

Abstract
Controlled release of non-steroidal anti-inflammatory drugs such as ibuprofen and naproxen could be beneficial for the treatment of inflammatory diseases while reducing the side effects resulting from their continuous use. Novel biodegradable polyesters solely comprised of biocompatible components (e.g., tartaric acid, 1,8-octanediol, and ibuprofen or naproxen as pendant groups) have been synthesized using tin (II) 2-ethylhexanoate as catalyst at 130 °C and subsequently characterized to determine their structures and physicochemical properties. The polymers release the free drug (ibuprofen or naproxen) in vitro in a controlled manner without burst release, unlike the release rates achieved when the drugs are encapsulated in other polymers. These new biomaterials are not cytotoxic towards mouse fibroblasts up to 0.10 mg/mL. The drugs retain their chemical structure following hydrolytic degradation of the polymer, suggesting that bioactivity is preserved.
Keywords: ibuprofen, naproxen, tartaric acid, biodegradable, polyester, prodrug
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs) have analgesic, antipyretic, and anti-inflammatory activity.1, 2 Ibuprofen (1) and naproxen (2), Figure 1, are propionic acid-derivative NSAIDs commonly used to treat pain and swelling associated with rheumatoid arthritis, osteoarthritis, psoriatic arthritis, and Ankylosing spondylitis.3-5 Administration of high systemic doses is often required to treat these chronic conditions because both 1 and 2 have relatively short half-life in plasma (2.1 and 14 hours, respectively).1 When repeatedly administered, severe gastrointestinal side effects such as stomach ulceration, bleeding, and perforation occur because the drug is distributed throughout the body to targeted and non-targeted sites. 6,7, 8 Therefore, the therapeutic potentials of 1 and 2 could be significantly enhanced by incorporating them into controlled-delivery systems.
Figure 1.
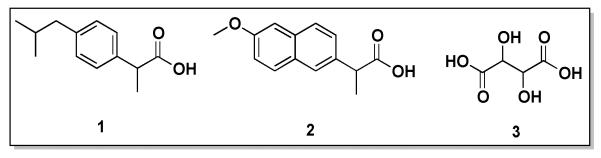
Chemical structures of ibuprofen (1), naproxen (2), and tartaric acid (3).
Drug delivery systems have been developed to localize drug release and prolong the duration of drug effect. The preparation of polymer microparticles encapsulating 1 or 2 has been studied.9-15 The major issues associated with this type of drug delivery system are low drug loading (less than 30 %), burst release and short-term (rapid) drug release. Acrylic and vinyl polymers have been widely studied to conjugate 1 or 2 onto the polymer backbones.7, 8, 16-20 Although these polymers are biocompatible, they are not biodegradable.21 Therefore, when the entire drug is released, the polymer would remain in the body which could cause patient discomfort and adverse effects.6 Despite the limitations, these drug delivery systems have been shown to lower the side effects associated with the systemic administration of the drug and to increase the duration of the drugs’ anti-inflammatory effects.7 To overcome these issues, we proposed chemical incorporation of bioactive molecules into biodegradable polymer backbones as a unique drug delivery method. Drugs containing only one reactive functional group can be incorporated onto a polymer as pendant groups; we explored this type of chemical incorporation with phenolic antiseptics, achieving high drug loading (48-58 wt.%).22
In this work, bioactives 1 and 2 were incorporated into biodegradable polyester backbones through their propionic acid moiety. Thus, we designed polymers containing 65-67 wt% of drug into the polymer that upon hydrolytic degradation, releases bioactives 1 and 2 in a controlled manner. This work presents the synthesis and characterization of biodegradable ibuprofen- and naproxen-based polyesters. Tartaric acid (Figure 1, 3), a naturally occurring and biocompatible compound that has antioxidant properties,23 was used as the polymer backbone. The polymers were synthesized at 130 °C catalyzed by tin (II) 2-ethylhexanoate. Chemical structures and physical properties of all compounds were measured, and in vitro release studies performed in phosphate buffered saline (PBS) at 37 °C to mimic physiological conditions. The cytocompatibilities of the polymers towards mouse fibroblasts at various concentrations were determined. Lastly, the structural integrities of the released drugs were studied.
Figure 3.

Infrared spectra of ibuprofen-based diacid 6a (top) and ibuprofen-based polyester 7a (bottom); key stretching frequencies are noted on the spectra.
