

ABSTRACT
The bacterial cell poles are emerging as subdomains where many cellular activities take place, but the mechanisms for polar localization are just beginning to unravel. The general phosphotransferase system (PTS) proteins, enzyme I (EI) and HPr, which control preferential use of carbon sources in bacteria, were recently shown to localize near the Escherichia coli cell poles. Here, we show that EI localization does not depend on known polar constituents, such as anionic lipids or the chemotaxis receptors, and on the cell division machinery, nor can it be explained by nucleoid occlusion or localized translation. Detection of the general PTS proteins at the budding sites of endocytotic-like membrane invaginations in spherical cells and their colocalization with the negative curvature sensor protein DivIVA suggest that geometric cues underlie localization of the PTS system. Notably, the kinetics of glucose uptake by spherical and rod-shaped E. coli cells are comparable, implying that negatively curved “pole-like” sites support not only the localization but also the proper functioning of the PTS system in cells with different shapes. Consistent with the curvature-mediated localization model, we observed the EI protein from Bacillus subtilis at strongly curved sites in both B. subtilis and E. coli. Taken together, we propose that changes in cell architecture correlate with dynamic survival strategies that localize central metabolic systems like the PTS to subcellular domains where they remain active, thus maintaining cell viability and metabolic alertness.
IMPORTANCE
Despite their tiny size and the scarcity of membrane-bounded organelles, bacteria are capable of sorting macromolecules to distinct subcellular domains, thus optimizing functionality of vital processes. Understanding the cues that organize bacterial cells should provide novel insights into the complex organization of higher organisms. Previously, we have shown that the general proteins of the phosphotransferase system (PTS) signaling system, which governs utilization of carbon sources in bacteria, localize to the poles of Escherichia coli cells. Here, we show that geometric cues, i.e., strong negative membrane curvature, mediate positioning of the PTS proteins. Furthermore, localization to negatively curved regions seems to support the PTS functionality.
Introduction
Almost all processes in eukaryotic cells are presumed to be spatiotemporally controlled, but only in recent years has subcellular organization been shown to be highly significant also for bacterial cells (1). The documentation of distinct distribution patterns for proteins, lipids, and even RNAs in bacterial cells suggests that spatial organization of macromolecules is a conserved phenomenon in all cell types (2). In rod-shaped bacteria, the poles, characterized by unique composition and topology, are emerging as specialized sites for a wide variety of cellular functions, ranging from chromosome segregation to signal transduction and virulence (3, 4). Although the cues that recruit most proteins to the poles are largely unknown, in few cases, certain properties of the poles were suggested as potential localization cues. Interaction with the anionic phospholipid cardiolipin, which is enriched in regions of cytoplasmic membrane near the poles and septa of growing Escherichia coli cells (5), has been suggested to account for polar localization of the osmosensory transporter ProP and the mechanosensitive channel MscS (6, 7). Strong negative curvature (concave), which characterizes the poles and the sites near the forming septum in dividing rod-shaped bacterial cells, has been suggested to be sensed by DivIVA, a membrane-binding protein that localizes to the septa and the poles in Bacillus subtilis cells (8, 9), and by MinD, a cell division protein that oscillates between the poles in E. coli (10). Notably, strong positive curvature (convex) was suggested to play a role in the localization of the SpoVM protein to the peripheral membrane of the forespore during sporulation of B. subtilis cells (11). On the other hand, the Tar receptors of the chemotaxis complex were suggested to localize by stochastic self-assembly of clusters (12).
A central signal transduction system that localizes to the poles in E. coli is the phosphoenolpyruvate-dependent phosphotransferase system (PTS), which governs hierarchal uptake of carbohydrates and adjusts cell metabolism accordingly. The PTS regulates global pathways, such as catabolite repression and inducer exclusion (13), and specialized pathways that enable sugar utilization (14) in Gram-negative and Gram-positive bacteria. It has recently been shown by our lab that the PTS is subjected to spatiotemporal regulation (15). Hence, the “control center” of the PTS, i.e., the general PTS proteins enzyme I (EI) and HPr, was shown to cluster mainly near the cell poles. Polar localization of each protein occurs independently, but HPr was shown to be released from the poles in an EI- and sugar-dependent manner.
The general PTS proteins were shown to also spatially regulate downstream auxiliary PTS components. Thus, BglG, a transcription factor that positively regulates transcription of the β-glucoside utilization operon (bgl) in E. coli, is sequestered near the membrane in an inactive form via interaction with the β-glucoside transporter, BglF (16); in the presence of β-glucosides, BglG is dephosphorylated by BglF and migrates to the cell poles, where it is activated by EI and HPr; consequently, the dimeric form of BglG is released to the cytoplasm, where it binds to the emerging bgl transcript and antiterminates transcription of the bgl operon (15). Similarly, LicT, a BglG homologue from B. subtilis, has recently been shown to be localized near the poles upon addition of β-glucosides to the medium (17). Spatiotemporal control of other transcription regulators by sugar permeases was also documented, i.e., repression of Mlc and MalT activity as positive regulators of glucose and maltose utilization genes in E. coli via interaction and membrane sequestration with the PTS glucose permease (18) and the maltose ABC transporter MalFGK2 (19), respectively, and activation of MtlR as a positive regulator of mannitol operon expression in B. subtilis via interaction with the mannitol permease (20). Hence, the distinctly localized PTS proteins, i.e., the general PTS at the poles and the sugar permeases at the cell circumference, control expression of the sugar utilization genes via a series of orchestrated spatial relocations of regulatory proteins. Still, the nature of the cues that recruit the general proteins to the poles and the spatiotemporal mechanisms that ensure efficient uptake of sugars by relocation of the other PTS components are currently unknown.
In this study, we set out to identify the cues that recruit the general PTS proteins to the poles of E. coli cells, focusing mainly on EI, which is steadily present in this microdomain. We show that the anionic lipid cardiolipin, which localizes preferentially to the poles, is not involved in recruiting EI. Components of the chemotaxis complex, which clusters at the poles, i.e., the monocyte chemoattractant protein (MCP) receptors and the CheA sensor, the latter previously shown to interact with EI, are also not involved in EI polar recruitment. Following inhibition of cell division, either by antibiotic treatment or by mutations that lead to the formation of filamentous cells with multiple chromosomes, EI was detected near the budding septum, whose maturation is inhibited. This localization pattern is not due to nucleoid occlusion, since the same pattern is observed after chromosome condensation. Furthermore, EI localization is not due to RNA targeting and localized translation, since the mRNA that codes for EI localizes to the cytoplasm rather than to the poles.
A clue to the rationale behind the PTS protein localization pattern was provided by imaging them in pole-lacking cells: in spherical E. coli cells, formed due to mutations in cytoskeletal proteins or in response to treatment with cytoskeleton-inhibiting antibiotics, EI still clustered into foci. Intriguingly, the foci localized near the budding site of the intracytoplasmic membrane-bound vesicles (IMVs), previously shown to form in such cells (21). These sites, like the poles, are strongly curved, and we show here that the membrane curvature sensor protein DivIVA from B. subtilis colocalizes with EI to these sites. The other polar PTS proteins, HPr and BglG, also colocalize with EI near the IMVs in spherical cells. In accord with the suggestion that the PTS proteins localize to sites of negative curvature, in a murein hydrolase E. coli mutant with unusual polar and septum architecture, EI localized mainly to the highly curved corners of the flat fused poles. Finally, we show that the native EI protein of B. subtilis also localizes to sites of negative curvature in B. subtilis and E. coli cells. Taken together, our results provide evidence that geometric cues play a role in the localization of the general PTS proteins and emphasize the importance of cell architecture in the regulation of metabolic systems.
RESULTS
Candidate system approach for identifying the PTS-polar localization machinery.
It has previously been shown by our lab that the general PTS proteins localize to the poles of E. coli cells (15). Using a functional fusion between the EI and the mCherry proteins (15), we observed EI at the poles regardless of the growth conditions. In an attempt to identify a cellular machinery that mediates polar localization of the PTS proteins, we examined the EI localization pattern in mutant E. coli cells that are impaired in machineries that were implicated in setting up cell polarity, i.e., those that mediate membrane composition, cell division, nucleoid occlusion, or localized translation. Before initiating this approach, we verified that our previous observation of EI-mCherry at the E. coli cell poles is not due to fluorescent protein-mediated clustering (22) by imaging EI fused to a highly monomeric derivative of green fluorescent protein (mGFP), which does not cause false localization patterns (23). The EI-mGFP fusion protein localized to the poles and to the division sites of E. coli cells, similarly to the EI-mCherry protein (see Fig. S1 in the supplemental material).
First, we asked if the polarly localized phospholipid cardiolipin (5, 24), which recruits polar proteins like ProP and MscS, is involved in EI polar positioning. To answer this, we compared the pattern of EI-mCherry distribution in wild-type and in cls mutant E. coli cells. The cls mutant cells have only trace amounts of cardiolipin (CL) (6), as verified by staining them with the 10-N-nonyl acridine orange (NAO) dye that binds to anionic phospholipids, preferably to CL (25) (see Fig. S2 in the supplemental material). The results, presented in Fig. 1, clearly indicate that the substantial decrease in CL levels in the cls mutant cells did not affect the polar and septal localization of the EI-mCherry protein (compare Fig. 1A and B), implying that CL is not involved in EI localization.
