest that the mechanism tipping the balance towards the Bcl-2 pro-apoptotic side may argue against the sensitizer model of Bcl-2 family signaling described in the introductory sections of this review. ABT-737 concentrations ranging from low nanomolar to low micromolar were effective in vitro against small-cell lung cancer, myeloid leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, lymphoma lines in vitro (Del Gaizo Moore et al. 2007; Kline et al. 2007; Konopleva et al. 2006; Lock et al. 2008; Trudel et al. 2007).
ABT-263
Notably whilst ABT-737 has not progressed to the clinic, its analogue ABT-263 (Figure 2F) has despite sharing a particularly low affinity for Mcl-1 (Lock et al. 2008) (Clinicaltrials.gov). ABT-263 has entered 15 clinical trials (Clinicaltrials.gov). These include studies on ABT-263 efficacy in chronic lymphocytic leukemia, lymphoma, small-cell lung cancer and solid tumor. The trials test the drug as single agent but also in combination with erlotinib (inhibitor of epidermal growth factor tyrosine kinase receptor), irinotecan (topoisomerase I inhibitor), ketoconazole (anti-fungal), fludarabine (purine analogue), anti-CD20 antibody rituximab, cyclophosphamide or bendamustine (alkylating agents), gemticabine (DNA synthesis inhibitor), DNA intercalating carboplatin, paclitaxel or docetaxel (mitotic inhibitors) and etoposide combined with cisplatin (Clinicaltrials.gov). Applied to in vivo xenograft models, ABT-263 displayed significant and even complete regression of small-cell lung cancer and acute lymphoblastic leukemia at doses of 100 mg/kg/d p.o. q.d. for 17-21 days (Shoemaker et al. 2008; Tse et al. 2008). In some of these experiments a persistence of inhibition remained for several weeks after cessation of treatment (Shoemaker et al. 2008). These observations are similar to the marked persistent effects of (-)-gossypol on tumor suppression after end of treatment (Wolter et al. 2006).
In some models of B-cell lymphoma and multiple myeloma the drug had little effect as single agent but significantly potentiated the effect of rituximab, CHOP regimen (common chemotherapeutics used clinically in lymphoma patients), rapamycin or bortezomib (Ackler et al. 2008; Tse et al. 2008). An in vitro and in vivo study of a large panel of tumor cell lines from 9 separate lineages indicated that sensitivity to ABT-263 was related to lower Mcl-1 and a generally higher expression of Bcl-2, Bcl-xL, Bid and NOXA. However, drug resistance was particularly marked in all tumors with higher Bcl-w expression (Lock et al. 2008). The authors of this study concluded that ABT-263 had primary application in acute lymphoblastic leukemia, based on their observation of low efficacy in tumors of other histologies although notably, given clinical trials underway, lung cancer lines were not included in this panel. Given the limited amount of in vitro information available directly comparing ABT-737 and ABT-263 efficacies and mechanisms it will be interesting to see whether the vast literature of the parent drug will be truly applicable to the function of the clinically tested drug. Whilst the overall molecular shape and modeled molecular target fit may be very similar, the modifications made on the three separate residues that produced the orally available version may yet be found to add or remove functionality from ABT-263 compared to ABT-737 (Park et al. 2008).
TW-37
As obatoclax was derived from bacterial streptorubin B, so TW-37 (Figure 2G) was derived from the natural product of cotton (-)-gossypol (Wang et al. 2006). Also, like obatoclax, TW-37 is another example of a pan-Bcl-2 inhibitor being able to inhibit all the major members of the Bcl-2 family including Mcl-1 (Wang et al. 2006). Although derived from AT-101 it has a distinctly different structure and a lower binding efficacy for Bcl-xL (IC50 = 1.1 μM, TW-37 v. 0.48 μM, AT-101) but similar binding capacity for Bcl-2 and Mcl-1, both in the low nanomolar range (Wang et al. 2006). TW-37 is well tolerated in vivo with maximum tolerated doses of up to 40 mg/kg/d for 20 days as single agent (5 days on, 1 day off) and 20 mg/kg/d for three days in conjunction with traditional chemotherapy such as CHOP (Mohammad et al. 2007; Verhaegen et al. 2006). Examined against malignant B-cell lines and primary lymphoma patient samples TW-37 had strong inhibitory activity in the very low nanomolar to high nanomolar range (Al-Katib et al. 2009). Notably the drug had no cytotoxic effect, over similar concentration and exposure time, on normal peripheral blood lymphocytes in the same study. This observation agrees with previous data in peripheral blood mononuclear cells and also human dermal fibroblasts (Mohammad et al. 2007; Zeitlin et al. 2006). In addition to lymphocytic cancer lines, TW-37 has also shown in vitro and in vivo efficacy against head and neck cancer lines, melanoma cell lines and pancreatic cancer lines (Ashimori et al. 2009; Verhaegen et al. 2006; Wang et al. 2008; Zeitlin et al. 2006). Efficacy in vivo has been significant but still relatively modest when given as single agent however in combination with co-administered chemotherapeutics such as cisplatin, mitogen activated protein kinase (MAPK) pathway inhibitors or CHOP regimen TW-37 has provided markedly significant tumor inhibition (Ashimori et al. 2009; Mohammad et al. 2007; Verhaegen et al. 2006).