EXPERIMENTAL SECTION
Materials
Naproxen and 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI) were purchased from Fisher Scientific (Pittsburg, PA). NCTC clone 929 (strain L) mouse areolar fibroblast cells were purchased from ATCC (Manassas, VA). Fetal bovine serum (FBS), penicillin/streptomycin (pen/strep), L-Glutamine, trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA), and Dulbecco’s modified eagle medium (DMEM) were obtained from GIBCO BRL (Rockville, MD). CellTiter 96® Aqueous One Solution Cell Proliferation Assay was obtained from Promega (Madison, WI). All other chemicals and reagents were purchased from Sigma-Aldrich (Milwaukee, WI) and used as received.
Proton and Carbon Nuclear Magnetic Resonance (1H- and 13C-NMR) spectroscopies
1H- and 13C-NMR spectra were obtained using a Varian 500 MHz spectrometer. Samples were dissolved (~ 5 mg/mL for 1H-NMR and ~ 20 mg/mL for 13C-NMR) in deuterated chloroform (CDCl3), with trimethylsilane as internal reference. Each spectrum was an average of 16 and 250 scans, respectively.
Infrared (IR) Spectroscopies
Fourier transform IR (FT-IR) spectra were obtained using a Thermo Nicolet/Avatar 360 FT-IR spectrometer. Samples (1 wt.%) were solvent-cast onto NaCl plates using dichloromethane (DCM). Each spectrum was an average of 32 scans.
Molecular Weight
Mass spectrometry (MS) was used to determine the molecular weights (MW) of polymer intermediates. A Finnigan LCQ-DUO equipped with Xcalibur software and an adjustable Atmospheric Pressure ionization Electrospray Ion Source (API-ESI) were used. Samples were dissolved in methanol (MeOH) and diluted to 10 μg/mL before injection using a glass syringe. Pressure during the experiments was 0.8×10−5 Torr and the API temperature was 150 °C.
Gel permeation chromatography (GPC) was used to determine weight-average molecular weight (Mw) and polydispersity index (PDI) of the polymers. Waters system consisting of a 515 HPLC pump, a 717plus autosampler, and a 410 refractive index (RI) detector was used. Waters Empower 2 software was used for data collection and analysis. Samples were dissolved in tetrahydrofuran (10 mg/mL), 20 μL aliquot was injected, and eluted through two PL gel columns 103 and 105 Å (Polymer Laboratories) in series at a flow rate of 1 mL/min. The Mw was calculated relative to narrow Mw polystyrene standards.
Thermal Analysis
TGA was used to obtain the decomposition temperatures (Td). TGA analysis was performed using a Perkin-Elmer TGA7 analyzer with TAC7/DX controller equipped with a Dell OptiPlex Gx 110 computer running Perkin-Elmer Pyris software. Samples (~10 mg) were heated under nitrogen at a rate of 10 °C/min from 25 to 400 °C. Td was defined as the onset of decomposition and represented by the beginning of a sharp slope on the thermogram.
Thermal analysis was performed using DSC to obtain the glass transition (Tg) and melting (Tm) temperatures. DSC was performed using a Thermal Advantage (TA) DSC Q200 running on an IBM ThinkCentre computer equipped with TA Instrument Explorer software for data collection and control. Samples (4-8 mg) were heated under nitrogen from −40 °C to 200 °C at a heating rate of 10 °C/min. Two heating/cooling cycles were used for each sample set. TA Universal Analysis 2000, version 4.5A was used to analyze the data. Tg was defined as the midpoint of the curve and Tm as the peak maximum.
Ibuprofen-tartrate Protected Diacid Synthesis (5a in Scheme 1)
Scheme 1.

Synthesis of ibuprofen- and naproxen-protected diacids (5a and 5b, respectively) by coupling of the drug’s (1 or 2) carboxylic acids to the hydroxyl groups of the dibenzyl protected tartaric acid (4). Deprotection to yield the diacids (6a and 6b) was performed using two different hydrogenation methods and synthesis of ibuprofen- and naproxen-tartaric polymers (7a and 7b) was performed using tin (II) 2-ethylhexanoate as catalyst.