FIG 1 .
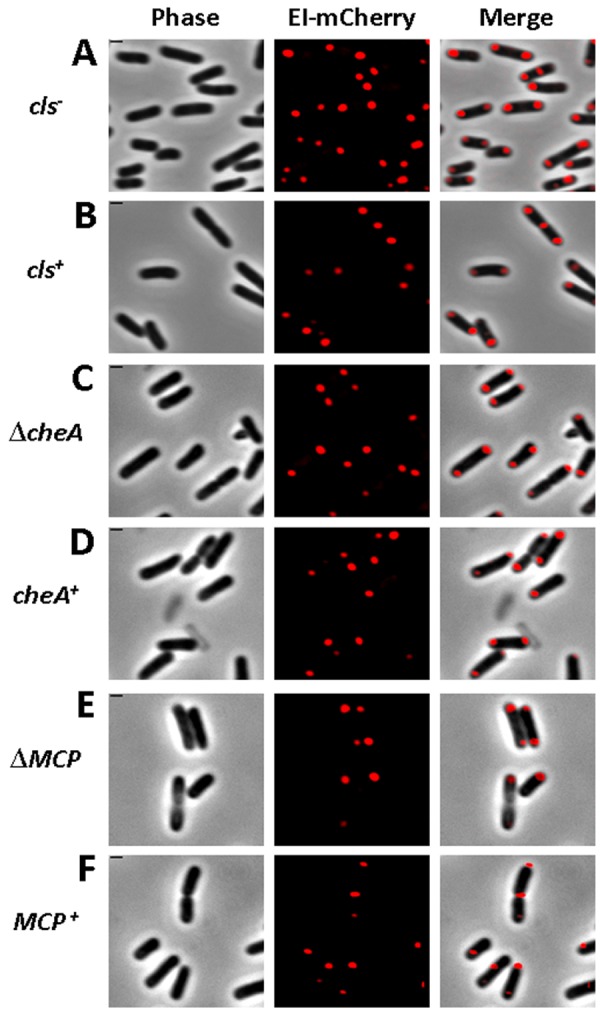
Polar localization of EI does not depend on cardiolipin and the chemotaxis complex. Images showing EI-mCherry distribution in WC3899 E. coli cells mutated in the cardiolipin synthase (cls) gene (A) compared to the parental W3899 cls+ strain (B) and in AW546 ∆cheA E. coli cells deleted for the cheA gene (C) or in UU2612 E. coli cells deleted of all chemotaxis receptors (E) compared to their respective parental strains AW546 and RP437 (D and F, respectively). The mCherry fusion protein was observed by fluorescence microscopy (red), and the cells were observed with phase microscopy (gray). Also, overlays of the signals from the fluorescence and phase microscopy are shown (merge). Scale bar corresponds to 1 µm.
The next candidate that we considered was the chemotaxis complex, whose spatiotemporal organization is reminiscent of that of the PTS (4, 26). Moreover, an interaction between CheA and EI, as well as cross talk between the two systems, has been reported (27, 28). Therefore, we tested the subcellular localization of EI-mCherry in nonchemotactic cheA mutant cells (29) and in cells that lack all chemoreceptors (ΔMCPs) (29). In both cases, polar localization of EI was not affected (compare Fig. 1C to D and E to F), suggesting that the polarly localized chemotaxis complex does not play a role in EI positioning.
The subsequent candidate whose involvement in PTS localization we explored was the cell division machinery. For this purpose, we examined the EI localization pattern in E. coli strains that are impaired in different stages of cell division. We found that in cells depleted for MinCDE or FtsQ, or in which FtsI was inhibited by cephalexin treatment, all growing as nonseptate filamentous cells, EI consistently localized to the poles, as well as to the budding nonmaturing division sites, detected after the addition of the membrane stain Mito Tracker Green (MTG) (Fig. 2A to C). Hence, EI localization does not depend on the key components of the cell division machinery.
FIG 2 .
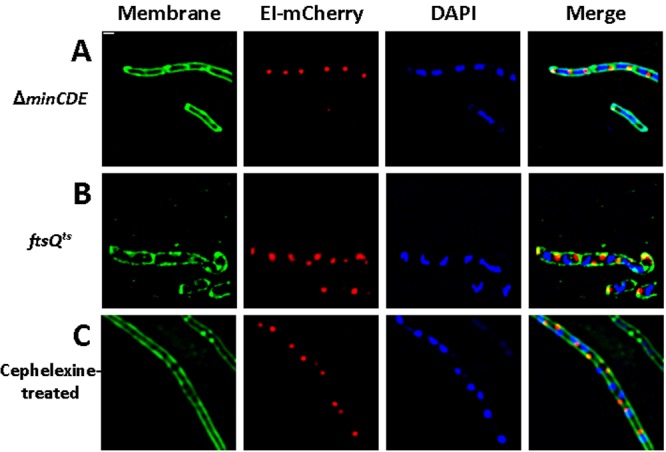
EI localizes to the budding site of the nonmature septum in filamentous nondividing cells. Fluorescence microscopy images showing EI-mCherry distribution (red) in PB114 E. coli cells carrying a ΔminCDE mutation (A), in MCQ1 E. coli cells carrying an ftsQ1ts mutation that were grown in the restrictive temperature (B), and in MG1655 E. coli wild-type cells treated with cephalexin (C). Membrane staining was with MTG (green), and DNA staining was with DAPI (blue). Overlays of the fluorescent signals are also shown (merge). Scale bar corresponds to 1 µm.
The following candidate that we tested was nucleoid occlusion (NO), a process that prevents Z ring assembly and septa formation at nucleoid-occupied places, which is also involved in localizing proteins to the DNA-free regions at the cell poles. To examine if NO mediates EI localization, we took advantage of the finding that blocking translation with antibiotics, e.g., chloramphenicol, causes nucleoid compaction (30, 31), leaving larger DNA-free regions and enabling proteins, whose polar localization is due to NO, to disperse throughout these regions. Hence, we examined the localization pattern of EI-mCherry in cells that have been treated with chloramphenicol. The results indicate that EI-mCherry still localized to foci near the poles and did not disperse throughout the space that had been created due to the compaction of the nucleoid (compare Fig. 3A and B, showing cells untreated and treated with chloramphenicol, respectively). An even more obvious result was obtained when EI-mCherry was imaged in filamentous ftsZ84 mutant cells, grown at the restrictive temperature, which had been treated with chloramphenicol (Fig. 3C). As a control, we imaged cells expressing PopZ-YFP, which had previously been shown to localize to the poles due to NO (32). The results in Fig. S3 in the supplemental material demonstrate that, in filamentous ftsZ84 cells treated with the translation inhibitor kasugamycin, PopZ-YFP largely spreads in the nucleoid-free space (see Fig. S3). Hence, EI is not confined to the poles due to NO.
FIG 3 .
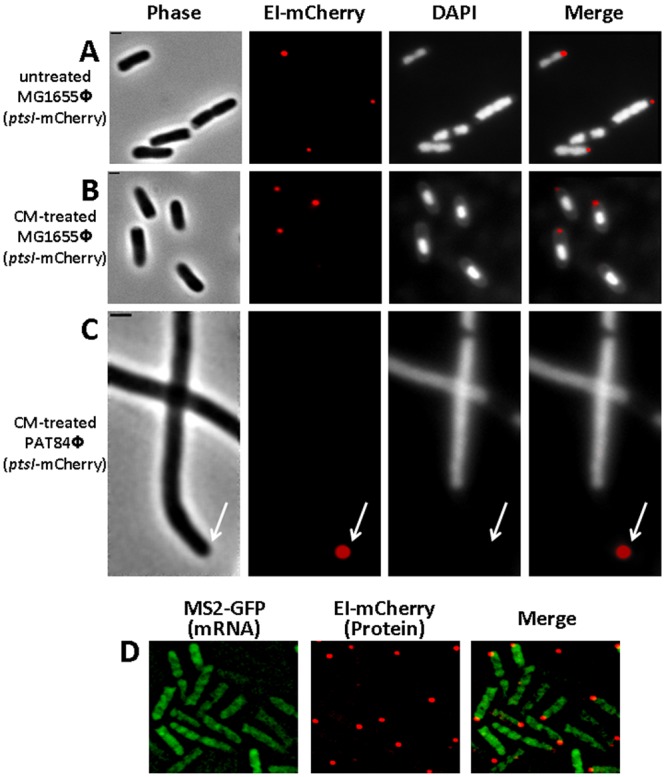
Nucleoid occlusion and localized translation do not account for EI polar localization. (A to C) Images showing EI-mCherry distribution in MG1655Φ(ptsI-mCherry) wild-type E. coli cells untreated (A) or treated with 150 µg/ml chloramphenicol for 20 min (B) and in PAT84Φ(ptsI-mCherry) ftsZ844ts mutant cells that were shifted to the nonpermissive temperature (42°C) and treated with 150 µg/ml chloramphenicol for 20 min. The cells were observed with phase microscopy (gray), the mCherry fusion protein was observed by fluorescence microscopy (red), and DNA staining was with DAPI (white). (D) Fluorescence microscopy images of cells expressing the MS2-GFP protein (green) and an EI-mCherry fusion (red), whose encoding RNA transcripts are tagged with six repeats of the MS2-binding sites. Overlays of the fluorescent signals in panels A to D are also shown (merge). The arrow indicates the pole region. The scale bar corresponds to 1 µm.