TW-37 has been shown to induce cell growth inhibition via Bcl-2 related pathways, separate from direct mitochondrial release of apoptosis signals. TW-37 induces S-phase cell cycle arrest in head and neck cells, pancreatic cancer cells and angiogenically activated endothelial cells. Notably, TW-37 at nanomolar concentrations prevented accumulation of cells in G2-phase in response to micromolar concentrations of cisplatin and instead caused S-phase accumulation (Ashimori et al. 2009). Whilst the literature appears to present mixed opinion over the effect of cell cycle arrest at various phases on the efficacy of cisplatin, in this study a concomitant significant increase in apoptosis was observed for cell populations accumulating in S-phase after treatment with TW-37 and cisplatin (Ashimori et al. 2009). These effects, seen in cancer cells, were mirrored in proliferating endothelial cells (Ashimori et al. 2009).
An interesting mechanistic reason was discovered for use of TW-37 in the treatment of certain melanomas. Verhaegen and colleagues noted that melanomas with upregulated MAPK pathway activity are resistant to some traditional chemotherapeutics and that MAPK pathway inhibition may constitute a secondary line of attack. However, they identified aberrant Bcl-2 pathway activity as a secondary line of resistance to MAPK pathway inhibitors themselves (Verhaegen et al. 2006). They successfully used this as rationale for use of TW-37 in the synergistic induction of apoptosis in otherwise resistant melanoma lines in the presence of the MAPK pathway inhibitor (Verhaegen et al. 2006). Notably, in this study, TW-37 was also used as a tool to dissect the apoptosis pathway further revealing a novel role for MAPK in control of reactive oxygen species and subsequent pro-apoptotic p53 pathway activity (Verhaegen et al. 2006). The potential for therapeutic drugs to be used in uncovering basic mechanisms can be lost in the initial focus on acquiring efficacy data, but this is still an important application for these molecules.
HA14-1
One inhibitor of Bcl-2, which has been highly successful as an experimental tool, is HA14-1 (Figure 2H). HA14-1 is arguably the oldest published synthetic small molecule inhibitor of Bcl-2 (Wang et al. 2000a). Discovered through a screen of 193,833 compounds for affinity and goodness of fit to the BH3 binding site on Bcl-2, HA14-1 showed nanomolar affinity binding for Bcl-2 and Bcl-xL (Wang et al. 2000a). After nearly 100 publications HA14-1 has not proceeded to the clinic, but whether or not it is a clinically viable drug HA14-1 has greatly enhanced our knowledge of Bcl-2 family mechanisms and interactions.