Ibuprofen (1, 3.21 g, 2.2 eq) was dissolved in anhydrous DCM and stirred under argon. Then 4-(dimethylamino)pyridine (DMAP, 1.9 g, 2.2 eq) was added to the reaction mixture. Dibenzyl-L-tartrate (4, 2.3 g, 1 eq) was dissolved in anhydrous DCM and added to the reaction mixture followed by the addition of EDCI (6.0 g, 4.4 eq). The resulting yellowish solution was stirred for 2 h. The reaction mixture was diluted with ethyl acetate (EtOAc), extracted with 10% KHSO4 and saturated NaHCO3. The organic layer was dried over MgSO4 and the solvent evaporated under reduced pressure to give a brown, viscous oil that was dried in vacuo at room temperature overnight. Yield: 93 %. 1H-NMR (CDCl3, 500 MHz, δ): 7.30 (6H, m, ArH), 7.16 (6H, m, ArH), 7.06 (6H, m, ArH), 5.67 (2H, split, CH), 5.05-4.53 (4H, split, CH2), 3.80-3.60 (2H, dm, CH), 2.41 (4H, m, CH2), 1.79 (2H, m, CH), 1.45 (6H, t, CH3), 0.86 (12H, d, CH). 13C-NMR (CDCl3, 500 MHz, δ): 173.5 (1C), 173.2 (1C), 165.7 (1C), 165.3 (1C), 140.9 (2C), 136.9 (2C), 135.0 (2C), 129.5 (6C), 128.6 (6C), 127.7 (6C), 71.1 (2C), 67.7 (2C), 45.1 (2C), 44.7 (2C), 30.4 (2C), 22.7 (4C), 18.5 (2C). IR: 1769 cm−1 (C=O ester) and 1751 cm−1 (C=O ester). MS: M/Z = 729 [M + Na]. Td = 237 °C.
Naproxen-tartrate Protected Diacid Synthesis (5b in Scheme 1)
Synthesis of 5b was performed using the procedure described for 5a in Section 2.5.1 using 2.2 eq of naproxen (2) instead of 1. Yield: 81 % (green foam). 1H-NMR (CDCl3, 500M Hz, δ): 7.64 (6H, t, ArH), 7.37 (2H, d, ArH), 7.18 (6H, m, ArH), 7.10 (2H, d, ArH), 7.03 (2H, d, ArH), 6.83 (4H, d, ArH), 5.62 (2H, s, CH), 4.57-4.29 (4H, dd, CH2), 3.93 (2H, m, CH), 3.88 (6H, s, OCH3), 1.52 (6H, d, CH3). 13C-NMR (CDCl3, 500 MHz, δ): 173.5 (2C), 165.4 (2C), 158.0 (2C), 135.1 (2C), 134.6 (2C), 134.0 (2C), 129.6 (2C), 129.1 (2C), 128.6 (4C), 128.5 (2C), 128.0 (6C), 127.4 (2C), 126.5 (2C), 119.3 (2C), 105.8 (2C), 71.2 (2C), 67.6 (2C), 55.5 (2C), 45.0 (2C), 18.4 (2C). IR: 1767 cm−1 (C=O ester) and 1748 cm−1 (C=O ester). MS: M/Z = 777 [M + Na]. T = 294 ° C.
Ibuprofen-tartaric Diacid Synthesis (6a in Scheme 1)
Anhydrous DCM and triethylamine (TEA, 3.0 mL, 2.5 eq) were added to palladium (II) acetate [Pd(OAc)2, 4.2 g, 2.5 eq] and the mixture stirred under argon. Ibuprofen-tartrate protected diacid (5a, 6.0 g, 1 eq) was dissolved in DCM and added dropwise to the reaction mixture. The solution was left stirring for 5 min then triethylsilane (Et3SiH, 34 mL, 25 eq) added dropwise via a syringe pump (over 1 h). The reaction was stirred at room temperature under argon overnight. MeOH (3 mL) was added and the mixture was filtered over celite to remove Pd catalyst. The filtrate was concentrated under reduced pressure and the orange residue diluted in EtOAc. The precipitate formed was removed via vacuum filtration. The filtrate was concentrated under reduced pressure; the orange liquid obtained was diluted in acetonitrile (ACN) and extracted with hexanes. The ACN layer was dried under reduced pressure. The orange residue was diluted in EtOAc and extracted with water. The organic layer was dried over MgSO4 and the solvent evaporated under reduced pressure to give a yellow foam that was dried in vacuo at room temperature overnight. Yield: 77 %. 1H-NMR (CDCl3, 500 MHz, δ): 7.18 (4H, d, ArH), 7.08 (4H, d, ArH), 5.68 (2H, split, CH), 3.79 (2H, t, CH), 2.5 (4H, m, CH2), 1.84 (2H, m, CH), 1.51 (6H, t, CH3), 0.88 (12H, d, CH3). 13C-NMR (CDCl3, 500 MHz,δ): 173.6 (1C), 173.3 (1C), 170.7 (1C), 170.2 (1C), 141.0 (2C), 136.7 (2C), 129.5 (4C), 127.6 (4C), 70.5 (2C), 45.1 (2C), 44.8 (2C), 30.4 (2C), 22.6 (4C), 18.4 (2C). IR: 1751 cm−1 (C=O ester), 1733 cm−1 (C=O acid), and 3231 cm−1 (OH acid). MS: M/Z = 549 [M + Na]. T = 224 ° C.
