

Abstract
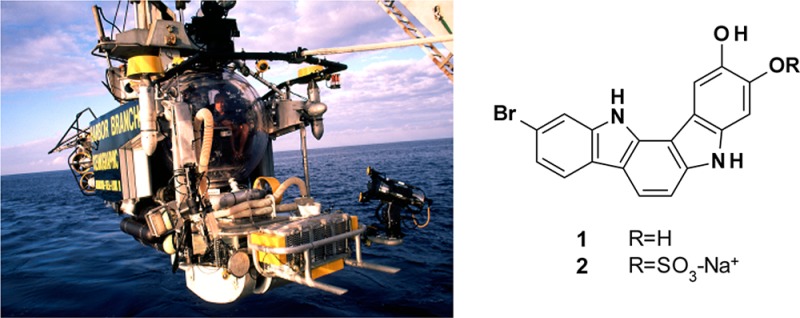
Two new indolo[3,2-a]carbazoles (1, 2) were isolated from a deep-water collection of a sponge of the genus Asteropus. The structures of 1 and 2 were determined through the analysis of spectroscopic data including mass spectrometry and 2D-NMR. Compound 1 showed minimum inhibitory concentrations of 25 μg/mL against the fungal pathogen Candida albicans and 50 μg/mL against methicillin-resistant Staphylococcus aureus (MRSA). Compounds 1 and 2 showed no cytotoxicity against the PANC1 human pancreatic carcinoma and NCI/ADR-RES ovarian adenocarcinoma cell lines at our standard test concentration of 5 μg/mL.
Marine organisms have been a source of novel natural products with unique chemical scaffolds possessing a variety of potent biological activities. As part of our work to provide novel marine natural products to the Molecular Libraries Small-Molecule Repository under the NIH Roadmap Initiative, we investigated a deep-water collection of a sponge of the genus Asteropus. Previous studies of sponges belonging to the genus Asteropus have led to the isolation of a number of different classes of organic compounds including saponins,1−5 sterols,6 pteridine derivatives,7 pyroglutamyl dipeptides,8 tetrahydro-β-carboline alkaloids,8 and a peptide.9 Reported herein is the isolation, structure elucidation, and initial biological testing of a new indolo[3,2-a]carbazole, 1, and its sulfonated derivative, 2, from this specimen of Asteropus sp. These compounds are structurally related to ancorinazole10 reported from two New Zealand collections of the sponge Ancorina sp. Both Asteropus and Ancorina belong to the sponge family Ancorinidae and are members of the polyphyletic Astrophorida grouping of sponges.

The sponge used in this study was collected using the Johnson-Sea-Link submersible at a depth of 131 m off Ocean Cay, Bahamas, approximately 20 nautical miles south of Bimini. The sample was frozen at −20 °C immediately after collection and stored frozen until workup. The frozen sponge was extracted exhaustively with EtOH. After concentration by distillation under reduced pressure, the extract was partitioned first between EtOAc and H2O, and then the aqueous phase was further partitioned with n-butanol. The EtOAc and n-butanol partitions were fractionated by medium-pressure liquid chromatography on a Combi-Flash Rf4x using a Redi-Sep Gold C-18 column and a linear gradient of CH3CN and H2O both containing 0.1% TFA. Final purification was achieved using mass-directed preparative HPLC on a Vydac C-18 column eluted with a gradient of CH3CN in H2O both containing 0.1% TFA to yield 1 (44.1 mg, 1.9 × 10–2 % of wet weight) and 2 (5.9 mg, 2.7 × 10–3 % of wet weight).
Compound 1 was isolated as a dark blue, amorphous solid. Inspection of the 13C NMR spectrum coupled with HRESIMS analysis of 1 suggested a molecular formula of C18H11BrN2O2, indicative of 14 degrees of unsaturation. The mass spectrum showed equal-intensity ions at m/z [M + H]+ 367 and 369 in positive ion mode, suggesting that 1 was monobrominated. Absorptions observed at 3399 and 3206 cm–1 in the IR spectrum suggested the presence of amine and alcohol functionalities, respectively.