Bcl-2 inhibitors and tumor angiogenesis
TW-37 has been shown to specifically inhibit angiogenic functions in vitro and reduce microvessel density in a tumor-free biodegradable scaffold model of angiogenesis in vivo (Zeitlin et al. 2006). Subsequently TW-37 was found to inhibit angiogenesis in tumor models in vivo resulting in significantly reduced tumor microvessel density (Ashimori et al. 2009). This is clearly in part due to increased apoptosis in the endothelial cells but may also be due to non-apoptotic mechanisms as TW-37 was found to inhibit cell migration and microvessel formation in vitro at drug concentrations well below those required to induce apoptosis (Zeitlin et al. 2006). These low dose specifically anti-angiogenic effects may inform studies examining alternative, non-MTD based, dosing regimens for Bcl-2 inhibitors. In very recent studies performed in our laboratories we have examined the therapeutic application of low concentration, metronomic dosing of TW-37 co-administered with either cisplatin or radiation (Zeitlin et al. 2010) (Imai et al 2010 submitted). This metronomic dosing regimen resulted in significant reduction in tumor growth and is encouraging for future combination studies where multiple drug toxicities may require administration of lower doses (Imai et al 2010 submitted). Notably the anti-angiogenic effect of Bcl-2 inhibition is not limited to the small molecule inhibitors of Bcl-2 but has also been reported in vitro and also in vivo in xenografted tumors derived from both melanoma and prostate cancer cells bearing anti-Bcl-2 antisense drugs (Anai et al. 2007; Del Bufalo et al. 2003). Whilst the Bcl-2 knock-down was targeted to the tumor cells in these studies the results were still seen in the endothelial fraction of the tumor. This may be due to therapeutic disruption of the pro-angiogenic chemokine crosstalk between tumor cells and tumor associated endothelial cells demonstrated in our laboratory (Kaneko et al. 2007).
The influence of choice of ligands on the drug discovery process
There is a broad range of activity in the class of drugs called “small molecule inhibitors of Bcl-2”. Whilst chance may play a part, differences in the design approaches used to create, identify or characterize the final active compound are virtually certain to have played a role in the variety of efficacies found within this class of drug. Indeed there is a remarkable diversity in structure of small molecule inhibitors of Bcl-2, with the notable exceptions of AT-101/(-)-gossypol) and its variant apogossypolone series and also ABT-737 and its two derivatives ABT-263 and A385358. Three factors narrowing specificity of the compound for the target protein species have clearly influenced the development of the final inhibitory compounds: Source, structural screen, target/displacing ligand. The latter factor is likely to introduce most variability with different Bcl-2 family proteins used as displacement ligand or binding target in the functional screening process.
In this discussion, the natural or synthetic derivation of the drugs is less important than the molecular screening process and in particular the ligand binding studies. For example gossypol was finally identified in a computer-based NMR-guided binding assay from a screen of a 50 molecule panel of natural products (Kitada et al. 2003). Notably the NMR modeling was based upon (-)-gossypol binding to Bcl-xL whilst an in vitro ligand binding screen for binding to Bcl-xL was performed with a Bad peptide as competitive ligand. On the other hand, computer based molecular docking studies indicated that Antimycin A3 and its analogues would bind directly to the hydrophobic grooves of both Bcl-2 and Bcl-xL (Kim et al. 2001; Tzung et al. 2001). However, ligand displacement studies were performed with a fluorescent BAK peptide (Kim et al. 2001; Tzung et al. 2001). As discussed above with the exception of the BH domains there is great variability in structure of Bcl-2 family proteins resulting in variability of recognition of the BH3-binding site on Bcl-2 or Bcl-xL. Thus, as expected, drugs developed to inhibit the binding of specific, different ligands to a particular binding site ultimately display different affinity and selectivity profiles. This is illustrated well in one of the studies discussed previously, comparing several small molecule inhibitors of the Bcl-2 family (Zhai et al. 2006). In that instance a peptide fragment of BH3-only Bid was used to compare a variety of small molecule inhibitors of Bcl-2. With a level playing field the assay indicated a nearly ten-fold lower inhibitory capacity for antimycin A compared to even the racemic preparation of gossypol, on the Bcl-2/Bid interaction (Zhai et al. 2006).