Naproxen-tartaric Diacid Synthesis (6b in Scheme 1)
Synthesis was performed using the procedure described for 6a in Section 2.5.1. Yield: 90 % (orange foam). 1H-NMR (CDCl3, 500 MHz, δ): 7.70 (4H, t, ArH), 7.38 (4H, d, ArH), 7.15 (4H, d, ArH), 5.57 (2H, s, CH), 4.00 (2H, m, CH), 3.91(6H, s, OCH3), 1.60 (6H, d, CH). 13C-NMR (CDCl3, 500 MHz, δ): 173.5 (2C), 160.0 (2C), 157.9 (2C), 135.0 (2C), 134.0 (2C), 129.5 (2C), 127.4 (2C), 126.4 (4C), 126.3 (2C), 119.3 (2C), 105.7 (2C), 71.0 (2C), 55.5 (2C), 44.9 (2C), 18.3 (2C). IR: 1748 cm−1 (C=O ester), 1733 cm−1 (C=O acid), and 3447 cm−1 (OH acid). MS: M/Z = 597 [M + Na]. T = 235 ° C.
Optimized Diacid Synthesis
Ibuprofen- or naproxen-tartrate protected diacid (5a or 5b, 1 eq) was dissolved in anhydrous DCM (10 mL/g of protected diacid) and 10% palladium on carbon (Pd/C, catalytic amount) was added. The reaction flask was evacuated by vacuum and purged with hydrogen gas (3×). The reaction was stirred at room temperature under hydrogen overnight. The mixture was filtered over celite to remove Pd/C. The filtrate was dried under reduced pressure to give a yellow or orange foam that was dried in vacuo at room temperature overnight. Yield: > 90 %. The characterization is described in sections 2.5.1 and 2.5.2.
Ibuprofen-tartaric Polymer Synthesis (7a in Scheme 1)
Ibuprofen-tartaric diacid (0.51 g, 1 eq), 1,8-octanediol (0.14 g, 1eq), and tin (II) 2-ethylhexanoate (26 μL, 5 wt.%) were added to a double-neck round-bottom flask and degassed through vacuum/argon cycles (3×). The mixture was heated to 130 °C under vacuum (< 2 mmHg), and stirred (100 rpm) using an overhead mechanical stirrer (T-line laboratory stirrer, Talboys Engineering Corp., Montrose, PA) for 6 h. The product was cooled and dissolved in DCM (minimal amount). The product was isolated by removing the DCM under reduced pressure and dried under vacuum at room temperature overnight. Yield: 0.42 g (82 %), orange paste. 1H-NMR (CDCl3, 500 MHz, δ): 7.20 (4H, b, ArH), 7.08 (4H, b, ArH), 5.60 (2H, b, CH), 4.10-3.6 (6H, b, CH, CH2), 2.44 (4H, b, CH2), 1.85 (2H, b, CH), 1.60-1.00 (18H, b, CH3, 3CH2), 0.89 (12H, b, CH). 13 C-NMR (CDCl3, 500 MHz, δ): 173.6 (2C), 165.3 (2C), 140.9 (2C), 136.9 (2C), 129.5 (4C), 127.6 (4C), 71.0 (2C), 66.3 (2C), 45.3 (2C), 44.9 (2C), 30.6 (2C), 29.3 (2C), 28.4 (2C), 25.7 (2C), 22.4 (4C), 18.3 (2C). IR: 1768 and 1750 cm−1 (C=O, ester). Mw = 11,200 Da, PDI = 1.4. Tg = −17 °C. Td = 289 °C.