The 1H NMR spectrum collected in DMSO-d6 showed four exchangeable resonances observed at δH 11.65, 11.00, 9.28, and 8.18 and seven methine protons all with chemical shifts between δH 7.96 and 6.92, consistent with a heterocyclic structure. The 13C NMR spectrum showed a total of 18 sp2-hybridized carbons with chemical shifts between δC 97.0 and 145.0 and included seven methine carbons and 11 quaternary carbons. The DQF-gCOSY spectrum revealed the presence of two spin systems. The first was assigned as a 1,2,4-trisubstituted aromatic ring based upon observation of a proton at δH 7.96 (d, J = 8.2 Hz), which was coupled to a proton observed at δH 7.24 (dd, J = 8.2, 1.4 Hz), which in turn showed meta coupling to a proton observed at δH 7.65 (d, J = 1.4 Hz). The second spin system was a mutually coupled two-proton spin system observed at δH 7.89 (d, J = 8.2 Hz) and 7.20 (d, J = 8.2 Hz). The final two methine protons were observed as singlets at δH 7.91 and 6.92.
Residual one-bond couplings observed in the 1H–15N g-HMBC spectrum allowed the exchangeable protons observed at δH 11.64 and 11.00 to be placed on nitrogen. Correlations observed in the HMBC spectra (both 1H–13C and 1H–15N) for the methine protons of the 1,2,4-trisubstituted aromatic ring and the exchangeable proton observed at δH 11.64 were consistent with the presence of a 6-bromoindolyl moiety in 1. All HMBC correlations typically observed for a 6-bromoindolyl moiety were observed (Figure 1 and Supporting Information Figure S1 and Table S5). Selected correlations supporting this assignment are as follows: The exchangeable resonance observed at δH 11.64 (NH-12) showed correlations to carbons observed at δC 133.5911 (C-12a), 113.26 (C-7a), 123.1 (C-7b), and 140.18 (C-11a). A correlation observed in the 1H–15N gHMBC spectrum between the proton observed at δH 7.65 (d J = 1.4 Hz, H-11) and N-12 supports the assignment of the indole ring and allows for placement of H-11 relative to the nitrogen atom. The protons observed at δH 7.96 (H-8), 7.24 (H-9), and 7.65 (H-11) all show correlations in the 1H–13C HMBC spectrum to the carbon observed at 123.1 (C-7b). These same protons also show correlations to the carbon observed at δC 115.9, which can be assigned as C-10. The chemical shift of C-10 is consistent with substitution with bromine, and therefore the bromine atom required by the molecular formula was placed at C-10. The second two-proton spin system defined by the DQF-gCOSY and gHSQC spectra was attached at C-7a of the indole moiety due to long-range couplings observed in the 1H–13C HMBC spectrum from the proton observed at δH 7.20 (H-6) to the carbon observed at δC 113.26 (C-7a) and from the proton observed at δH 7.89 (H-7) to the carbons observed at δC 133.57 (C-12a), 113.26 (C-7a), and123.1 (C-7b).
Figure 1.

Key correlations observed in the HMBC and 2D-NOESY spectra for compound 1.
The remaining atoms in the molecule are two aromatic methine groups both appearing as singlets, six quaternary sp2 carbons, one exchangeable proton attached to nitrogen, two oxygen atoms, and two exchangeable protons whose chemical shifts are consistent with assignment as phenolic protons. Interpretation of the gHMBC spectra suggested the presence of a second indole ring (Figure 1 and Supporting Information Figure S2 and Table S5). Key couplings are as follows: the exchangeable proton observed at δH 11.00 (H-5) showed correlations to four carbons at δC 139.1 (C-5a), 106.9 (C-12b), 113.24 (C-12c), and 133.52 (C-4a). The singlet resonance observed at δH 6.92 showed a correlation in the 1H–15N HMBC to the indole nitrogen N-5, suggesting it is H-4. This same proton showed correlations in the 1H–13C HMBC to δC 113.24 (C-12c), 140.13 (C-2), 145.0 (C-3), and 133.52 (C-4a). The singlet methine observed at δH 7.91 showed correlations to δC 133.5 (C-4a), 113.2 (C-12c), 140.13 (C-2), and 145.0 (C-3). The exchangeable phenolic proton observed at δH 8.18 showed long-range coupling in the HMBC spectrum to the carbons observed at δC 107.2 (C-1), 140.13 (C-2), and 145.0 (C-3), suggesting its assignment as a phenolic proton at C-2. The exchangeable proton observed at δH 9.28 showed correlations in the HMBC spectrum to the carbons observed at δC 140.13 (C-2), 145.0 (C-3), and 97.0 (C-4), suggesting its assignment as a phenolic proton at C-3. The chemical shifts of C-2 and C-3 were consistent with hydroxy substitution, indicating the presence of a 2,3-dihydroxy indole moiety in 1. The HMBC data do not allow for relative assignment of the chemical shifts for C-2 and C-3, and therefore the assignment was made based upon comparison of observed values to values calculated using ACD C13 Predictor v11.