These differences are amplified by use of ligand binding interactions to model greater specificity in development of the synthetic inhibitors of Bcl-2. TW-37 was developed by rational design using AT-101 structural binding to the hydrophobic groove on Bcl-2 as a starting point. Computational docking was employed to model the interaction and combinatorial chemistry used to modify the original structure (Wang et al. 2006). The authors reviewed AT-101 binding and compared it to Bcl-2 BH-3-binding domain interaction with peptide fragments from Bid, Bim and Bad. TW-37 was investigated for functional affinity to Bcl-2 by both fluorescent polarization-based binding assay and immunosorbant assay (ELISA). This approach to screen for broad functional inhibition of BH3-only binding led to the development of a true pan-Bcl-2 inhibitor. In similar fashion, Obatoclax (GX15-070) was rationally designed to fit the Bcl-2 BH3-binding domain in competition with pro-apoptotic Bax (Daïri et al. 2007). It was developed using structural data obtained from the bacterial derivative Streptorubin B. Like TW-37 it is also a pan-Bcl-2 inhibitor effective against Bcl-2, Bcl-w, Bcl-xLand Mcl-1 mediated protection (Nguyen et al. 2007). In contrast, ABT-737 was developed using high-throughput NMR screen of a chemical library for best fit to the hydrophobic groove of Bcl-xL, instead of Bcl-2, followed by chemical modification to increase that fit to specifically mimic Bak binding to the groove. As has been discussed it displays a notable lack of affinity to Mcl-1 and Bfl1/A1, compared to TW-37 or Obatoclax. Although variations in affinity are impossible to ascribe solely to initial modeling and screening differences these factors remain clear and logical variables that should be noted by those interested in developing novel Bcl-2 inhibitors, or in comparing functionality of existing drugs.
Conclusion
Most of the drugs profiled in this review have good efficacy and activity at pharmacologically relevant concentrations in sensitive cancer cells. It seems clear that we can design new molecules or screen for existing molecules that perform well as inhibitors of Bcl-2 family proteins in vitro. The new challenge will be in turning these pharmacologically interesting molecules into therapeutically useful drugs. In the case of AT-101, oral bioavailability is already present. However, without this level of fortune, turning effective experimental tools into clinical weapons will commonly require further study and modification, as is evidenced by ABT-737 and its orally available progeny ABT-263. The discovery of a new molecular structure that blocks the BH3/BH3 binding site interaction is the first step up a mountain whose summit is an effective drug. Along the way we are likely to learn much more about the function of the Bcl-2 family of proteins and, in this new century, with the spread of novel technologies for analysis of protein/protein interactions we can expect those existing summits, currently few in number, to expand into a more impressive range of peaks.
Acknowledgments
Grant support: P50-CA97248 (University of Michigan Head & Neck SPORE), R01-DE14601, R01-DE15948, R01-DE16586, and R21-DE19279 from the NIH to J. Nör.
References
- Ackler S, Xiao Y, Mitten MJ, et al. ABT-263 and rapamycin act cooperatively to kill lymphoma cells in vitro and in vivo. Mol Cancer Ther. 2008;7:3265–3274. doi: 10.1158/1535-7163.MCT-08-0268. [DOI] [PubMed] [Google Scholar]
- Ahmad N, Adhami VM, Gupta S, Cheng P, Mukhtar H. Role of the retinoblastoma (pRb)-E2F/DP pathway in cancer chemopreventive effects of green tea polyphenol epigallocatechin-3-gallate. Arch Biochem Biophys. 2002;398:125–131. doi: 10.1006/abbi.2001.2704. [DOI] [PubMed] [Google Scholar]
- Al-Katib AM, Sun Y, Goustin AS, et al. SMI of Bcl-2 TW-37 is active across a spectrum of B-cell tumors irrespective of their proliferative and differentiation status. J Hematol Oncol. 2009;2:8. doi: 10.1186/1756-8722-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anai S, Goodison S, Shiverick K, Hirao Y, Brown BD, Rosser CJ. Knock-down of Bcl-2 by antisense oligodeoxynucleotides induces radiosensitization and inhibition of angiogenesis in human PC-3 prostate tumor xenografts. Mol Cancer Ther. 2007;6:101–111. doi: 10.1158/1535-7163.MCT-06-0367. [DOI] [PubMed] [Google Scholar]