Naproxen-tartaric Polymer Synthesis (7b in Scheme 1)
Synthesis was performed using the procedure described in Section 2.7.1. Yield: 0.19 g, 95 %), yellow foam. 1H-NMR (CDCl3, 500 MHz, δ): 7.70 (4H, b, ArH), 7.39 (4H, b, ArH), 7.13 (4H, b, ArH), 5.56 (2H, b, CH), 3.97 (2H, b, CH), 3.91(6H, b, OCH3), 3.60-3.16 (4H, b, CH2) 1.58 (6H, b, CH3), 1.57-0.64 (12H, b, 3CH). 13C-NMR (CDCl3, 500 MHz, δ): 173.5 (2C), 165.6 (2C), 157.9 (2C), 135.0 (2C), 134.0 (2C), 129.6 (2C), 127.4 (2C), 126.4 (4C), 126.3 (2C), 119.3 (2C), 105.7 (2C), 71.2 (2C), 66.2 (2C), 55.4 (2C), 44.9 (2C), 29.1 (2C), 28.1 (2C), 25.4 (2C), 18.3 (2C). IR: 1768 and 1747 cm−1 (C=O, ester). Mw = 6,000 Da, PDI = 1.2. Tg = 23 °C. Td = 260 °C.
In Vitro Drug Release Studies
Drug (1 and 2) release from their respective polymer (7a and 7b) was studied at 37 °C in PBS (pH 7.4) with agitation (60 rpm) to mimic physiological conditions. Triplicate samples of each polymer (50.0 mg powder) were placed in 20 mL scintillation vials (Fisher, Fair Lawn, NJ) with 15 mL of PBS. At predetermined time points, samples were centrifuged at 3,000 rpm for 5 min (Hettich Zentrifugen EBA12) to isolate the polymer. All the degradation media (15 mL) was collected and replaced with fresh PBS (15 mL) at each time point. Samples were immediately analyzed using HPLC as described below.
High-Performance Liquid Chromatography (HPLC)
Quantitative analysis of the in vitro degradation products was performed via HPLC using an XTerra® RP18 5 μm 4.6 × 150 mm column (Waters, Milford, MA) on a Waters 2695 Separations Module equipped with a Waters 2487 Dual λ Absorbance Detector. The system was connected to a Dell computer running Empower software. Samples were filtered using 0.22 μm poly(vinylidine fluoride) syringe filters (Fisher). The HPLC method was adapted from previously published methods.17, 24 The mobile phase was 10 mM KH2PO4, 70 % ACN, and 30 % water at pH 3.5. Samples (20 μL) were run at 25 °C at a flow rate of 1 mL/min. Absorbance was monitored at λ = 265 nm for both drugs. The instrument was calibrated using standard 1 and 2 solutions of known concentrations.
Structure Determination of Released Drugs
Release media from day 5 (time point with the highest drug concentration) were freeze-dried for 24 h at −40 °C and 133×10−3 mBar (LABCONO Freeze Dry System/Freezon 4.5). The resulting white powder was dissolved in acidic water (5mL, pH ~1) and extracted with DCM (5 × 3mL). DCM was evaporated under reduced pressure and the samples dried under vacuum for 2 days. Solutions of the free drugs (1 and 2) were prepared and treated as described above for the release media. 1H-NMR spectroscopy was used to confirm the chemical structures of the released drugs.
Cytocompatibility Studies
In vitro cytocompatibility studies were performed by culturing NCTC clone 929 (strain L) mouse areolar fibroblast cells (L929 cells) in cell media (DMEM supplemented with 10% FBS, 1% pen/strep) containing the dissolved diacids (6a and 6b) and polymers (7a and 7b). Polymers and diacids were separately dissolved in dimethyl sulfoxide (DMSO) and diluted with cell media to reach concentrations of 0.10 and 0.05 mg/mL. These solutions were sterilized under UV at λ = 254 nm for 900 s (Spectronics Corporation, Westbury, NY) and then allocated to wells in a 96-well plate with 2000 cells/well. DMSO (0.5 %) in cell media was used as negative control.
Cell viability was determined using CellTiter 96® Aqueous One Solution Cell Proliferation Assay. After 24 h, 48 h, and 72 h incubation with polymers or diacids, 20 μL of (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) reagent was added to each well and further incubated for 2 h at 37 °C. The absorbance was then recorded with a microplate reader (Coulter, Boulevard Brea, CA) at 492 nm.