The final connectivities were made based upon the HMBC and NOE data (Figure 1). Correlations in the HMBC spectra between H-6 and both N-5 and C-12b (δC 106.9) and between H-7 and C-5a (δC 139.1) suggested the bond between C-6 and C-5a. As all atoms required by the molecular formula have been incorporated and the molecular formula requires one additional ring, a bond was drawn between C-12a and C-12b to form the final six-membered ring, thus completing the structure assignment of 1. This ring closure was supported by a correlation observed in the NOESY experiment between NH-12 and H-1. Key correlations in the NOESY spectrum supporting the overall structure assignment were H-6 → NH-5 → H-4 → OH-3; and H-11 → NH-12 → H-1.
Compound 2 was isolated as a dark brown, amorphous solid. The mass spectrum observed in negative ion mode showed equal-intensity ions at m/z [M – H]− 445 and 447, suggesting that the compound contains one bromine atom. HRESIMS analysis suggested a molecular formula of C18H11BrN2O5S for 2. An absorption observed at 3390 cm–1 in the IR spectrum is characteristic of an amine functionality. The NMR data of 2 were very similar to those observed for 1 and supported the assignment of an indolo[3,2-a]carbazole structure as found in 1. A notable difference was the observation of only three exchangeable resonances observed at δH 11.75, 11.22, and 8.37 in 2. The mass difference of approximately 80 amu between the two compounds along with the molecular formula was consistent with sulfonation of one of the hydroxy groups. The methine protons observed at δH 8.06 and 7.06 could be assigned as H-1 and H-4, respectively, based upon interpretation of the 1H–13C HMBC spectrum (Figure S16 and Table S20) with a key distinguishing correlation being that observed from H-1 to C-12b. The NOESY data confirmed the assignments with NOEs observed between NH-12 and H-1 and between NH-5 and H-6 as observed for 1. The phenolic proton observed in the 1H NMR spectrum of 2 (δH 8.37) showed long-range coupling in the 1H–13C gHMBC spectrum to C-1 (δC 108.0), C-2 (δC 143.4), and C-3 (δC 140.1), assigning it as the C-2-OH. Therefore the sulfate moiety must be attached at C-3. Prior reports indicate that the resonances for carbons and protons ortho to the position substituted by a sulfate group shift downfield and the carbon directly attached to the sulfate shifts upfield.12 In 2, the H-4 methine resonance (ortho to the sulfate moiety) shifts from δH 6.92 to 7.26 as expected if the C-3 phenol is substituted by a sulfate group in 2 (Figure S17).
Compounds 1 and 2 were screened for their activity against the microorganisms Candida albicans and methicillin-resistant Staphylococcus aureus (MRSA). Compound 1 showed a minimum inhibitory concentration of 25 μg/mL against C. albicans and 50 μg/mL against MRSA. Compound 2 showed no growth inhibitory effects against either of the organisms at concentrations up to 50 μg/mL. Both compounds 1 and 2 showed no cytotoxicity toward the PANC1 human pancreatic carcinoma or NCI/ADR-Res ovarian adenocarcinoma cell lines when tested at a concentration of 5 μg/mL. Compounds 1 and 2 represent the second and third naturally occurring indolo[3,2-a]carbazoles. No biological activity was reported for ancorinazole, but a series of synthetic indolo[3,2-a]carbazoles have been predicted to block binding of benzodiazepine to the γ-aminobutyric acid A/benzodiazepine receptor.13,14 Screening of these naturally occurring metabolites in such an assay system could identify new lead structures for the development of medicines to target neurological and psychiatric disorders.