- Ashimori N, Zeitlin BD, Zhang Z, et al. TW-37, a small-molecule inhibitor of Bcl-2, mediates S-phase cell cycle arrest and suppresses head and neck tumor angiogenesis. Mol Cancer Ther. 2009;8:893–903. doi: 10.1158/1535-7163.MCT-08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan K, Burger JA, Wierda WG, Gandhi V. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood. 2009;113:149–153. doi: 10.1182/blood-2008-02-138560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgrass R, Weiwad M, Erdmann F, et al. Reversible inhibition of calcineurin by the polyphenolic aldehyde gossypol. J Biol Chem. 2001;276:47914–47921. doi: 10.1074/jbc.M103273200. [DOI] [PubMed] [Google Scholar]
- Benz CC, Keniry MA, Ford JM, et al. Biochemical correlates of the antitumor and antimitochondrial properties of gossypol enantiomers. Mol Pharmacol. 1990;37:840–847. [PubMed] [Google Scholar]
- Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campas C, Cosialls AM, Barragan M, et al. Bcl-2 inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Exp Hematol. 2006;34:1663–1669. doi: 10.1016/j.exphem.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Cao X, Rodarte C, Zhang L, Morgan CD, Littlejohn J, Smythe WR. Bcl2/bcl-xL inhibitor engenders apoptosis and increases chemosensitivity in mesothelioma. Cancer Biol Ther. 2007;6:246–252. doi: 10.4161/cbt.6.2.3626. [DOI] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho EM, Athayde C, Atta G, et al. Gossypol blood levels and inhibition of spermatogenesis in men taking gossypol as a contraceptive. A multicenter, international, dose-finding study. Contraception. 2000;61:61–67. doi: 10.1016/s0010-7824(99)00117-1. [DOI] [PubMed] [Google Scholar]
- Croce CM, Thierfelder W, Erikson J, et al. Transcriptional activation of an unrearranged and untranslocated c-myc oncogene by translocation of a C lambda locus in Burkitt. Proc Natl Acad Sci U S A. 1983;80:6922–6926. doi: 10.1073/pnas.80.22.6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daïri K, Yao Y, Faley M, et al. A Scalable Process for the Synthesis of the Bcl inhibitor Obatoclax. Org Process Res Dev. 2007;11:1051–1054. [Google Scholar]
- Del Bufalo D, Trisciuoglio D, Scarsella M, Zangemeister-Wittke U, Zupi G. Treatment of melanoma cells with a bcl-2/bcl-xL antisense oligonucleotide induces antiangiogenic activity. Oncogene. 2003;22:8441–8447. doi: 10.1038/sj.onc.1206999. [DOI] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009;122:2801–2808. doi: 10.1242/jcs.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erikson J, Finan J, Tsujimoto Y, Nowell PC, Croce CM. The chromosome 14 breakpoint in neoplastic B cells with the t(11;14) translocation involves the immunoglobulin heavy chain locus. Proc Natl Acad Sci U S A. 1984;81:4144–4148. doi: 10.1073/pnas.81.13.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175:409–416. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford JM, Hait WN, Matlin SA, Benz CC. Modulation of resistance to alkylating agents in cancer cell by gossypol enantiomers. Cancer Lett. 1991;56:85–94. doi: 10.1016/0304-3835(91)90198-q. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- Goldsmith KC, Lestini BJ, Gross M, et al. BH3 response profiles from neuroblastoma mitochondria predict activity of small molecule Bcl-2 family antagonists. Cell Death Differ. 2010;17:872–882. doi: 10.1038/cdd.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidarelli A, Clementi E, Brambilla L, Cantoni O. Mechanism of the antimycin A-mediated enhancement of t-butylhydroperoxide-induced single-strand breakage in DNA. Biochem J. 1997;328(Pt 3):801–806. doi: 10.1042/bj3280801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc Natl Acad Sci U S A. 1991;88:6961–6965. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YW, Wang LS, Dowd MK, Wan PJ, Lin YC. (-)-Gossypol reduces invasiveness in metastatic prostate cancer cells. Anticancer Res. 2009;29:2179–2188. [PubMed] [Google Scholar]
- Huang YW, Wang LS, Chang HL, et al. Molecular mechanisms of (-)-gossypol-induced apoptosis in human prostate cancer cells. Anticancer Res. 2006;26:1925–1933. [PubMed] [Google Scholar]