RESULTS AND DISCUSSION
Synthesis: Polymers Precursors
A published procedure25 for the synthesis of chicoric acid was adapted to synthesize the polymer precursors ibuprofen- and naproxen-based diacids (6a and 6b, respectively), Scheme 1. This synthetic procedure was chosen because of the structural similarities between chicoric acid and the diacids 6a and 6b (Figure 1S). Dibenzyl-protected tartaric acid (4) was used for the synthesis of ibuprofen- and naproxen-protected diacids (5a and 5b, respectively), to couple the NSAID (1 or 2) to the hydroxyl groups of the tartrate backbone using EDCI (first step Scheme 1). Selective deprotection to obtain the diacids 6a and 6b was performed using silane-promoted palladium-mediated hydrogenation (second step Scheme 1). This debenzylation method is known to preserve sensitive functional groups and the newly formed ester linkages. As expected, compounds 6a and 6b were successfully synthesized using this method. However, the product isolation process was complicated and tedious (comprised of multiple extractions). Therefore, the use of H2 and Pd/C was explored. This common hydrogenation method yielded the pure products (6a and 6b) after an easy isolation comprised of filtration of the Pd/C and evaporation of the solvent and byproducts. All compounds were obtained in high yields (i.e., more than 77 %).
Characterization
The chemical structures of the compounds were confirmed by 1H- and 13C-NMR and IR spectroscopies. Figure 2 shows the 1H-NMR spectra of the ibuprofen-containing compounds 5a and 6a. All the expected peaks are shown in the spectra (Figure 2 labeled a-k top, a-i middle) with no unexpected peaks observed. The spectra confirms successful coupling of the drug to the tartrate backbone and subsequent deprotection. The debenzylation was successful as demonstrated by the disappearance of the benzylic protons (i-k, Figure 2 top). For the naproxen-containing compounds 5b and 6b, the debenzylation was also demonstrated by 1H-NMR spectra (Figure 2S). The 13C-NMR specta showed the presence of all carbons and no extra peaks were observed, also supporting successful deprotection. As further characterization, the IR spectra of 5a and 5b show the formation of the ester bonds by the presence of the ester carbonyls (C=O) at ~ 1770 and 1750 cm−1. The IR spectra of 6a and 6b indicate that deprotection was successful with the presence of the ester carbonyl stretch at ~ 1760 and terminal carboxylic acid carbonyl stretch at ~ 1730 cm−1 (Figures 3 top and 3S top). The molecular weight of the intermediates were confirmed and corresponded to [M + Na]. All compounds were viscous oils or foams and did not display melting points; the decomposition temperatures ranged between 224-294 °C. These high decomposition temperatures are important when polymerizing at high temperatures.
Figure 2.

1H-NMR spectra of compounds 5a (top) and 6a (middle) showing the presence and disappearance of the benzyl protecting groups and polymer 7a (bottom).
Polymers Synthesis and Characterization
The polyesters were prepared by reacting the diacids 6a and 6b (respectively) with 1,8-octanediol using tin (II) 2-ethylhexanoate as catalyst at 130 °C (step 3, Scheme 1). Polyesters containing tartaric acid and 1,8-octainediol have been previously reported with tin (II) 2-ethylhexanoate as catalyst.26 The 1,8-octanediol is generally regarded as safe and has known bacteriostatic, bactericidal, and preservative properties.27 In addition, tin (II) 2-ethylhexanoate is the catalyst of choice for many polymerizations due to its low cost, low toxicity, and high efficiency.28-30 The 1H-NMR spectra for the polymers (7a and 7b) show broadening of the peaks and the presence of all the peaks expected (Figure 2 bottom and Figure 2S bottom). The IR spectra of 7a and 7b indicate the presence of the ester carbonyl at ~ 1770 and 1750 cm−1 (Figures 3 and 3S, respectively). Polymers with moderate Mw (11,200 and 6,000 Da) and low PDI values (1.2-1.4) were obtained. These polymers decomposed at temperatures above 250 °C and have low Tg values (−17 °C for 7a and 23 °C for 7b).
In Vitro Drug Release
After successfully synthesizing the polymers, their ability to release the free drug was studied in vitro. Polymer samples (7a and 7b) were incubated in buffered media (pH 7.4) mimicking physiological conditions (37 °C and 60 rpm). At predetermined time points, the media was collected and analyzed using HPLC. The retention time (Rt) for 1 was 3.08 min and for 2, 2.40 min, the diacids 6a and 6b had Rt of 4.61 and 3.17 min, respectively. During the studies, oligomers were not detected, and diacid peaks were observed in trace amounts. Figure 4 shows the in vitro drug release profiles for 1 and 2 during 30 days. No burst release was observed in the degradation profiles; the drugs were released from the respective polymers in a controlled manner. Both drugs were released at approximately the same rate for the first 10 days, as expected due to the structural similarities between the two polymers. Polymer 7a continued to release 1 at a constant rate from day 10 to day 30. However, release of 2 began to plateau after day 10. Further studies will be performed to study the polymer degradation and in vivo drug release mechanisms. After 30 days, polymer 7a released ~ 14 % of 1 and 7b released ~ 8 % of 2 (based on calculated theoretical values). At this rate, 100 % drug release –and corresponding polymer degradation - is expected over 7 to 12 months.
