

ABSTRACT
Cultivation-independent surveys of microbial diversity have revealed many bacterial phyla that lack cultured representatives. These lineages, referred to as candidate phyla, have been detected across many environments. Here, we deeply sequenced microbial communities from acetate-stimulated aquifer sediment to recover the complete and essentially complete genomes of single representatives of the candidate phyla SR1, WWE3, TM7, and OD1. All four of these genomes are very small, 0.7 to 1.2 Mbp, and have large inventories of novel proteins. Additionally, all lack identifiable biosynthetic pathways for several key metabolites. The SR1 genome uses the UGA codon to encode glycine, and the same codon is very rare in the OD1 genome, suggesting that the OD1 organism could also transition to alternate coding. Interestingly, the relative abundance of the members of SR1 increased with the appearance of sulfide in groundwater, a pattern mirrored by a member of the phylum Tenericutes. All four genomes encode type IV pili, which may be involved in interorganism interaction. On the basis of these results and other recently published research, metabolic dependence on other organisms may be widely distributed across multiple bacterial candidate phyla.
IMPORTANCE
Few or no genomic sequences exist for members of the numerous bacterial phyla lacking cultivated representatives, making it difficult to assess their roles in the environment. This paper presents three complete and one essentially complete genomes of members of four candidate phyla, documents consistently small genome size, and predicts metabolic capabilities on the basis of gene content. These metagenomic analyses expand our view of a lifestyle apparently common across these candidate phyla.
Introduction
The current number of bacterial phyla recognized by rRNA databases is between 63 and 84 (SILVA and GreenGenes, accessed August 2013), although the true count is almost certainly higher, with some estimates as high as 100 phyla (N. R. Pace, personal communication, 2013) (1). A careful examination of naming conventions across databases and phylogeny allowed the dereplication of the list of phyla and suggested that there are at least 38 without cultivated representatives (2); these are referred to as candidate divisions (3, 4) or candidate phyla (CP). The increase in the number of CP over the last 10 years can be attributed in part to the subdivision of one major clade, initially referred to as OP11 (5), that is now recognized to comprise several phyla, including OP11, OD1, and SR1 (6). Some CP, such as TM7, are relatively well defined (5), while others, including WWE3 (7), and PER (8), have only recently been proposed.
CP organisms have been detected by 16S rRNA gene sequencing surveys spanning a wide array of environment types, including the human oral microbiome (9, 10), the mammalian gut (11), bioreactors (7, 12, 13), freshwater lakes (14, 15), hypersaline microbial mats (1), and deep-sea vents (16). Targeted 16S rRNA gene primer assays have documented diversity within the CP and assessed the abundance of these organisms in certain environments, especially under anoxic and sulfidic conditions (6, 7, 13–15, 17, 18). However, the full diversity and roles of CP organisms in the environment remain unclear. These questions have motivated the use of two cultivation-independent approaches to the sequencing of CP genomes, (i) single-cell flow systems coupled to multiple-displacement amplification and (ii) metagenomics. Single-cell sequencing has generated genomic information for representatives of several CP (10, 19–23), but genomes are typically highly incomplete and amplification bias affects the sequenced gene copy number (24). Metagenomic methods have yielded nearly complete and complete genomic sequences of uncultivated groups in natural environments (8, 25–29). However, an impediment to the metagenomic reconstruction of genomes is the relatively low abundance of CP cells in environmental samples. Biostimulation is one way to alter the profile of a microbial community, enriching for certain metabolic capabilities (e.g., see reference 30). For example, acetate amendment of groundwater combined with filtering (enriching for cells <1.2 µm in diameter) at an aquifer in Rifle, CO, recently led to the successful reconstruction of 49 partial and nearly complete genomes (8) of members of several CP, including OD1, OP11, BD1-5, and PER. Such strategies have the additional advantage that they can potentially illuminate organism responses to specific stimuli, as well as correlations in organism abundance patterns.
Here, we applied improved metagenomic methods to Illumina sequence data sets recovered from a series of biostimulated sediment communities and assembled complete genomes corresponding to four CP: SR1, WWE3, TM7, and OD1. We report genome characteristics, metabolic potential, and abundance across a range of geochemical conditions and community compositions Analysis of these new genomes and several other recently published genomes from the same CP indicates a surprisingly consistent life strategy and provides insight into why members of these phyla remain cultivated.