Experimental Section
General Experimental Procedures
The UV spectra were collected on a Hitachi U-3010 spectrophotometer. The IR spectra were collected on a Perkin-Elmer Spectrum 100 instrument with universal ATR. NMR data were collected on a JEOL ECA-600 spectrometer operating at 600.17 MHz for 1H, 150.9 MHz for 13C, and 60.8 MHz for 15N (instrument reference set to liquid NH3). The edited-gHSQC was optimized for 140 Hz, and the long-range JH,C (gHMBC and band selective gHMBC) were optimized for 8 Hz. Chemical shifts are referenced to solvent (DMSO-d6 δH observed at 2.50 ppm and δC observed at 39.5 ppm). Chemical shifts for 15N were referenced to liquid NH3 with long-range JH,N optimized for 6 Hz. The ESIMS spectra were measured on a Finnigan LTQ mass spectrometer located at HBOI. The HRESIMS spectra were measured using a Kratos MS50TC mass spectrometer at the University of California, Riverside. Medium-pressure liquid chromatography was conducted on an Isco-Teledyne Combi-Flash Rf4x system. HPLC was performed using a Hitachi LaChrom quaternary gradient HPLC system equipped with diode array detector (L-7455) monitoring at 230 nm and ELSD detection. Preparative fractionation was conducted using mass-directed collection on a Waters Autopurification system equipped with a Waters 2535 quaternary gradient module, a Waters 2998 PDA detector, a Waters 515 HPLC pump, a Waters SFO system fluidics organizer, a Waters 2767 sample manager, and a Waters 3100 mass detector operating in positive and negative ion ESI mode. All solvents used were HPLC grade.
Biological Materials
The sponge (HBOI specimen number 22-X-00-4-010) was collected using the Johnson-Sea-Link human-occupied submersible at a depth of 131 m off Ocean Cay, Bahamas, approximately 20 nautical miles south of Bimini (latitude: 25 24.21′ N; longitude: 79 14.21′ W). It is an unidentified species of Asteropus (phylum Porifera, class Demospongiae, subclass Heteroscleromorpha, order Tetractinellida, suborder Astrophorina, family Ancorinidae).15 The sponge is most closely related to Asteropus niger Hajdu and Van Soest, 1992.16 The sample was massive-spherical in shape with clusters of oscules distributed over the surface. Its ectosome and choanosome were black inside both in situ and in air; it is dark brown in EtOH. While the spicule complement is similar to that described for A. niger, that species is characterized by two size categories of megasclere oxeas, one of which is less than 500 μm long.16 The smaller category of megasclere oxeas in the material studied is rare, and the dimensions are much longer and thicker than those reported for A. niger. A taxonomic voucher specimen preserved in EtOH is deposited in the Harbor Branch Oceanographic Museum (catalog number 003:01087).
Extraction and Isolation
The frozen sponge Asteropus (211 g) was diced and extracted exhaustively by macerating with EtOH (3 × 200 mL) using a Waring blender. The combined filtered extracts were concentrated by distillation under reduced pressure to yield 11.10 g of crude residue. The residue was partitioned between EtOAc (300 mL) and H2O (3 × 200 mL) to give after removal of solvent 1.44 g of EtOAc extract. The aqueous phase was further partitioned with n-butanol (3 × 200 mL) to afford, after removal of solvent, 7.11 and 1.94 g of aqueous and n-butanol extracts, respectively. The n-butanol extract was fractionated using a Combi-Flash Rfx4 system equipped with a 15.5 g Redi-Sep RP-18 Rf gold ISCO column operating at 200 psi with a flow rate of 30 mL/min. Fractions were eluted using a linear gradient of A [H2O/CH3CN (95:5 v/v + 0.1% TFA)] and B [CH3CN (+0.1% TFA)]; t = 0 min A:B (95:5 v/v), t = 23 min A:B 0:100 v/v, hold for 2 min. Further purification of a fraction eluting at 10 column volumes (approximately 4 min) (200 mg) was carried out on a Waters Autopure LCMS mass-directed preparative purification system using a Vydac 218TP protein and peptide C-18 preparative column (150 × 22 mm, 10 μm particle size) eluted with a linear gradient of A [H2O/CH3CN (95:5 v/v + 0.1% TFA)] and B [CH3CN (+0.1% TFA)], t = 0 min A:B (95:5 v/v), t = 23 min A:B (0:100 v/v), hold for 2 min, and selecting mass collection at m/z 367 and 447. This led to the isolation of 1 (44.1 mg, 1.9 × 10–2 % of wet weight) and 2 (5.9 mg, 2.7 × 10–3 % of wet weight).