Figure 4.

In vitro ibuprofen (1, filled diamonds) and naproxen (2, filled circles) release profiles from polymers 7a and 7b, respectively (± standard error).
Following polymerization and in vitro release, the chemical composition of the released bioactives was monitored by 1H-NMR spectroscopy. No changes in chemical shifts and integration were observed in comparing the drugs (1 and 2) released at day 5 and the free drugs (Figures 4S and 5S). These results suggest that the structure of both drugs (1 and 2) were preserved, which implies that the released drugs retain all the properties and activities of the unprocessed drug (Figures 4S and 5S).
Cell Cytocompatibility Studies
Cytocompatibility of the diacids and polymers were evaluated using L929 mouse fibroblasts, a commonly used cell type to test toxicity of new biomaterials.31 The diacids (6a and 6b) and the polymers (7a and 7b), separately, were dissolved in DMSO and then diluted with cell culture media to concentrations of 0.10 and 0.05 mg/mL to mimic late and early polymer degradation, with final DMSO concentration 0.5%. Cell viability was evaluated at 24, 48, and 72 h. Figure 5 shows cell viability for all samples and the DMSO-containing media control. All samples resulted in normal cell proliferation with the exception that polymer 7a at 0.10 mg/mL resulted in a much lower cell proliferation rate. This data indicates that these materials are mostly cytocompatible within the concentration range tested.
Figure 5.

Normalized L929 cell viability in culture media containing polymers and diacids (top: 0.05 mg/mL; bottom: 0.10 mg/mL) at 24, 48, and 72 h. Data represent mean and standard deviation of six samples.
CONCLUSIONS
In this work, we presented the synthesis and characterization of novel biodegradable polyesters comprised of all biocompatible elements: tartaric acid, 1,8-octanediol, and an NSAID (1 or 2). With these polymers, the duration of drug release can be prolonged, (more than 1 month) with no burst release. The released drugs retained their chemical structure, suggesting that bioactivity is preserved. These polymers can be used to deliver 1 and 2 in a prolonged and controlled manner, thus have the potential to treat inflammatory diseases. Our future work includes the incorporation of other propionic acid-derivative NSAIDs as pendant groups to polyesters and in vivo anti-inflammatory activity testing.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the National Institutes of Health (NIH 5 R01DE0132070-09 and NIH 1 R01DE019926-01) for financial support. Dr. Indubhusan Deb, Michael Drahl, Dr. Dawanne Poree, Dr. Sarah Hehir, Nicholas Stebbins, and Dr. Bryan Langowski (Rutgers, Department of Chemistry & Chemical Biology) are thanked for intellectual discussions and assistance.
Footnotes
Notes
The authors declare no competing financial interest.