- Huerta-Yepez S, Vega M, Jazirehi A, et al. Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-kappa B and inhibition of Bcl-xl expression. Oncogene. 2004;23:4993–5003. doi: 10.1038/sj.onc.1207655. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Zhang Z, Mantellini MG, et al. Bcl-2 orchestrates a cross-talk between endothelial and tumor cells that promotes tumor growth. Cancer Res. 2007;67:9685–9693. doi: 10.1158/0008-5472.CAN-07-1497. [DOI] [PubMed] [Google Scholar]
- Karl E, Warner K, Zeitlin B, et al. Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kappaB and CXC chemokines. Cancer Res. 2005;65:5063–5069. doi: 10.1158/0008-5472.CAN-05-0140. [DOI] [PubMed] [Google Scholar]
- Karsan A, Yee E, Kaushansky K, Harlan JM. Cloning of human Bcl-2 homologue: inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 1996;87:3089–3096. [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kim KM, Giedt CD, Basanez G, et al. Biophysical characterization of recombinant human Bcl-2 and its interactions with an inhibitory ligand, antimycin A. Biochemistry. 2001;40:4911–4922. doi: 10.1021/bi002368e. [DOI] [PubMed] [Google Scholar]
- Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- Kline MP, Rajkumar SV, Timm MM, et al. R-(-)-gossypol (AT-101) activates programmed cell death in multiple myeloma cells. Exp Hematol. 2008;36:568–576. doi: 10.1016/j.exphem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline MP, Rajkumar SV, Timm MM, et al. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21:1549–1560. doi: 10.1038/sj.leu.2404719. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Watt J, Contractor R, et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax) Cancer Res. 2008;68:3413–3420. doi: 10.1158/0008-5472.CAN-07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Kumar B, Cordell KG, D'Silva N, et al. Expression of p53 and Bcl-xL as predictive markers for larynx preservation in advanced laryngeal cancer. Arch Otolaryngol Head Neck Surg. 2008;134:363–369. doi: 10.1001/archotol.134.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lama D, Sankararamakrishnan R. Identification of core structural residues in the sequentially diverse and structurally homologous Bcl-2 family of proteins. Biochemistry. 2010;49:2574–2584. doi: 10.1021/bi100029k. [DOI] [PubMed] [Google Scholar]
- Leone M, Zhai D, Sareth S, Kitada S, Reed JC, Pellecchia M. Cancer prevention by tea polyphenols is linked to their direct inhibition of antiapoptotic Bcl-2-family proteins. Cancer Res. 2003;63:8118–8121. [PubMed] [Google Scholar]
- Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- Li J, Viallet J, Haura EB. A small molecule pan-Bcl-2 family inhibitor, GX15-070, induces apoptosis and enhances cisplatin-induced apoptosis in non-small cell lung cancer cells. Cancer Chemother Pharmacol. 2008;61:525–534. doi: 10.1007/s00280-007-0499-3. [DOI] [PubMed] [Google Scholar]
- Liu G, Kelly WK, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res. 2009;15:3172–3176. doi: 10.1158/1078-0432.CCR-08-2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Kolesar J, McNeel DG, et al. A phase I pharmacokinetic and pharmacodynamic correlative study of the antisense Bcl-2 oligonucleotide g3139, in combination with carboplatin and paclitaxel, in patients with advanced solid tumors. Clin Cancer Res. 2008;14:2732–2739. doi: 10.1158/1078-0432.CCR-07-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Carol H, Houghton PJ, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1181–1189. doi: 10.1002/pbc.21433. [DOI] [PubMed] [Google Scholar]
- Manion MK, O'Neill JW, Giedt CD, Kim KM, Zhang KY, Hockenbery DM. Bcl-XL mutations suppress cellular sensitivity to antimycin A. J Biol Chem. 2004;279:2159–2165. doi: 10.1074/jbc.M306021200. [DOI] [PubMed] [Google Scholar]
- Meng Y, Tang W, Dai Y, et al. Natural BH3 mimetic (-)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol Cancer Ther. 2008;7:2192–2202. doi: 10.1158/1535-7163.MCT-08-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad RM, Goustin AS, Aboukameel A, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 2007;13:2226–2235. doi: 10.1158/1078-0432.CCR-06-1574. [DOI] [PubMed] [Google Scholar]
- Mohammad RM, Wang S, Aboukameel A, et al. Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-X(L) [(-)-gossypol] against diffuse large cell lymphoma. Mol Cancer Ther. 2005;4:13–21. [PubMed] [Google Scholar]