RNA targeting and localized translation were the final candidates we examined for a possible role in PTS localization. A recent study from our lab provided evidence that bacterial mRNAs localize to the regions, where their protein products are required, in a translation-independent manner, suggesting that localized translation may exist in bacteria as a mechanism for protein localization (33). To examine the possibility that EI polar localization is due to localized translation, we aimed at imaging the ptsI mRNA and its encoded EI protein simultaneously. To this end, we fused the ptsI gene to mCherry, tagged the resulting ptsI-mCherry mRNA with six repeats of the MS2 coat protein binding sites, and coexpressed it with the MS2-GFP fusion protein (34). The results in Fig. 3D show that, whereas the EI-mCherry protein localized to foci near the cell pole, the RNA transcripts that encode this protein were distributed in the cytoplasm. Hence, localization of the ptsI mRNA and its encoded EI protein do not correlate, precluding the possibility that localized translation, due to mRNA targeting, is involved in EI polar localization.
The general PTS proteins localize to regions of strong negative curvature in spherical E. coli cells.
The rod shape of bacterial cells is maintained by several proteins, including the prokaryotic actin homolog MreB (35). Mutations in their encoding genes or antibiotics that affect the function of these proteins result in the conversion of the cells from rod shaped to spherical (21). MreBCD has also been implicated in polar localization of proteins (36, 37). To examine whether MreB is important for EI localization, we imaged EI-mCherry in cells in which the MreB cytoskeletal system has been disrupted either by treatment with the MreB inhibitor A22 or by the mreBCD mutation. The results in Fig. 4A show that EI-mCherry did not spread out in A22-treated spherical cells but was rather clustered into foci, mostly one focus per cell. In 79% of the cells (n = 451), the EI-mCherry focus was adjacent to the budding site of the intracytoplasmic membrane-bound vesicles (IMVs), previously shown to form in such cells due to an imbalance between membrane synthesis and the cell surface requirement (21). The IMVs can be observed by differential interference contrast (DIC) and MTG membrane staining and are typified by the sharp angles (“corners”) created between them and the cell membrane on both their sides. Similar results were obtained in spherical ∆mreBCD cells, expressing EI-mCherry from a plasmid (Fig. 4B), or in A22-treated cells, expressing EI-mCherry from the chromosome (Fig. 4C), ruling out the possibility that this pattern of localization was caused by the antibiotic treatment or by EI overexpression, respectively. Importantly, when the N-terminal domain of EI fused to mCherry (EI-N-mCherry), previously shown not to localize to the poles (15), was expressed in A22-treated spherical cells, the fusion protein was distributed throughout the cell rather than localizing near the IMVs (Fig. 4D). Together, these results indicate that there is a significant correlation between EI localization and the highly curved sites in spherical cells, suggesting that EI is recruited to sites of strong negative membrane curvature.
FIG 4 .
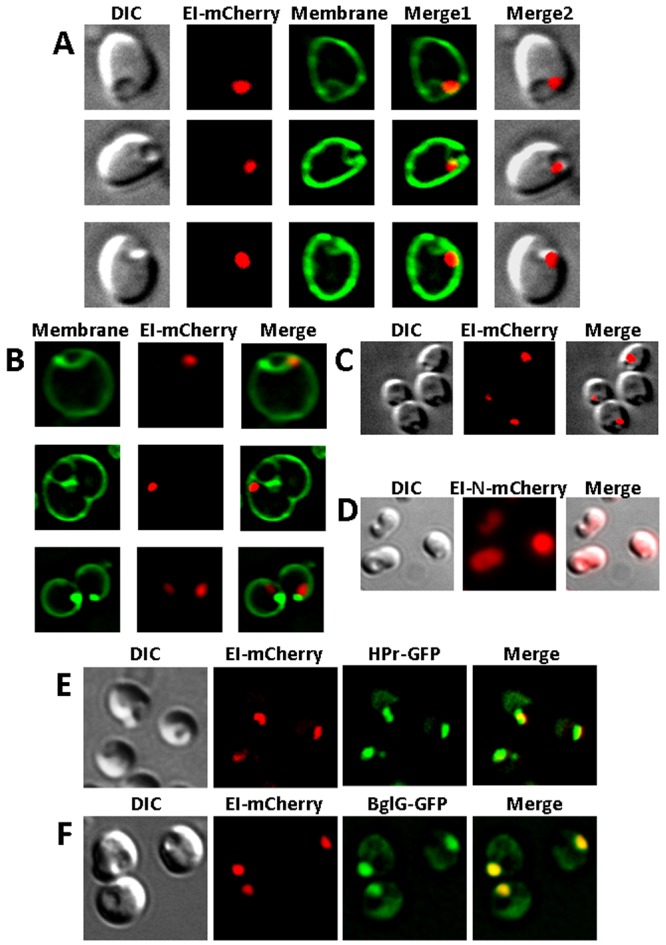
Spatial distribution of EI, HPr, and BglG in spherical E. coli cells and their colocalization. (A to C) Images showing the following E. coli strains expressing EI-mCherry from a plasmid (A and B) or from the chromosome (C): A22-treated PA340 (A), PA340-678 ΔmreBCD (B), A22-treated MG1655Φ(ptsI-mCherry) (C). (D) Images of A22-treated MG1655 E. coli cells expressing EI-N-mCherry. (E and F) Images of MG1655 Δpts E. coli cells expressing EI-mCherry together with either HPr-GFP (E) or BglG-GFP (F). The mCherry and GFP fusion proteins were observed by fluorescence microscopy (red and green, respectively), and the cells were observed with DIC (gray). Membrane staining was with MTG (green). Overlays of the fluorescent signals and DIC microscopy (A, C, and D) or of the fluorescent signals (E and F) are also shown (merge).
Because inactivation of MreB affects not only cell shape but also other molecular processes that are spatially synchronized, we asked if disruption of the cell shape has an effect on the subcellular organization of the PTS as a system and on its ability to function. First, we examined the localization patterns of known EI interaction partners, HPr and BglG, previously shown to colocalize with it, in spherical cells. In wild-type rod-shaped E. coli cells, a functional fusion between the phosphor carrier small protein HPr and GFP was shown to colocalize with EI-mCherry at the poles under nonstimulatory conditions, i.e., in the absence of PTS sugars, although localization of the two proteins was not interdependent (15). To test whether a similar mechanism prevails in spherical cells, we observed the localization of EI-mCherry and HPr-GFP, expressed at comparable levels, in A22-treated ∆pts spherical cells. The results in Fig. 4E demonstrate that, except for a small fraction of HPr-GFP dispersed in the cytoplasm, the two-fluorescent proteins colocalize near the IMVs, suggesting that each of the general PTS proteins potentially recognizes the IMVs. Notably, we did not observe spherical cells in which the HPr-GFP and EI-mCherry foci localized differently near IMVs (n = 518). Next, we coexpressed the auxiliary PTS transcription factor BglG fused to GFP, which has previously been shown to colocalize with the general PTS proteins at the poles (15), together with EI-mCherry and observed the distribution of the two proteins in A22-treated ∆pts spherical cells. The results in Fig. 4F show that BglG-GFP and EI-mCherry colocalize, as in rod-shaped cells. Finally, we compared the rate of glucose uptake by rod-shaped and spherical E. coli cells; the latter shape formed as a result of A22 treatment or due to a deletion of the mreBCD gene. As shown in Fig. S3 in the supplemental material, glucose uptake by spherical and rod-shaped cells was at comparable efficiencies, implying that the PTS system functions appropriately in spherical cells. Together, these results indicate that the changes in cell shape do not affect spatial arrangement of the PTS system, nor does it affect its ability to function. The general PTS proteins localize to negatively curved regions, be it the cell poles of rod-shaped cells or the IMV budding sites of spherical cells.
The E. coli EI protein colocalizes with the negative membrane curvature sensor DivIVA protein from B. subtilis.