Compound 1:
dark blue solid; UV (MeOH) λmax (log ε) 209 (3.82), 270 (3.89), and 295 (3.63); IR (film) 3399, 3206, 1611, 1568 cm–1; 1H NMR (DMSO-d6) see Tables 1 and S5; 13C NMR (DMSO-d6) see Tables 1 and S5; HRESIMS m/z [M + H]+ 367.0081 (calcd for C18H12BrN2O2, 367.0082).
Table 1. 1H and 13C NMR Data for 1 and 2 (DMSO-d6, 600 MHz)a.
|
1 |
2 |
|||||
|---|---|---|---|---|---|---|
| position | δC, mult | δN | δH (J in Hz) | δC mult | δN | δH (J in Hz) |
| 1 | 107.2, CH | 7.91, s | 108.0, CH | 8.06, s | ||
| 2 | 140.13, Cb | 143.4, C | ||||
| OH-2 | 8.18, s | 8.37, s | ||||
| 3 | 145.0, C | 140.1, C | ||||
| OH-3 | 9.28, s | |||||
| 4 | 97.0, CH | 6.92, s | 105.3, CH | 7.26, s | ||
| 4a | 133.52, C | 132.9, C | ||||
| 5 | 113 | 11.00, bs | 114 | 11.22, s | ||
| 5a | 139.1, C | 140.2, Cc | ||||
| 6 | 104.1, CH | 7.20, d (8.2) | 104.3, CH | 7.26, m | ||
| 7 | 116.0, CH | 7.89, d (8.2) | 117.6, CH | 8.00, d (8.2) | ||
| 7a | 113.26, Cb | 113.4, Cc | ||||
| 7b | 123.1, C | 122.9, C | ||||
| 8 | 120.3, CH | 7.96, d (8.0) | 120.4, CH | 7.99, d (8.2) | ||
| 9 | 121.3, CH | 7.24, dd (8.0, 1.4) | 121.5, CH | 7.26, m | ||
| 10 | 115.3, C | 115.4, C | ||||
| 11 | 113.33, CHb | 7.65, d (1.4) | 113.3, CHc | 7.65, d (1.4) | ||
| 11a | 140.18, Cb | 140.3, Cd | ||||
| 12 | 112 | 11.64, bs | 113 | 11.75, s | ||
| 12a | 133.59, C | 134.1, C | ||||
| 12b | 106.9, C | 106.2, C | ||||
| 12c | 113.24, Cb | 118.0, C | ||||
1H and 13C NMR data were measured at 600.2 and 150.9 MHz, respectively.
Congested areas of the spectrum; assignments were made on the basis of a band-selective 1H–13C gHMBC experiment. Two decimal places are shown to distinguish resonances of very similar chemical shifts, but that could be clearly resolved in the band-selective experiment.
Assignments are interchangeable.
Compound (2):
dark brown solid; UV (MeOH) λmax (log ε) 202 (3.44) and 268 (3.39); IR (film) 3390, 2924, 1587, 1471, 1438 cm–1; 1H NMR (DMSO-d6) see Tables 1 and S20; 13C NMR (DMSO-d6) see Tables 1 and S20; HRESIMS m/z [M – H]− 444.9489 (calcd for C18H10BrN2O5 S, 444.9499).