RESULTS
We conducted a time course biostimulation experiment with flowthrough sediment columns suspended within an aquifer at Rifle, CO. Thirteen columns were pumped with acetate-amended groundwater for 13 to 63 days, and individual columns were sacrificed at points of geochemical interest (K. M. Handley et al., unpublished). Metagenomic sequencing of DNA from acetate-amended column sediment and an unamended background sediment sample revealed the presence of a variety of bacteria, including diverse members of the CP (I. Sharon et al., unpublished data).
Genomic sequences of four CP organisms of interest were present in multiple samples. The fragmentation patterns of each genome differed across the metagenomic data sets, making it possible to identify overlaps in scaffolds from different samples and to generate high-quality draft genomes. Differential organism abundance patterns across the samples were used to confirm that all fragments >3 kb in length were correctly assigned to the four genomes (Fig. 1). Draft genomes were subsequently curated to complete and nearly complete genomes by using paired-end read information (see Materials and Methods). The reconstructed genomes are presented here as RAAC1 to RAAC4 for Rifle acetate amendment columns 1 to 4. On the basis of phylogenetic analysis, the RAAC1 genome falls within SR1, RAAC2 falls within WWE3, a clade sister to OP11 (7), RAAC3 falls into TM7 group 3, and RAAC4 falls into OD1 (Fig. 1; see Fig. S1 in the supplemental material). Notably, all four genomes are very small, 0.7 to 1.17 Mb in length (Table 1), within the range typically seen only in obligate symbionts (Fig. 2). Only the RAAC4 (OD1) genome is not circularized. Attempts to circularize this genome by PCR amplification failed, despite control reactions demonstrating that other segments of the OD1 genome could be amplified from the sediment column extract. Closely related genomes of similar sizes have been recovered from the same site, suggesting that the genome is likely to be nearly complete (unpublished data). While each genome represents the composite sequence of a population, very little strain variation was observed, with the exception of TM7, for which several closely-related strains were observed.
FIG 1 .

Complete and nearly complete genomes from four CP (RAAC1 to RAAC4) were reconstructed. (Left) Phylogeny of selected CP based on 16S rRNA genes. The maximum-likelihood tree shown was constructed from an alignment containing 111 taxa and 1,565 unambiguously aligned positions. Bootstrap values of >80% are displayed as filled circles (for accession numbers, see Fig. S1 in the supplemental material). Also noted are previously sequenced partial and complete genomes for related members of the CP for which full-length 16S rRNA gene sequences were available (8, 10, 12, 19, 20, 22). (Right) ESOMs made by using differential coverage across the 14 sediment columns confirmed genome binning.
TABLE 1 .
Genome information for the four CP genomes examined in this study
| Parameter | RAAC1 (SR1) | RAAC2 (WWE3) | RAAC3 (TM7) | RAAC4 (OD1) |
|---|---|---|---|---|
| Completeness | Circular, closed | Circular, closed | Circular, closed | Not circular, 3 gaps |
| Length (bp) | 1,177,760 | 878,109 | 845,464 | 693,528 |
| Relative GC content | 0.31 | 0.43 | 0.49 | 0.31 |
| Avg ORF length (bp) | 1,011 | 926 | 837 | 908 |
| Avg intergenic distance (bp)a | 92 | 66 | 72 | 86 |
| Relative protein coding density | 0.91 | 0.92 | 0.91 | 0.9 |
| No. of tRNAs | 37 | 45 | 43 | 42 |
| No. of protein-coding genes | 1,059 | 874 | 921 | 687 |
| No. (%) of ORFs with predicted function | 596 (56) | 618 (71) | 701 (76) | 504 (73) |
| No. (%) of ORFs with domain-only predictionb | 115 (11) | 49 (6) | 38 (4) | 33 (5) |
| No. (%) of conserved hypothetical ORFsc | 106 (10) | 121 (14) | 143 (16) | 96 (14) |
Average intergenic distance was calculated by including all nonzero distances between protein- and RNA-coding sequences.