The membrane-anchored protein DivIVA from B. subtilis was the first bacterial protein shown to sense negative membrane curvature. In B. subtilis cells, DivIVA localizes predominantly to the division septum and to a lesser extent to the poles. However, when expressed in E. coli cells, DivIVA localized primarily to the poles, compared to the septum (38, 39). The difference in DivIVA localization patterns between B. subtilis and E. coli is explained by the difference in septum formation during cell division in these two organisms, i.e., whereas division of B. subtilis cells results in areas of extreme concave curvature at the two points where the division septum meets the lateral edge of the cell, which are more negatively curved than the hemispherical poles, E. coli cell division is mediated by gradual constriction of the cell membrane, creating angles that are less negatively curved than the poles (8, 39). To test the hypothesis that EI is recruited to sites of strong negative curvature, we compared its localization pattern to that of DivIVA. To this end, we coexpressed EI-mCherry and DivIVA-GFP in rod-shaped as well as in spherical E. coli cells. In dividing rod-shaped cells, the two fluorescent proteins colocalized at the poles and near the forming septum, both showing the highest intensity at the poles (Fig. 5A). In A22-treated spherical E. coli cells, EI-mCherry and DivIVA-GFP colocalize near IMV budding sites, although, unlike EI, some DivIVA-GFP was spread out in the cell (Fig. 5B). To rule out the possibility that DivIVA spreading is due to competition with EI for negatively curved sites, we expressed DivIVA-GFP in ∆pts E. coli cells, which are deleted for the genes expressing EI and HPr. The results (see Fig. S4 in the supplemental material) show that DivIVA-GFP distribution is similar in the presence and absence of EI, that is, it localizes mainly to foci, but also spreads out to a certain extent in the cytoplasm. Hence, EI and the negative membrane curvature sensor DivVIA exhibit the same localization patterns in spherical cells.
FIG 5 .
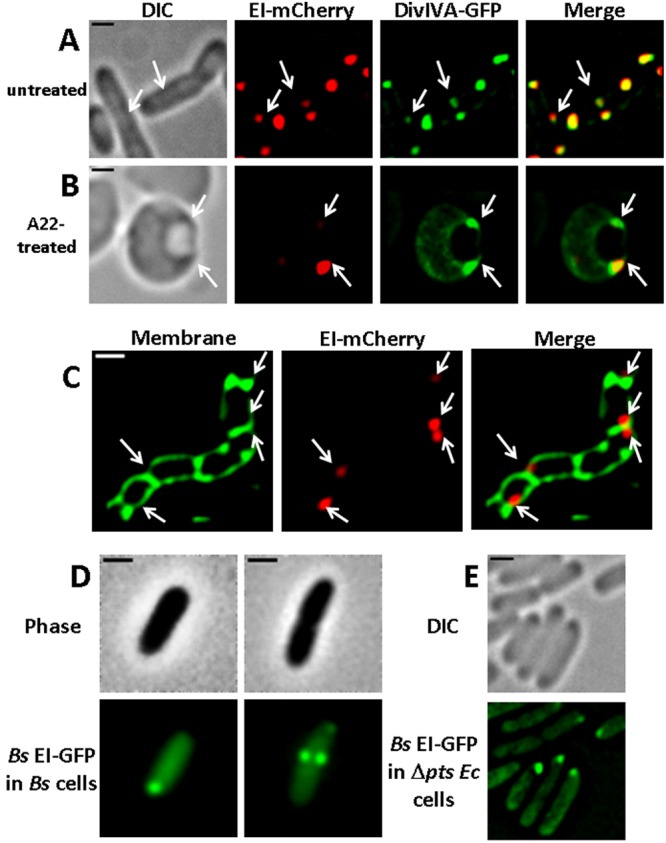
The E. coli EI protein and the B. subtilis DivIVA and EI proteins show similar localization patterns. (A and B) Images showing the distribution pattern of the E. coli EI protein fused to mCherry and the B. subtilis DivIVA protein fused to GFP in wild-type E. coli cells untreated (A) or treated with A22 (B). (C) Images showing EI-mCherry in MHD63, a murein hydrolase E. coli mutant. (D and E) Images showing the distribution pattern of the B. subtilis EI protein fused to GFP in wild-type B. subtilis (Bs) cells (D) and in ∆pts E. coli (Ec) cells (E). The cells were observed with phase microscopy or DIC (gray), and the mCherry and GFP fusion proteins were observed by fluorescence microscopy (red and green, respectively). Overlays of the fluorescent signals in panels A to C are also shown (merge). Arrows indicate negatively curved regions. The scale bar corresponds to 1 µm.
Next, we examined EI localization in murein hydrolase E. coli mutant cells, which cannot separate after division, thus forming long chains of cells divided by relatively flat septa, reminiscent of B. subtilis cell chains (40). When the B. subtilis DivIVA protein was expressed in this E. coli mutant, it was observed mainly at the corners formed between the flat septa and the lateral cell membrane (39). When EI-mCherry was expressed in this mutant, it localized mainly to one of the highly curved corners of the flat fused poles (Fig. 5C). The difference in the average number of foci formed by the two proteins per cell is discussed below. Based on the results in Fig. 5, we conclude that the soluble EI protein, like the membrane-anchored DivIVA protein, is recruited to strongly curved subcellular sites.
Membrane curvature-mediated localization of EI is conserved across species.
The major PTS protein EI is a widely conserved protein in both Gram-positive and Gram-negative bacteria. To understand if negative curvature-mediated localization is a conserved property of EI, we examined the localization of the B. subtilis EI protein, which shares a significant homology with the E. coli EI. For this purpose, we tagged the B. subtilis EI protein with GFP at its N terminus and introduced it into the amyE site in the B. subtilis chromosome. The resulting EI-GFP fusion protein, expressed from a xylose-inducible promoter, was functional and could complement a ∆ptsI B. subtilis strain grown in minimal medium containing a PTS sugar as a sole carbon source, provided that xylose was added. When expressed in rich medium, such as LB, EI-GFP was distributed throughout the cell (data not shown). However, when expressed in cells grown in C medium supplemented with mannitol as a sole carbon source, EI-GFP localized in a pattern which is very similar to that reported for DivIVA, i.e., in nondividing B. subtilis cells, EI-GFP localized mainly to the cell pole, whereas in dividing cells it was detected as two bright spots near the forming septum and a weaker spot at the pole (Fig. 5D). Furthermore, when the B. subtilis EI-GFP protein was expressed in ∆pts E. coli cells, i.e., cells lacking the endogenous general PTS proteins, it localized near the cell pole in 41% of the cells (n = 412) (Fig. 5E). Of note, the B. subtilis EI-GFP protein can complement an E. coli strain with a mutation in the ptsI gene (see Fig. S6), in agreement with the ability of the B. subtilis EI to substitute for the E. coli EI in vitro (41). Taken together, we conclude that localization of EI to sites of strong negative curvature is a conserved property of this protein.
DISCUSSION
Molecular processes are often confined to subcellular compartments, thus circumventing the problem of intracellular molecular crowding (42, 43). Even in the absence of membrane barriers, many soluble enzymes that carry out distinct processes are integral parts of macromolecular assemblies, in both prokaryotic and eukaryotic cells (44, 45). The composition of macromolecular assemblies is dynamic, as certain components may participate in forming various assemblies at different subcellular regions or move between them, depending on environmental conditions. Such assemblies appear to have particular significance in compartmentalizing and regulating metabolic pathways (46, 47). There are obvious advantages to bringing together sequentially acting enzymes of specific metabolic pathways, as exemplified by the bacterial microcompartments (BMCs) that encapsulate metabolic reactions within a proteinaceous shell, thus enhancing catalytic activity, protecting vulnerable enzymes from degradation, and sequestering toxic intermediates (48). It is reasonable to assume that organizing signaling systems that respond to metabolites in macromolecular assemblies increases the efficiency of pathway flux and helps in generating optimal responses to environmental cues.
The poles of rod-shaped bacteria are emerging as important subdomains to which various molecular assemblies, which sense metabolites and adjust cell metabolism and behavior accordingly, localize. Examples include the chemotaxis complex in E. coli (49, 50) and in Caulobacter crescentus (51), the flagellum-dependent motility system in Vibrio, Pseudomonas, Helicobacter, and Campylobacter species, the flagellum-independent gliding motility in Myxococcus xanthus (52), and the control center of the PTS system in E. coli, which plays a central role in controlling the metabolic state in most bacterial cells depending on carbohydrate availability (15). Except for the PTS, all other polarly localized signaling systems have a membrane-anchored component(s). The PTS proteins known to localize to the poles are all soluble. They include EI, which remains at the poles independent of environmental conditions, HPr, which discharges from the poles in response to the presence of PTS sugars in the growth medium, and transcription factors that regulate PTS operons, i.e., BglG in E. coli and LicT in B. subtilis, which are recruited to the poles depending on the availability of sugars that are transported and metabolized by the products of the operons that they regulate (15, 17). The membrane-bound components of the PTS, the sugar permeases, do not localize specifically to the poles; rather, they are distributed around the cell circumference (15, 16). Hence, identifying the cues that recruit the soluble PTS proteins to the poles is important not only to improve our understanding of this central system, which has important implications on most other cellular pathways, but also to reveal fundamental mechanisms underlying organization and regulation of soluble systems in the bacterial cell.