Antimicrobial Assays
The minimum inhibitory concentrations (MICs) were determined for C. albicans and methicillin-resistant S. aureus as described by Chen et al.17
Cytotoxicity Assays
Compounds 1 and 2 were evaluated for their cytotoxicity toward the PANC1 human pancreatic cancer (ATCC No. CRL1469) and NCI/ADR-Res ovarian adenocarcinoma cell lines using a method previously described in the literature.18
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (1P41GM089163, 1RC2AT005917, and 2R01CA093455) and the State of Florida Center of Excellence in Biomedical and Marine Biotechnology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. This is Harbor Branch Oceanographic Institute contribution number 1905.
Supporting Information Available
Full tables of NMR data, 1H NMR, 13C NMR, and selected 2D NMR spectra in DMSO-d6 for 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kitagawa I.; Kobayashi M.; Okamoto Y.; Yoshikawa M.; Hamamoto Y. Chem. Pharm. Bull. 1987, 35, 5036–5039. [DOI] [PubMed] [Google Scholar]
- Ksebati M. B.; Schmitz F. J.; Gunasekera S. P. J. Org. Chem. 1988, 53, 3917–3921. [Google Scholar]
- Schmitz F. J.; Ksebati M. B.; Gunasekera S. P.; Agarwal S. J. Org. Chem. 1988, 53, 5941–5947. [Google Scholar]
- Kobayashi M.; Okamoto Y.; Kitagawa I. Chem. Pharm. Bull. 1991, 39, 2867–2877. [Google Scholar]
- Espada A.; Jimenez C.; Rodriguez J.; Crews P.; Riguera R. Tetrahedron 1992, 48, 8685–8696. [Google Scholar]
- Bohlin L.; Gehrken H. P.; Scheuer P. J.; Djerassi C. Steroids 1980, 35, 295–304. [DOI] [PubMed] [Google Scholar]
- Murayama S.; Nakao Y.; Matsunaga S. Tetrahedron Lett. 2008, 49, 4186–4188. [Google Scholar]
- Li H.; Dang H. T.; Li J.; Sim C. J.; Hong J.; Kim D.-K.; Jung J. H. Biochem. Syst. Ecol. 2010, 38, 1049–1051. [Google Scholar]
- Takada K.; Hamada T.; Hirota H.; Nakao Y.; Matsunaga S.; van Soest R. W.; Fusetani N. Chem. Biol. 2006, 13, 569–574. [DOI] [PubMed] [Google Scholar]
- Meragelman K. M.; West L. M.; Northcote P. T.; Pannell L. K.; McKee T. C.; Boyd M. R. J. Org. Chem. 2002, 67, 6671–6677. [DOI] [PubMed] [Google Scholar]
- A band-selective HMBC experiment was used to distinguish between carbons having chemical shifts within ±0.09 ppm.
- Gulavita N. K.; Wright A. E.; McCarthy P. J.; Pomponi S. A.; Kelly-Borges M.; Chin M.; Sills M. A. J. Nat. Prod. 1993, 56, 1613–1617. [DOI] [PubMed] [Google Scholar]
- Urbano C. M.; Luque R. I.; Gomez-Nieto M. A. J. Chem. Inf. Model. 2006, 46, 2022–2029. [DOI] [PubMed] [Google Scholar]
- Xia B.; Ma W.; Zheng B.; Zhang X.; Fan B. Eur. J. Med. Chem. 2008, 43, 1489–1498. [DOI] [PubMed] [Google Scholar]
- Hajdu E.; Van Soest R. W. M. Bijdr Dierk 1992, 62, 3–19. [Google Scholar]
- Morrow C. C.; Picton B. E.; Erpenbeck D.; Boury-Esnault N.; Maggs C. A.; Allcock A. L. Mol. Phylogenet. Evol. 2012, 62, 174–190. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Killday K. B.; McCarthy P. J.; Schimoler R.; Chilson K.; Selitrennikoff C.; Pomponi S. A.; Wright A. E. J. Nat. Prod. 2001, 64, 262–264. [DOI] [PubMed] [Google Scholar]
- Gunasekera S. P.; Zuleta I. A.; Longley R. E.; Wright A. E.; Pomponi S. A. J. Nat. Prod. 2003, 66, 1615–1617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.