Using IPRSCAN software version 4.6, data version 35.0.
Based on best-hit annotation to Uniref90 database 2012_9 with sequences from reference 8 removed to simplify post-processing.
FIG 2 .
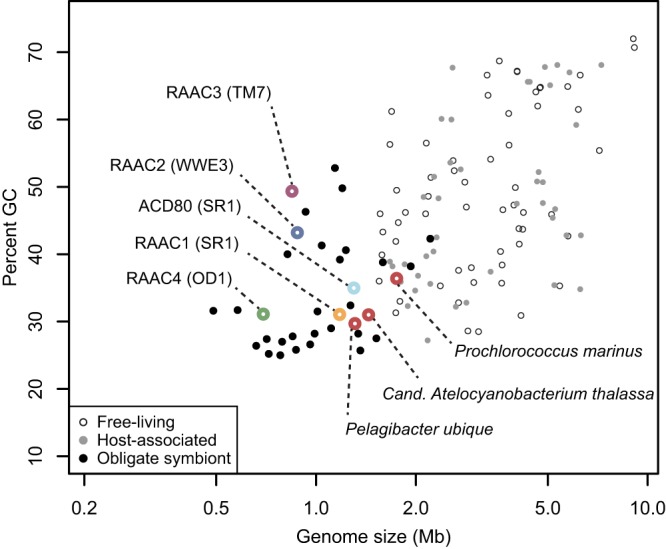
GC content versus genome size for the RAAC1 to RAAC4 genomes with reference data from NCBI. Biotic relationship categories are as described by Giovannoni et al. (49). Open red circles represent the following marine bacteria with small genomes (left to right): Pelagibacter ubique (free living), Candidatus Atelocyanobacterium thalassa (UCYN-A, a symbiont), and Prochlorococcus marinus (free living).
A previous study of planktonic cells from the same site reconstructed genomes related to those reported here (8), but lack of associated 16S rRNA gene sequences complicated classification efforts. The current genomes allow phylogenetic resolution via 16S rRNA and protein trees (Fig. 1; see Fig. S2 in the supplemental material). One partial genome, ACD25, previously classified as OP11 (8), matches the WWE3 genome (RAAC2) at the species level and has an identical protein sequence for the DNA-directed RNA polymerase beta subunit (see Fig. S2). On the basis of analyses reported here, ACD22 and ACD24 are also reassigned to phylum WWE3. Another genome, ACD80, previously classified as a distant BD1-5 relative, is reclassified as belonging to phylum SR1, in agreement with Campbell et al. (10), and a tentatively binned scaffold containing a 16S rRNA gene was confirmed as belonging to that genome (Fig. 1; see Fig. S1) (8).
Time series relative abundance.
The four CP genomes exhibit distinct enrichment patterns across the 14 samples (Fig. 3). Acetate stimulation resulted in the early increase and then decline of betaproteobacteria, followed by an increase in members of the class Clostridia (Fig. 3), a pattern that largely paralleled the shift in terminal-electron-accepting processes during acetate amendment (31) (Handley et al., unpublished). The change from iron reduction to sulfate reduction is reflected in the exponential variation in the groundwater sulfide concentration across samples (see Fig. S3A in the supplemental material). The SR1 genome rose in relative abundance by as much as 500-fold as sulfide concentrations increased; its abundance also correlated strongly with that of a member of the phylum Tenericutes (see Fig. S3B). The log relative abundance of the SR1 genome is directly correlated with log sulfide concentration until peak sulfate reduction (see Fig. S3A) (R2 =0.78, P < 0.0005), after which abundance continued to rise and sulfide levels began to decline. This correlation could be due to a variety of factors, including metabolic relationships between the members of SR1 and organisms performing sulfate reduction. Of the CP genomes, the WWE3 genome appears to be the most stably abundant (at 0.13 to 0.58%) from mid-iron reduction through sulfate reduction. The TM7 and OD1 genomes had the highest relative abundance in samples 3 and 7, before notable sulfate reduction occurred (Fig. 3).
FIG 3 .