- Nguyen M, Marcellus RC, Roulston A, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. P NATL ACAD SCI USA. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nor JE, Christensen J, Liu J, et al. Up-Regulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001;61:2183–2188. [PubMed] [Google Scholar]
- Nor JE, Christensen J, Mooney DJ, Polverini PJ. Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol. 1999;154:375–384. doi: 10.1016/S0002-9440(10)65284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien SM, Claxton DF, Crump M, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CL, Bauer JA, Wolter KG, et al. In vitro effects of the BH3 mimetic, (-)-gossypol, on head and neck squamous cell carcinoma cells. Clin Cancer Res. 2004;10:7757–7763. doi: 10.1158/1078-0432.CCR-04-0551. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Paik PK, Rudin CM, Brown A, et al. A phase I study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in solid tumor malignancies. Cancer Chemother Pharmacol. 2010 doi: 10.1007/s00280-010-1265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoluzzi L, Gonen M, Gardner JR, et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood. 2008;111:5350–5358. doi: 10.1182/blood-2007-12-129833. [DOI] [PubMed] [Google Scholar]
- Park CM, Bruncko M, Adickes J, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. 2008;51:6902–6915. doi: 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]
- Patra SK, Rizzi F, Silva A, Rugina DO, Bettuzzi S. Molecular targets of (-)-epigallocatechin-3-gallate (EGCG): specificity and interaction with membrane lipid rafts. J Physiol Pharmacol. 2008;59(9):217–235. [PubMed] [Google Scholar]
- Peng G, Dixon DA, Muga SJ, Smith TJ, Wargovich MJ. Green tea polyphenol (-)-epigallocatechin-3-gallate inhibits cyclooxygenase-2 expression in colon carcinogenesis. Mol Carcinog. 2006;45:309–319. doi: 10.1002/mc.20166. [DOI] [PubMed] [Google Scholar]
- Perez-Galan P, Roue G, Lopez-Guerra M, et al. BCL-2 phosphorylation modulates sensitivity to the BH3 mimetic GX15-070 (Obatoclax) and reduces its synergistic interaction with bortezomib in chronic lymphocytic leukemia cells. Leukemia. 2008;22:1712–1720. doi: 10.1038/leu.2008.175. [DOI] [PubMed] [Google Scholar]
- Perez-Galan P, Roue G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 2007;109:4441–4449. doi: 10.1182/blood-2006-07-034173. [DOI] [PubMed] [Google Scholar]
- Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Pianetti S, Guo S, Kavanagh KT, Sonenshein GE. Green tea polyphenol epigallocatechin-3 gallate inhibits Her-2/neu signaling, proliferation, and transformed phenotype of breast cancer cells. Cancer Res. 2002;62:652–655. [PubMed] [Google Scholar]
- Qiu J, Levin LR, Buck J, Reidenberg MM. Different pathways of cell killing by gossypol enantiomers. Exp Biol Med (Maywood) 2002;227:398–401. doi: 10.1177/153537020222700605. [DOI] [PubMed] [Google Scholar]
- Reed JC. Mechanisms of apoptosis. Am J Pathol. 2000;157:1415–1430. doi: 10.1016/S0002-9440(10)64779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata R, Ueno T, Nakamura T, Sakamoto M, Torimura T, Sata M. Green tea polyphenol epigallocatechin-3-gallate inhibits platelet-derived growth factor-induced proliferation of human hepatic stellate cell line LI90. J Hepatol. 2004;40:52–59. doi: 10.1016/s0168-8278(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Schimmer AD, O'Brien S, Kantarjian H, et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:8295–8301. doi: 10.1158/1078-0432.CCR-08-0999. [DOI] [PubMed] [Google Scholar]
- Schmitt E, Beauchemin M, Bertrand R. Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) during G2/M cell-cycle checkpoint. Oncogene. 2007;26:5851–5865. doi: 10.1038/sj.onc.1210396. [DOI] [PubMed] [Google Scholar]
- Shoemaker AR, Mitten MJ, Adickes J, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14:3268–3277. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- Shore GC, Viallet J. Modulating the bcl-2 family of apoptosis suppressors for potential therapeutic benefit in cancer. Hematology Am Soc Hematol Educ Program. 2005:226–230. doi: 10.1182/asheducation-2005.1.226. [DOI] [PubMed] [Google Scholar]