To identify the cues that recruit the PTS proteins to the poles, we monitored the distribution pattern of the PTS proteins in cells in which known factors that set up cell polarity or that are tightly linked to it were systematically disrupted. We focused mainly, although not only, on EI, whose distribution pattern does not change much over time, and environmental conditions. We could thus show that EI localization does not depend on interaction with the phospholipid cardiolipin. Of note, proteins shown thus far to localize to the poles in a cardiolipin-dependent manner were integral membrane proteins and, hence, were more likely to interact with a phospholipid than the soluble EI protein. We also ruled out the possibility that the polar chemotaxis complex is required for recruiting EI to the poles, although the opposite scenario, i.e., that the PTS affects the organization of the chemotaxis complex, remains open. Because cell polarization and cell division are highly interrelated processes that regulate one another in a spatiotemporal manner (53) and because the cell poles are derived from the division septum, we could not rule out the possibility that the cell division machinery provides a mechanistic basis for the positioning of proteins at the poles. Indeed, birth scar proteins, formed at the division site that later establish polarity, have been reported in C. crescentus (54, 55). Still, despite the detection of EI near the forming septum in dividing cells, our results rule out the possibility that the cell division machinery is involved in recruiting EI to the poles. Additionally, we show that localization of EI to the poles is not due to nucleoid occlusion, since EI consistently localized to the poles of nucleoid-compacted cells, rather than spreading out in the nucleoid-free space, as observed for PopZ. Notably, the bacterial chromosome is not arbitrarily arranged and packaged, and certain regions are always positioned near the poles (56). Thus, the visualization of EI foci near the poles of nucleoid-compacted cells also precludes the possibility that EI interacts with a pole-proximal region in the bacterial chromosome or with certain proteins that direct this chromosomal region to the poles.
Targeting of mRNAs to the site where their protein products are required to enable localized translation is a well-established mechanism of protein localization in eukaryotic cells (57, 58). Until recently, it was believed that localization of bacterial proteins depends solely on features within the proteins and, hence, occurs either co- or posttranslationally (1). Recently, the possibility that bacterial proteins can be localized via mRNA targeting and localized translation, similarly to higher-order cells, has been suggested (33). Based on the results presented here, this is not the case for EI, since its encoding mRNA is spread in a typical distribution pattern visualized with cytoplasmic RNAs (see reference 33), whereas the protein itself is clearly observed near the poles. This result supports our previous suggestion that, similar to eukaryotic cells, certain mRNAs in bacteria localize to the future destination of their encoded proteins, whereas others do not (2), implying that enzymes like EI localize after being translated.
The nature of the cues that affect EI localization was unveiled when we visualized EI in E. coli cells that lost the ability to form a rod shape and are supposedly poleless. Notably, such so-called spherical cells, obtained by the disruption of the cytoskeletal or cell wall synthetic machineries via mutations or antibiotic treatments, do not occupy a perfect sphere shape. Rather, due to the imbalance between membrane synthesis and cell surface requirements, the cells produce phospholipid membrane in excess, leading to the formation of IMVs (21). The regions where the lateral cell membrane meets these protruding membrane involutions represent sites of high negative curvature, thus bearing geometric resemblance to the poles of rod-shaped cells. The IMVs were shown to be implicated in the localization of cell division proteins, including MinD (21), which oscillates between the poles of rod-shaped cells and has been suggested to sense curved regions (10). In light of all this information, our observation that EI localizes next to the IMV budding sites in spherical E. coli cells suggests that EI is recruited to sites of negative membrane curvature.
This hypothesis is reinforced by our observation that DivIVA, a negative membrane curvature sensor protein from B. subtilis, colocalizes with EI next to the IMVs in spherical E. coli cells. DivIVA emerges as a useful marker for tracking membrane organization in vivo, since a recent study also used DivIVA-GFP to detect sharply bent membrane regions in antibiotic-treated cells (59). The use of DivIVA-GFP as a “molecular sensor” for membrane curvature not only validates that the regions next to the IMVs are negatively curved, but it also is helpful in identifying regions of negative curvature when IMVs are not readily resolvable by DIC. Consistent with our assumption about the role of geometric cues in EI localization, in dividing rod-shaped E. coli cells, EI-mCherry and DivIVA-GFP formed less intense foci near the forming division septum (less curved regions) and a more intense foci near the poles (more curved regions). Still, DivIVA and EI differed in their localization patterns in a murein hydrolase E. coli mutant, which forms long chains of cells divided by rather flat septa; that is, whereas DivIVA was observed mainly in both negatively curved sites formed between the flat septa and the lateral cell membrane (39), EI-mCherry was observed mainly in one such site per cell. This difference might reflect differences in the oligomeric state and higher-order structure of the two proteins. Alternatively, it might be due to differences in their interacting partners, since DivIVA is bound to the membrane, whereas the soluble EI protein is probably associated with factors that maintain it at the poles.
The finding of Bendezu and de Boer that the sites in which MinD accumulates and between which it oscillates invariably coincide with the sites of IMV presence in spherical E. coli cells (21) raises the question of whether the IMV budding sites bear functional, and not only geometric, resemblance to the poles of rod-shaped cells. The results presented herein support this notion; not only are the known PTS interaction partners, HPr and BglG, colocalized with EI to these sites, but the kinetics of sugar uptake by spherical cells and normal rods are comparable. In light of our observations and previously published data, we propose that the IMV budding sites in poleless spherical E. coli cells represent pole-like regions, which maintain not only the geometric but also the functional features of the poles.
Recruitment to regions of high negative curvature seems to be a feature of the general PTS proteins which is conserved across species. Thus, when EI from B. subtilis was expressed in E. coli cells, it localized to the poles, which are the sites of highest negative curvature in these cells, similarly to the localization preference of the endogenous E. coli EI protein. When the same protein was expressed in B. subtilis cells, it localized preferentially to the two points where the division septum meets the lateral edge of the cell, which are the sites of highest negative curvature in these cells, but in the absence of forming septum, it localized to the cell pole. Still, one difference between the two organisms is that in E. coli cells, the EI proteins from both species localize to the cell pole independent of growth conditions, whereas in B. subtilis cells, EI localization seems to depend on growth conditions. Thus, when grown in LB, the native B. subtilis EI showed very little if any distinct localization (data not shown), similar to the distribution documented previously (17). The reason for the dependence of the B. subtilis EI localization pattern on the medium, which is in contrast to the lack of such dependence for E. coli EI (15), is currently not known. Since our results suggest that in addition to membrane curvature, more factors are involved in localizing EI to the poles (see below), it is likely that there are differences between EI pole-tethering factors in the two organisms.
The few proteins suggested thus far to be recruited to negatively curved regions contain a membrane-tethering component (8). The mechanisms that tether soluble proteins to the poles are less known. It has been suggested that PopZ in Caulobacter relies on multimerization in chromosome-free regions (32, 60, 61), but this does not seem applicable for EI and HPr, which have never been suggested to multimerize beyond dimers. The question then arises: does EI have other properties that direct it to the membrane, or does it localize via a partner that tethers it to curved membrane regions? To discern between these possibilities, we compared the EI localization pattern when expressed at a physiological level and when overexpressed from a plasmid in an E. coli Δpts strain. In the first case, EI was detected only as a spot (Fig. 4C), whereas in the second case, EI was detected as a rather large focus with the excess spreading out in the cytoplasm (see Fig. S5B in the supplemental material). These observations exclude the possibility that EI is able to recognize the membrane by itself, since in such a case we would expect to detect foci in other curved membrane locations upon overexpression rather than spread out in the cytoplasm, and suggest that other factors are involved in anchoring it to curved regions. What are these factors? The gap between the growing knowledge on bacterial macromolecular assemblies and the limited knowledge on the mechanisms that underlie their association led to the suggestion that bacterial cells have various kinds of structuring proteins, often associated with or penetrating into the cytoplasmic membrane (62). We speculate that this group includes proteins that sense membrane curvature and provide the bacterial cells with means to sort and position soluble proteins in highly curved regions.
The implications of cellular architecture on metabolic regulation.
Undoubtedly, cell shape influences many processes, including cell survival and growth (63). Under routine laboratory growth conditions, changes in cell shape are not commonly observed. However, under conditions of environmental insults, including oxygen limitation, the presence of antibiotics, growth under competition, or intracellular growth in host cells, bacteria many times undergo changes in cell shape, including formation of spherical and filamentous cells (64, 65). Changes in cell shape often correlate with changes in localization of the cell division and cytoskeletal proteins (21, 66), ultimately resulting in a decrease or cessation of cell division. Under such unfavorable conditions, the cell division proteins are maintained in a nonfunctional state, rather than being degraded, making them available for the resumption of cell division when favorable environmental conditions are restored (66, 67). Negative regulation of protein activity by transient storage at the poles and relocation into the active pool upon requirement has been reported for MurG, an enzyme involved in cell wall biosynthesis (68). Our study suggests that cellular architecture is also involved in the regulation of major metabolic systems in bacteria, such as the PTS. However, unlike the cell division and cell wall biosynthesis proteins, changes in cellular morphology correlate with localization of the PTS proteins to subcellular domains that maintain their activity.
Combining the previously published data with the results reported here, we propose that changes in cell physiology induce dynamic survival strategies that involve reorganization of central cellular systems and, possibly, changes in cell morphology. However, whereas the cell division proteins are temporarily stored in an inactive form, thus saving the limited energy of the cells for processes essential for survival, the PTS and potentially other metabolic systems localize to subcellular domains, where they are active in sensing the environmental conditions and in the uptake of available nutrients, thus maintaining both cell viability and metabolic alertness. Upon encounter with favorable conditions, changes in cell morphology correlate with relocation of the cell division and metabolic machineries and reestablishment of normal cellular architecture. Our model hypothesizes that relocations of central cellular systems will be discovered in the future in organisms that undergo a reversible switch between rods and cocci upon encounter of unfavorable conditions, e.g., Paenibacillus dendritiformis (69). All together, we suggest that spatial regulation of central systems, including metabolic machineries, not only maintains optimal cell growth under favorable conditions but also provides a novel survival strategy that helps bacteria cope with certain unfavorable conditions.