The relative abundance of the RAAC1 to RAAC4 genomes varies across samples representing a range of geochemical conditions and microbial community compositions. (Left) Percent relative abundance (y axis) across the 14 independent sediment columns (x axis). All four genomes were found at <1% relative abundance in every sample. (Right) Endpoint ferrous iron and sulfate concentrations measured in column effluent and community composition. (A discussion will appear elsewhere [Handley et al., unpublished].) Sample 1 represents unamended background sediment, and the dashed line roughly divides samples from columns undergoing iron reduction (samples 2 to 8) versus sulfate reduction (samples 9 to 14). Samples 13 and 14 were taken after peak sulfate reduction had occurred.
Novel proteins and metabolic characterization.
On the basis of annotations, each of the four CP genomes contains a large fraction of proteins lacking functional predictions and proteins whose only homologs are hypothetical proteins (Table 1) (8, 10, 12, 19, 20). When the four genomes were searched against hidden Markov models (HMMs) representing all known protein families (called “sifting families” or “SFams”) (see reference 32), between 39% (OD1) and 53% (SR1) of the CP sequences were not matched to any family (see Fig. S4 in the supplemental material). Attempts to cluster novel proteins in order to identify previously unknown protein families were largely unsuccessful, likely because of the wide phylogenetic distances among the four genomes (data not shown).
Analyses by using KAAS (KEGG Automatic Annotation Server [33]), independent BLAST searches against the CP genomes, and gene-by-gene manual analysis for all genomes were performed to assess the completeness of central metabolic pathways. Consistent with previous analyses of other bacteria from these CP (8, 10, 12, 20), all of the genomes examined support fermentative metabolisms (Fig. 4) and lack tricarboxylic acid cycle genes. Superoxide dismutase and alkyl hydroperoxide reductase genes are present, presumably involved in resistance to oxidative damage. The four genomes encode distinct fermentation pathways, different abilities to use and store complex carbon, different electron-carrying proteins, and some key unique pathways, although consistency was observed within the individual CP (see Fig. S5 in the supplemental material).
FIG 4 .

Cell diagrams depicting central carbon metabolism, proteins putatively involved in electron transfer, and transporters in the CP genomes. Panels: A, RAAC1, SR1; B, RAAC2, WWE3; C, RAAC3, TM7; D, RAAC4, OD1. Boxes represent enzymes and are color coded as follows: green, identified; yellow, homology unclear; red, not identified. The numbers correspond to the enzymes listed in Table S1 in the supplemental material. Transporters: if unmarked, parallel ovals represent members of the major facilitator superfamily while circles and ovals together indicate predicted ABC transporters.
RAAC1 (SR1).
The SR1 genome lacks genes involved in the initial steps of the Embden-Meyerhof-Parnas (EMP), pentose phosphate, and Entner-Doudoroff pathways, consistent with our analysis of ACD80 and SR1-OR1 (8, 10). In RAAC1, genes were identified that encode triose phosphate isomerase and the lower portion of the EMP pathway, from the conversion of glyceraldehyde-3-phosphate to 3-phosphoglycerate by a possible nonphosphorylating glyceraldehyde 3-phosphate dehydrogenase (GapN) through the formation of pyruvate (Fig. 4A; see Fig. S5 in the supplemental material). The other two SR1 genomes, ACD80 and SR1-OR1, do not appear to contain these genes. The potential for gluconeogenesis is indicated by the presence of pyruvate phosphate dikinase, but the other gene involved in this pathway, fructose 1,6-bisphosphatase, was not identified in any SR1 genome. The RAAC1 genome possesses a gene cluster responsible for the fermentation of pyruvate to acetate and formate via pyruvate formate lyase (PFL), an alternative phosphotransacetylase (PduL) (34), acetate kinase, and a putative formate transporter. This pathway is also found in SR1-OR1, though not in a gene cluster. The organism may also degrade complex carbon by using a dockerin-like protein, a cellulosome anchoring protein, and several cell surface glucanases and pectin lyases, as observed in the other SR1 genomes.