- Skommer J, Wlodkowic D, Deptala A. Larger than life: Mitochondria and the Bcl-2 family. Leuk Res. 2007;31:277–286. doi: 10.1016/j.leukres.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Smoot RL, Blechacz BR, Werneburg NW, et al. A Bax-mediated mechanism for obatoclax-induced apoptosis of cholangiocarcinoma cells. Cancer Res. 2010;70:1960–1969. doi: 10.1158/0008-5472.CAN-09-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941–53. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- Tachibana H, Koga K, Fujimura Y, Yamada K. A receptor for green tea polyphenol EGCG. Nat Struct Mol Biol. 2004;11:380–381. doi: 10.1038/nsmb743. [DOI] [PubMed] [Google Scholar]
- Trudel S, Stewart AK, Li Z, et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res. 2007;13:621–629. doi: 10.1158/1078-0432.CCR-06-1526. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science. 1984;224:1403–1406. doi: 10.1126/science.6610211. [DOI] [PubMed] [Google Scholar]
- Tzung SP, Kim KM, Basanez G, et al. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol. 2001;3:183–191. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Verhaegen M, Bauer JA, Martin de la Vega C, et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006;66:11348–11359. doi: 10.1158/0008-5472.CAN-06-1748. [DOI] [PubMed] [Google Scholar]
- Vogler M, Butterworth M, Majid A, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–4413. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- Wang G, Nikolovska-Coleska Z, Yang CY, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem. 2006;49:6139–6142. doi: 10.1021/jm060460o. [DOI] [PubMed] [Google Scholar]
- Wang H, Li M, Rhie JK, et al. Preclinical pharmacology of 2-methoxyantimycin A compounds as novel antitumor agents. Cancer Chemother Pharmacol. 2005;56:291–298. doi: 10.1007/s00280-004-0978-8. [DOI] [PubMed] [Google Scholar]
- Wang JL, Liu D, Zhang ZJ, et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2000a;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang J, Wong SC, et al. Cytotoxic effect of gossypol on colon carcinoma cells. Life Sci. 2000b;67:2663–2671. doi: 10.1016/s0024-3205(00)00857-2. [DOI] [PubMed] [Google Scholar]
- Wang Z, Song W, Aboukameel A, et al. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and invasion in pancreatic cancer. Int J Cancer. 2008;123:958–966. doi: 10.1002/ijc.23610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wendt MD, Shen W, Kunzer A, et al. Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopotentiation activity in vitro and in vivo. J Med Chem. 2006;49:1165–1181. doi: 10.1021/jm050754u. [DOI] [PubMed] [Google Scholar]
- Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res. 2010;16:1701–1708. doi: 10.1158/1078-0432.CCR-09-1692. [DOI] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Wang SJ, Henson BS, et al. (-)-Gossypol Inhibits Growth and Promotes Apoptosis of Human Head and Neck Squamous Cell Carcinoma in Vivo. Neoplasia. 2006;8:163–172. doi: 10.1593/neo.05691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Yang D, Wang S, et al. (-)-Gossypol enhances response to radiation therapy and results in tumor regression of human prostate cancer. Mol Cancer Ther. 2005;4:197–205. [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yu Y, Deck JA, Hunsaker LA, et al. Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem Pharmacol. 2001;62:81–89. doi: 10.1016/s0006-2952(01)00636-0. [DOI] [PubMed] [Google Scholar]
- Zeitlin BD, Spalding AC, Campos MS, et al. Metronomic Small Molecule Inhibitor of Bcl-2 (TW-37) Is Antiangiogenic and Potentiates the Antitumor Effect of Ionizing Radiation. Int J Radiat Oncol Biol Phys. 2010 doi: 10.1016/j.ijrobp.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin BD, Zeitlin IJ, Nor JE. Expanding circle of inhibition: small-molecule inhibitors of Bcl-2 as anticancer cell and antiangiogenic agents. J Clin Oncol. 2008;26:4180–4188. doi: 10.1200/JCO.2007.15.7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin BD, Joo E, Dong Z, et al. Antiangiogenic effect of TW37, a small-molecule inhibitor of Bcl-2. Cancer Res. 2006;66:8698–8706. doi: 10.1158/0008-5472.CAN-05-3691. [DOI] [PubMed] [Google Scholar]
- Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 2006;13:1419–1421. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
- Zhang M, Liu H, Guo R, et al. Molecular mechanism of gossypol-induced cell growth inhibition and cell death of HT-29 human colon carcinoma cells. Biochem Pharmacol. 2003;66:93–103. doi: 10.1016/s0006-2952(03)00248-x. [DOI] [PubMed] [Google Scholar]