MATERIALS AND METHODS
Bacterial strains and growth media.
Bacterial strains used in this study are listed in Table S1 in the supplemental material. Unless otherwise indicated, cells were grown in LB at 30°C. PAT84Φ(ptsI-mCherry), expressing an EI-mCherry chromosomal fusion in an ftsZ844ts mutant background, was constructed by transferring ptsI-mCherry from MG1655Φ(ptsI-mCherry) to the chromosome of PAT84 by P1 transduction, as described before (70). YE101, a B. subtilis strain that expressed an EI-GFP fusion from its chromosome, was constructed by integrating the pEA18-EI-GFP plasmid into the chromosome of GR39, a ∆ptsI B. subtilis strain obtained by transforming chromosomal DNA of GR26 (71), at the amyE locus, as described before (72). When appropriate, antibiotics were added at the following concentrations: 30 µg/ml kanamycin, 100 µg/ml ampicillin, and 25 µg/ml chloramphenicol for E. coli strains and 100 µg/ml spectinomycin and 6 µg/ml chloramphenicol for B. subtilis strains.
Plasmids.
Construction of pBADLLEI-mCherry, which encodes translational fusion of EI to mCherry, pBADANSEI-mCherry&HPr-GFP, which encodes both EI-mCherry and HPr-GFP from the same promoter, and pJS185, which encodes GFP fused to BglG, has been described before (15). pBADGSEI-mGFP, which expresses EI fused to monomeric GFP, was constructed by replacing the mCherry-encoding sequence with mGFP as follows: mGFP, which carries an A206K substitution in the GFPmut2 sequence, was amplified by PCR from plasmid pAR100 (73) using forward and reverse primers that introduced SacI and XbaI sites, respectively; the amplified DNA was ligated to the pBADLLEI-mCherry plasmid, which was digested with SacI and XbaI. Construction of pGSDivIVA-GFP, which encodes DivIVA fused with GFP, was carried out in two steps. First, the GFP-encoding sequence was amplified by PCR from pBADANSEI-mCherry&HPr-GFP with primers that introduced SalI (forward) and HindIII (reverse) sites and inserted into pQE32 (Qiagene), which was digested with SalI and HindIII, to generate pGSlacGFP. Second, the divIVA gene was amplified from B. subtilis PY79 genomic DNA, using primers that introduced SacI (forward) and SalI (reverse) sites and a sequence encoding a three alanine linker, and the resulting amplicon was ligated to pGSlacGFP, digested with SacI and SalI.
Construction of pGSbsEI-GFP, which encodes EI from B. subtilis fused with GFP, was carried out by amplifying the ptsI gene from the chromosome of the B. subtilis PY79 strain, using BamHI forward and SalI reverse primers, and ligating the resultant amplicon to the pGSlacGFP plasmid, digested with the same enzymes. pZE12EImCherry6xbs was constructed by amplifying EI-mCherry along with its ribosome binding site (RBS) from pBADLLEI-mCherry with primers that introduced BglII and EagII sites. The fragment was ligated to pZE126xbs (46), which was cleaved by the same enzymes. pEA18-gfp-ptsI was constructed by amplifying the ptsI gene from B. subtilis PY79 genomic DNA, using forward and reverse primers that introduced NotI and BamHI sites, respectively, and the resultant amplicon was ligated to pEA18 (74), digested with NotI and BamHI.
Fluorescence microscopy.
Cells, supplemented with the appropriate antibiotics, were grown in LB medium at 30°C till mid-log phase. To obtain spherical E. coli cells, A22 (2 µg/ml) was added from the beginning of the growth. For obtaining filamentous cells, cephalexin (20 µM) was added from the beginning of the growth. To induce nucleoid compaction, cells were treated with chloramphenicol (150 µg/ml) or kasugamycin (300 µg/ml) for 20 min. To express EI-mCherry, HPr-GFP, and EI-mGFP from the pBAD promoter, 0.1% arabinose was added at an optical density at 600 nm (OD600) of 0.3 for 1 h for rod-shaped cells and for 10 to 20 min in spherical cells. To express BglG-GFP and DivIVA-GFP, 0.1 mM and 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) were added for 1 h, respectively. B. subtilis cells expressing a chromosomally integrated EI-GFP were grown in C medium supplemented with 1% mannitol till an OD600 of 0.4. Expression of the EI-GFP fusion protein was induced in these cells by adding 0.01% xylose from the beginning of growth. To express the B. subtilis EI protein fused to GFP from pGSbsEI-GFP in E. coli cells, 1 mM IPTG was added for 1 to 2 h after cells reached an OD600 of 0.3.
Fluorescence microscopy was carried out as described previously (75). In brief, 0.5 ml of cells was centrifuged, washed with 1× phosphate-buffered saline (PBS), and finally resuspended in 10 µl of PBS. When indicated, the membrane was stained with MTG (Molecular Probes, Invitrogen) at a final concentration of 10 µM, and DNA was stained with DAPI (4′,6-diamidino-2-phenylindole; Sigma) at a final concentration of 2 µg/ml. For cardiolipin staining, cells were washed and resuspended in 1× PBS that contained 100 nM nonyl acridine orange (NAO) and incubated at 37°C for 15 min. Cells were visualized and photographed using an Axioplan2 microscope (Zeiss) equipped with a CoolSnap HQ camera (Photometrics, Roper Scientific), Axiovert 200 M (Zeiss) equipped with a CoolSnap HQ camera (Photometrics, Roper Scientific), or Nikon Eclipse Ti equipped with an ORCA R2 camera (Hamamatsu Photonics). Images were processed using Metamorph Software (Molecular Devices).
Glucose consumption assay.
Wild-type, A22-treated wild-type (2 µg/ml), ∆mreBCD, and ∆pts cells were grown in LB supplemented with 25 mM glucose, and the measurements of glucose consumption were performed as described before (76).
SUPPLEMENTAL MATERIAL
EI fused to monomeric GFP localizes to the E. coli cell poles. DIC and fluorescence microscopy images of E. coli cells expressing EI-mGFP and an overlay of the signals (green, gray, and merge, respectively). The scale bar corresponds to 1 µm. Download
Cardiolipin staining in cls mutant and wild-type cells. E. coli cells mutated in the cardiolipin synthase (cls) gene (A) and the parental cls+ strain (B) were stained with NAO (10-N-nonyl acridine orange), which binds to anionic lipids, preferably to CL. NAO was observed by fluorescence microscopy (green), and the cells were observed with phase microscopy (gray). Overlays of the signals are also shown (merge). The scale bar corresponds to 1 µm. Download
Localization of PopZ in nucleoid-compacted cells. Images showing PopZ-YFP in PAT84 ftsZ844ts mutant cells that were shifted to the nonpermissive temperature (42°C) and treated with 300 µg/ml kasugamycin for 20 min. The cells were observed with DIC microscopy (gray), the YFP fusion protein was observed by fluorescence microscopy (green), and DNA staining was with DAPI (white). An overlay of the fluorescent signals is also shown (merge). An arrow indicates the nucleoid-free region in a nucleoid-compacted cell. An asterisk indicates the pole region in a cell in which the nucleoid has not been compacted. Download
Glucose consumption by rod-shaped and spherical E. coli cells. MG1655 E. coli cells (wild type), untreated (blue) or treated with A22 (red), ΔmreBCD E. coli cells (green), and MG1655 Δpts E. coli cells (purple) were grown in LB medium containing 25 mM glucose, and glucose consumption measurements were performed as described in Materials and Methods in the presence of 1 mM glucose. Download
Localization of DivIVA-GFP and EI-mCherry in E. coli Δpts cells. Images showing the distribution pattern of B. subtilis DivIVA protein fused with GFP (top) and of overexpressed E. coli EI protein fused to mCherry (bottom) in E. coli Δpts cells. The cells were observed with DIC (gray), and the GFP fusion protein (green) or mCherry fusion protein (red) were observed by fluorescence microscopy. Download
Complementation of a ptsI E. coli mutant with the B. subtilis EI protein. E. coli ptsI19ts cells (A) and ptsI19ts cells containing either pBADLLEI-mCherry, which expresses the E. coli EI protein fused to mCherry (B), or pGSbsEI-GFP, which expresses the B. subtilis EI protein fused to GFP (C), were streaked on a MacConkey plate containing 1% mannitol and 1 mM IPTG and incubated overnight at the restrictive temperature (42°C). Download
Bacterial strains used in this study and their relevant phenotype.
Plasmids used in this study and the proteins they encode.
ACKNOWLEDGMENTS
This research was supported by the Israel Science Foundation founded by the Israel Academy of Sciences and Humanities.