Relevant figures for the characterization of naproxen-based diacid and polymer are included in the supplementary information. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Brooks PM, Day RO. N. Engl. J. Med. 1991;324 doi: 10.1056/NEJM199106133242407. [DOI] [PubMed] [Google Scholar]
- 2.Laine L, Smith R, Min K, Chen C, Dubois RW. Aliment. Pharmacol. Ther. 2006;24:751–767. doi: 10.1111/j.1365-2036.2006.03043.x. [DOI] [PubMed] [Google Scholar]
- 3.Crielaard BJ, Lammers T, Schiffelers RM, Storm G. J. Controlled Release. 2012;161:225–234. doi: 10.1016/j.jconrel.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Sieper J, Braun J, Rudwaleit M, Boonen A, Zink A. Ann. Rheum. Dis. 2002;61(suppl 3):iii8–iii18. doi: 10.1136/ard.61.suppl_3.iii8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braun J, Sieper J. Lancet. 2007;369:1379–1390. doi: 10.1016/S0140-6736(07)60635-7. [DOI] [PubMed] [Google Scholar]
- 6.Jain JP, Modi S, Domb AJ, Kumar N. J. Controlled Release. 2005;103:541–563. doi: 10.1016/j.jconrel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 7.Khan MSY, Akhter M. Eur. J. Med. Chem. 2005;40:371–376. doi: 10.1016/j.ejmech.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Mirzaagha B. Int. J. Pharm. 2008;356:167–173. [Google Scholar]
- 9.Arica B, Ccedil; ali S, Atlla P, Durlu NT, akar N, Ka HS, Hincal AA. J. Microencapsulation. 2005;22:153–165. doi: 10.1080/02652040400026319. [DOI] [PubMed] [Google Scholar]
- 10.Thompson CJ, Hansford D, Higgins S, Rostron C, Hutcheon GA, Munday DL. J. Microencapsulation. 2009;26(8):676–683. doi: 10.3109/02652040802656333. [DOI] [PubMed] [Google Scholar]
- 11.Fernández-Carballido A, Herrero-Vanrell R, Molina-Martínez IT, Pastoriza P. Int. J. Pharm. 2004;279:33–41. doi: 10.1016/j.ijpharm.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Bozdag S, Calis S, Kas HS, Ercan MT, Peksoy I, Hincal AA. J. Microencapsulation. 2001;18:443. doi: 10.1080/02652040010018641. [DOI] [PubMed] [Google Scholar]
- 13.Castelli F, Conti B, Maccarrone DE, Conte U, Puglisi G. Int. J. Pharm. 1998;176:85–98. [Google Scholar]
- 14.Borovac T, Pelage JP, Kasselouri A, Prognon P, Guiffant G, Laurent A. J. Controlled Release. 2006;115:266–274. doi: 10.1016/j.jconrel.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Thompson CJ, Hansford D, Higgins S, Rostron C, Hutcheon GA, Munday DL. Int. J. Pharm. 2007;329:53–61. doi: 10.1016/j.ijpharm.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 16.Gallardo A, Parejo C, San Román J. J. Controlled Release. 2001;71:127–140. doi: 10.1016/s0168-3659(01)00212-7. [DOI] [PubMed] [Google Scholar]
- 17.Mizrahi B, Domb A. AAPS PharmSciTech. 2009;10:453–458. doi: 10.1208/s12249-009-9228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liso PA, Rebuelta M, Román JS, Gallardo A, Villar AM. J. Controlled Release. 1995;33:429–436. [Google Scholar]
- 19.Babazadeh M. Int. J. Pharm. 2006;316:68–73. doi: 10.1016/j.ijpharm.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 20.Davaran S, Entezami A, A. Anglais. 1998;34:187–192. [Google Scholar]
- 21.Pillai O, Panchagnula R. Curr. Opin. Chem. Biol. 2001;5:447–451. doi: 10.1016/s1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- 22.Prudencio A, Carbone AL, Griffin J, Uhrich KE. Macromol. Rapid Commun. 2009;30:1101–1108. doi: 10.1002/marc.200900059. [DOI] [PubMed] [Google Scholar]
- 23.DeBolt S, Cook DR, Ford CM. Proc. Natl. Acad. Sci. 2006;103:5608–5613. doi: 10.1073/pnas.0510864103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basheer C, Chong HG, Hii TM, Lee HK. Anal. Chem. 2007;79:6845–6850. doi: 10.1021/ac070372r. [DOI] [PubMed] [Google Scholar]
- 25.Lamidey A-M, Fernon L, Pouységu L, Delattre C, Quideau S, Pardon P. Helv. Chim. Acta. 2002;85:2328–2334. [Google Scholar]
- 26.Borzacchiello A, Ambrosio L, Nicolais L, Huang SJ. J. Bioact. Compat. Polym. 2000;15:60–71. [Google Scholar]
- 27.Frankenfeld JW, Wright DL. Exxon Research and Engineering Comapny: United States. 1976;3:970. [Google Scholar]
- 28.Storey RF, Sherman JW. Macromol. 2002;35:1504–1512. [Google Scholar]
- 29.Schwach G, Coudane J, Engel R, Vert M. Polym. Bull. 1994;32:617–623. [Google Scholar]
- 30.Schwach G, Coudane J, Engel R, Vert M. Biomaterials. 2002;23:993–1002. doi: 10.1016/s0142-9612(01)00209-5. [DOI] [PubMed] [Google Scholar]
- 31.Dumitriu S. Polym. Biomater. Second ed. Mercel Dekker Inc.; New York, NY: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.