We thank Sigal Ben-Yehuda and her lab members, Albert Taraboulos and his lab members, and Zakhariya Manevitch for help with fluorescence microscopy. We acknowledge the Coli Genetic Stock Center (CGSC) and the following individuals for gifts of strains: Masaaki Wachi, Janet Wood, Sandy Parkinson, Waldemar Vollmer, Sigal Ben-Yehuda, Piet A. J. de Boer, Josef Deutscher, Christine Jacobs-Wagner, and Michael Eisenbach. We appreciate helpful discussions with Anat Nussbaum-Shochat, Moti Baum, Shanmugapriya Kannaiah, Aya Khwaja, Avi-ad Buskila, and Nitsan Albocher.
Footnotes
Citation Govindarajan S, Elisha Y, Nevo-Dinur K, Amster-Choder O. 2013. The general phosphotransferase system proteins localize to sites of strong negative curvature in bacterial cells. mBio 4(5):e00443-13. doi:10.1128/mBio.00443-13.
REFERENCES
- 1. Rudner DZ, Losick R. 2010. Protein subcellular localization in bacteria. Cold Spring Harb. Perspect. Biol. 2:a000307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nevo-Dinur K, Govindarajan S, Amster-Choder O. 2012. Subcellular localization of RNA and proteins in prokaryotes. Trends Genet. 28:314–322 [DOI] [PubMed] [Google Scholar]
- 3. Toro E, Shapiro L. 2010. Bacterial chromosome organization and segregation. Cold Spring Harb. Perspect. Biol. 2:a000349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Govindarajan S, Nevo-Dinur K, Amster-Choder O. 2012. Compartmentalization and spatio-temporal organization of macromolecules in bacteria. FEMS Microbiol. Rev. 36:1005–1022 [DOI] [PubMed] [Google Scholar]
- 5. Mileykovskaya E, Dowhan W. 2000. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 182:1172–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Romantsov T, Helbig S, Culham DE, Gill C, Stalker L, Wood JM. 2007. Cardiolipin promotes polar localization of osmosensory transporter ProP in Escherichia coli. Mol. Microbiol. 64:1455–1465 [DOI] [PubMed] [Google Scholar]
- 7. Romantsov T, Battle AR, Hendel JL, Martinac B, Wood JM. 2010. Protein localization in Escherichia coli cells: comparison of the cytoplasmic membrane proteins ProP, LacY, ProW, AqpZ, MscS, and MscL. J. Bacteriol. 192:912–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramamurthi KS, Losick R. 2009. Negative membrane curvature as a cue for subcellular localization of a bacterial protein. Proc. Natl. Acad. Sci. U. S. A. 106:13541–13545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eswaramoorthy P, Erb ML, Gregory JA, Silverman J, Pogliano K, Pogliano J, Ramamurthi KS. 2011. Cellular architecture mediates DivIVA ultrastructure and regulates min activity in Bacillus subtilis. mBio 2(6):e00257-11. 10.1128/mBio.00257-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Renner LD, Weibel DB. 2011. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc. Natl. Acad. Sci. U. S. A. 108:6264–6269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramamurthi KS, Lecuyer S, Stone HA, Losick R. 2009. Geometric cue for protein localization in a bacterium. Science 323:1354–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Greenfield D, McEvoy AL, Shroff H, Crooks GE, Wingreen NS, Betzig E, Liphardt J. 2009. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 7:e1000137. 10.1371/journal.pbio.1000137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deutscher J. 2008. The mechanisms of carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 11:87–93 [DOI] [PubMed] [Google Scholar]
- 14. Lengeler JW, Jahreis K. 2009. Bacterial PEP-dependent carbohydrate: phosphotransferase systems couple sensing and global control mechanisms. Contrib. Microbiol. 16:65–87 [DOI] [PubMed] [Google Scholar]
- 15. Lopian L, Elisha Y, Nussbaum-Shochat A, Amster-Choder O. 2010. Spatial and temporal organization of the E. coli PTS components. EMBO J. 29:3630–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopian L, Nussbaum-Shochat A, O’Day-Kerstein K, Wright A, Amster-Choder O. 2003. The BglF sensor recruits the BglG transcription regulator to the membrane and releases it on stimulation. Proc. Natl. Acad. Sci. U. S. A. 100:7099–7104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rothe FM, Wrede C, Lehnik-Habrink M, Görke B, Stülke J. 2013. Dynamic localization of a transcription factor in Bacillus subtilis: the LicT antiterminator relocalizes in response to inducer availability. J. Bacteriol. 195:2146–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanaka Y, Itoh F, Kimata K, Aiba H. 2004. Membrane localization itself but not binding to IICB is directly responsible for the inactivation of the global repressor Mlc in Escherichia coli. Mol. Microbiol. 53:941–951 [DOI] [PubMed] [Google Scholar]
- 19. Richet E, Davidson AL, Joly N. 2012. The ABC transporter MalFGK(2) sequesters the MalT transcription factor at the membrane in the absence of cognate substrate. Mol. Microbiol. 85:632–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bouraoui H, Ventroux M, Noirot-Gros MF, Deutscher J, Joyet P. 2013. Membrane sequestration by the EIIB domain of the mannitol permease MtlA activates the Bacillus subtilis mtl operon regulator MtlR. Mol. Microbiol. 87:789–801 [DOI] [PubMed] [Google Scholar]
- 21. Bendezú FO, de Boer PA. 2008. Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J. Bacteriol. 190:1792–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. 2012. Segregation of molecules at cell division reveals native protein localization. Nat. Methods 9:480–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zacharias DA, Violin JD, Newton AC, Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 [DOI] [PubMed] [Google Scholar]
- 24. Cronan JE. 2003. Bacterial membrane lipids: where do we stand? Annu. Rev. Microbiol. 57:203–224 [DOI] [PubMed] [Google Scholar]
- 25. Petit JM, Maftah A, Ratinaud MH, Julien R. 1992. 10N-nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur. J. Biochem. 209:267–273 [DOI] [PubMed] [Google Scholar]
- 26. Kentner D, Sourjik V. 2006. Spatial organization of the bacterial chemotaxis system. Curr. Opin. Microbiol. 9:619–624 [DOI] [PubMed] [Google Scholar]
- 27. Lux R, Jahreis K, Bettenbrock K, Parkinson JS, Lengeler JW. 1995. Coupling the phosphotransferase system and the methyl-accepting chemotaxis protein-dependent chemotaxis signaling pathways of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 92:11583–11587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neumann S, Grosse K, Sourjik V. 2012. Chemotactic signaling via carbohydrate phosphotransferase systems in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 109:12159–12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parkinson JS, Houts SE. 1982. Isolation and behavior of Escherichia coli deletion mutants lacking chemotaxis functions. J. Bacteriol. 151:106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Helvoort JM, Kool J, Woldringh CL. 1996. Chloramphenicol causes fusion of separated nucleoids in Escherichia coli K-12 cells and filaments. J. Bacteriol. 178:4289–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun Q, Margolin W. 2004. Effects of perturbing nucleoid structure on nucleoid occlusion-mediated toporegulation of FtsZ ring assembly. J. Bacteriol. 186:3951–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ebersbach G, Briegel A, Jensen GJ, Jacobs-Wagner C. 2008. A self-associating protein critical for chromosome attachment, division, and polar organization in Caulobacter. Cell 134:956–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nevo-Dinur K, Nussbaum-Shochat A, Ben-Yehuda S, Amster-Choder O. 2011. Translation-independent localization of mRNA in E. coli. Science 331:1081–1084 [DOI] [PubMed] [Google Scholar]
- 34. Golding I, Cox EC. 2004. RNA dynamics in live Escherichia coli cells. Proc. Natl. Acad. Sci. U. S. A. 101:11310–11315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cabeen MT, Jacobs-Wagner C. 2005. Bacterial cell shape. Nat. Rev. Microbiol. 3:601–610 [DOI] [PubMed] [Google Scholar]
- 36. Cowles KN, Gitai Z. 2010. Surface association and the MreB cytoskeleton regulate pilus production, localization and function in Pseudomonas aeruginosa. Mol. Microbiol. 76:1411–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nilsen T, Yan AW, Gale G, Goldberg MB. 2005. Presence of multiple sites containing polar material in spherical Escherichia coli cells that lack MreB. J. Bacteriol. 187:6187–6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edwards DH, Thomaides HB, Errington J. 2000. Promiscuous targeting of Bacillus subtilis cell division protein DivIVA to division sites in Escherichia coli and fission yeast. EMBO J. 19:2719–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lenarcic R, Halbedel S, Visser L, Shaw M, Wu LJ, Errington J, Marenduzzo D, Hamoen LW. 2009. Localisation of DivIVA by targeting to negatively curved membranes. EMBO J. 28:2272–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Höltje JV. 2001. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 41:167–178 [DOI] [PubMed] [Google Scholar]
- 41. Reizer J, Sutrina SL, Wu LF, Deutscher J, Reddy P, Saier MH., Jr. 1992. Functional interactions between proteins of the phosphoenolpyruvate:sugar phosphotransferase systems of Bacillus subtilis and Escherichia coli. J. Biol. Chem. 267:9158–9169 [PubMed] [Google Scholar]
- 42. Ellis RJ. 2001. Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11:114–119 [DOI] [PubMed] [Google Scholar]
- 43. Lewitzky M, Simister PC, Feller SM. 2012. Beyond “furballs” and “dumpling soups”—towards a molecular architecture of signaling complexes and networks. FEBS Lett. 586:2740–2750 [DOI] [PubMed] [Google Scholar]
- 44. Wilson MZ, Gitai Z. 2013. Beyond the cytoskeleton: mesoscale assemblies and their function in spatial organization. Curr. Opin. Microbiol. 16:177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cioce M, Lamond AI. 2005. Cajal bodies: a long history of discovery. Annu. Rev. Cell Dev. Biol. 21:105–131 [DOI] [PubMed] [Google Scholar]
- 46. Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O’Connell JD, Mirrielees J, Ellington AD, Marcotte EM. 2009. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. U. S. A. 106:10147–10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ovádi J, Saks V. 2004. On the origin of intracellular compartmentation and organized metabolic systems. Mol. Cell. Biochem. 256:5–12 [DOI] [PubMed] [Google Scholar]
- 48. Yeates TO, Crowley CS, Tanaka S. 2010. Bacterial microcompartment organelles: protein shell structure and evolution. Annu. Rev. Biophys. 39:185–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maddock JR, Shapiro L. 1993. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science 259:1717–1723 [DOI] [PubMed] [Google Scholar]
- 50. Sourjik V, Berg HC. 2000. Localization of components of the chemotaxis machinery of Escherichia coli using fluorescent protein fusions. Mol. Microbiol. 37:740–751 [DOI] [PubMed] [Google Scholar]
- 51. Alley MR, Maddock JR, Shapiro L. 1992. Polar localization of a bacterial chemoreceptor. Genes Dev. 6:825–836 [DOI] [PubMed] [Google Scholar]
- 52. Kirkpatrick CL, Viollier PH. 2011. Poles apart: prokaryotic polar organelles and their spatial regulation. Cold Spring Harb. Perspect. Biol. 3:a006809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bowman GR, Lyuksyutova AI, Shapiro L. 2011. Bacterial polarity. Curr. Opin. Cell Biol. 23:71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huitema E, Pritchard S, Matteson D, Radhakrishnan SK, Viollier PH. 2006. Bacterial birth scar proteins mark future flagellum assembly site. Cell 124:1025–1037 [DOI] [PubMed] [Google Scholar]
- 55. Lam H, Schofield WB, Jacobs-Wagner C. 2006. A landmark protein essential for establishing and perpetuating the polarity of a bacterial cell. Cell 124:1011–1023 [DOI] [PubMed] [Google Scholar]
- 56. Ptacin JL, Shapiro L. 2013. Chromosome architecture is a key element of bacterial cellular organization. Cell. Microbiol. 15:45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kindler S, Wang H, Richter D, Tiedge H. 2005. RNA transport and local control of translation. Annu. Rev. Cell Dev. Biol. 21:223–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Donnelly CJ, Fainzilber M, Twiss JL. 2010. Subcellular communication through RNA transport and localized protein synthesis. Traffic 11:1498–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pogliano J, Pogliano N, Silverman JA. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 194:4494–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saberi S, Emberly E. 2010. Chromosome driven spatial patterning of proteins in bacteria. PLoS Comput. Biol. 6:e1000986. 10.1371/journal.pcbi.1000986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laloux G, Jacobs-Wagner C. 2013. Spatiotemporal control of PopZ localization through cell cycle-coupled multimerization. J. Cell Biol. 201:827–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Spitzer J. 2011. From water and ions to crowded biomacromolecules: in vivo structuring of a prokaryotic cell. Microbiol. Mol. Biol. Rev. 75:491–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Young KD. 2006. The selective value of bacterial shape. Microbiol. Mol. Biol. Rev. 70:660–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Givskov M, Eberl L, Møller S, Poulsen LK, Molin S. 1994. Responses to nutrient starvation in Pseudomonas putida KT2442: analysis of general cross-protection, cell shape, and macromolecular content. J. Bacteriol. 176:7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Justice SS, Hunstad DA, Cegelski L, Hultgren SJ. 2008. Morphological plasticity as a bacterial survival strategy. Nat. Rev. Microbiol. 6:162–168 [DOI] [PubMed] [Google Scholar]
- 66. Strahl H, Hamoen LW. 2010. Membrane potential is important for bacterial cell division. Proc. Natl. Acad. Sci. U. S. A. 107:12281–12286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bi E, Lutkenhaus J. 1993. Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J. Bacteriol. 175:1118–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Michaelis AM, Gitai Z. 2010. Dynamic polar sequestration of excess MurG may regulate enzymatic function. J. Bacteriol. 192:4597–4605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Be'er A, Florin EL, Fisher CR, Swinney HL, Payne SM. 2011. Surviving bacterial sibling rivalry: inducible and reversible phenotypic switching in Paenibacillus dendritiformis. mBio 2(3):e00069-11. 10.1128/mBio.00069-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Singaravelan B, Roshini BR, Munavar MH. 2010. Evidence that the supE44 mutation of Escherichia coli is an amber suppressor allele of glnX and that it also suppresses ochre and opal nonsense mutations. J. Bacteriol. 192:6039–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Darbon E, Galinier A, Le Coq D, Deutscher J. 2001. Phosphotransfer functions mutated Bacillus subtilis HPr-like protein Crh carrying a histidine in the active site. J. Mol. Microbiol. Biotechnol. 3:439–444 [PubMed] [Google Scholar]
- 72. Meyerovich M, Mamou G, Ben-Yehuda S. 2010. Visualizing high error levels during gene expression in living bacterial cells. Proc. Natl. Acad. Sci. U. S. A. 107:11543–11548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Segev E, Rosenberg A, Mamou G, Sinai L, Ben-Yehuda S. 2013. Molecular kinetics of reviving bacterial spores. J. Bacteriol. 195:1875–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Quisel JD, Lin DC, Grossman AD. 1999. Control of development by altered localization of a transcription factor in B. subtilis. Mol. Cell 4:665–672 [DOI] [PubMed] [Google Scholar]
- 75. Bejerano-Sagie M, Oppenheimer-Shaanan Y, Berlatzky I, Rouvinski A, Meyerovich M, Ben-Yehuda S. 2006. A checkpoint protein that scans the chromosome for damage at the start of sporulation in Bacillus subtilis. Cell 125:679–690 [DOI] [PubMed] [Google Scholar]
- 76. Aké FM, Joyet P, Deutscher J, Milohanic E. 2011. Mutational analysis of glucose transport regulation and glucose-mediated virulence gene repression in Listeria monocytogenes. Mol. Microbiol. 81:274–293 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
EI fused to monomeric GFP localizes to the E. coli cell poles. DIC and fluorescence microscopy images of E. coli cells expressing EI-mGFP and an overlay of the signals (green, gray, and merge, respectively). The scale bar corresponds to 1 µm. Download
Cardiolipin staining in cls mutant and wild-type cells. E. coli cells mutated in the cardiolipin synthase (cls) gene (A) and the parental cls+ strain (B) were stained with NAO (10-N-nonyl acridine orange), which binds to anionic lipids, preferably to CL. NAO was observed by fluorescence microscopy (green), and the cells were observed with phase microscopy (gray). Overlays of the signals are also shown (merge). The scale bar corresponds to 1 µm. Download
Localization of PopZ in nucleoid-compacted cells. Images showing PopZ-YFP in PAT84 ftsZ844ts mutant cells that were shifted to the nonpermissive temperature (42°C) and treated with 300 µg/ml kasugamycin for 20 min. The cells were observed with DIC microscopy (gray), the YFP fusion protein was observed by fluorescence microscopy (green), and DNA staining was with DAPI (white). An overlay of the fluorescent signals is also shown (merge). An arrow indicates the nucleoid-free region in a nucleoid-compacted cell. An asterisk indicates the pole region in a cell in which the nucleoid has not been compacted. Download
Glucose consumption by rod-shaped and spherical E. coli cells. MG1655 E. coli cells (wild type), untreated (blue) or treated with A22 (red), ΔmreBCD E. coli cells (green), and MG1655 Δpts E. coli cells (purple) were grown in LB medium containing 25 mM glucose, and glucose consumption measurements were performed as described in Materials and Methods in the presence of 1 mM glucose. Download
Localization of DivIVA-GFP and EI-mCherry in E. coli Δpts cells. Images showing the distribution pattern of B. subtilis DivIVA protein fused with GFP (top) and of overexpressed E. coli EI protein fused to mCherry (bottom) in E. coli Δpts cells. The cells were observed with DIC (gray), and the GFP fusion protein (green) or mCherry fusion protein (red) were observed by fluorescence microscopy. Download
Complementation of a ptsI E. coli mutant with the B. subtilis EI protein. E. coli ptsI19ts cells (A) and ptsI19ts cells containing either pBADLLEI-mCherry, which expresses the E. coli EI protein fused to mCherry (B), or pGSbsEI-GFP, which expresses the B. subtilis EI protein fused to GFP (C), were streaked on a MacConkey plate containing 1% mannitol and 1 mM IPTG and incubated overnight at the restrictive temperature (42°C). Download
Bacterial strains used in this study and their relevant phenotype.
Plasmids used in this study and the proteins they encode.