Also as reported previously for the two partially reconstructed SR1 genomes (8, 10), the SR1 genome presented here harbors a type II/III intermediate form of ribulose 1,5-bisphosphate carboxylase/oxygenase (RubisCO). This type of RubisCO is implicated in the AMP salvage pathway (35) identified in RAAC1 (Fig. 4A) and both of the other SR1 genomes sequenced to date. RAAC1 RubisCO possesses conserved catalytic residues and an additional 29-amino-acid sequence unique to this form of RubisCO. The sequence identity across all three SR1 RubisCOs is high (72%), suggesting that they play similar roles in the metabolism of these organisms. As suggested by Sato et al. (35), the 3-phosphoglycerate produced from the AMP salvage pathway may enter glycolysis for energy generation.
Lastly, the RAAC1 SR1 genome encodes several electron-carrying proteins, including a cytochrome b5, three Fe-S cluster proteins of unknown function, a ferredoxin reductase-like protein, three flavodoxins, and a rubrerythrin. Some of these may be important for the response to oxidative stress and/or reoxidation of reduced ferredoxin or NADH.
RAAC2 (WWE3).
The WWE3 genome possesses most of the genes for the EMP pathway, pyruvate ferredoxin oxidoreductase (PFOR, or possibly 2-oxoglutarate ferredoxin oxidoreductase subunits alpha and beta), which converts pyruvate to acetyl coenzyme A (acetyl-CoA), and an acetyl-CoA synthetase (ADP forming, EC 6.2.1.13) that produces acetate and ATP (Fig. 4B). There is also a gene cluster for the lower portion of the pentose phosphate pathway, converting ribulose-5-phosphate to glyceraldehyde-3-phosphate. Located in the same gene cluster is a protein identified as belonging to the d-isomer-specific 2-hydroxy-acid dehydrogenases, a superfamily that contains glyoxylate reductase and d-lactate dehydrogenase (36). The presence of phosphoenolpyruvate synthase suggests that the WWE3 organism may engage in gluconeogenesis, but it lacks fructose 1,6-bisphosphatase. These observations are consistent with the analysis of the newly reclassified WWE3, ACD22, ACD24, and ACD25 genomes (see Fig. S5 in the supplemental material), although these genomes are fragmented and incomplete and may be subject to binning error. The RAAC2 WWE3 organism may also be capable of synthesizing and utilizing glycogen, a common energy storage polysaccharide, via an operon containing a predicted galactose-1-phosphate uridylyltransferase, two glycogen synthase genes, an alpha-amylase-like gene, and a glycosyl hydrolase of family 57, the family containing the branching enzyme required to form glycogen.
The ATP synthase operon in the WWE3 genome contains alpha, beta, gamma, and delta/epsilon subunits; however, the adjacently located a, b, and c subunits lack significant homology to known ATPase subunits. Instead, the putative c subunit, responsible for ion translocation, bears homology to a similar transmembrane protein found in the ATPase operons of the OP11 and WWE3 genomes, ACD38 and ACD24 (8). Given the lack of homology to characterized ATPases (37), it remains unclear whether the ATPase, if functional, operates primarily in the forward or reverse direction and whether it pumps protons or sodium ions. This genome also contains a membrane-bound proton/sodium-pumping pyrophosphatase, which could be used to generate membrane potential. Electron carriers in the WWE3 organism include a ferredoxin oxidoreductase-like protein, a cytochrome b5, and a plastocyanin-like blue copper protein encoded in the genome adjacent to a predicted membrane-associated ferric reductase-like protein (Fig. 4B). Additional electron flow may proceed through a type 3B-like cytoplasmic nickel-dependent hydrogenase homologous to those found in another WWE3 genome, ACD22, and several OD1 genomes (8).
RAAC3 (TM7).
The pentose phosphate pathway and most of the EMP pathway are present in the TM7 genome (Fig. 4C), but enolase was not detected, nor was a means for converting pyruvate to acetyl-CoA (e.g., PFOR, PFL, or pyruvate dehydrogenase) or for using acetyl-CoA. We confirmed that enolase is not annotated or identifiable in any of the currently available TM7 genome sequences (12, 19, 20) (see Fig. S5 in the supplemental material). RAAC3 contains phosphoketolase but not acetate kinase, needed for the generation of ATP from this branch off the pentose phosphate pathway (Fig. 4C, box 19). Although both genes were identified in a gene cluster in a related genome (12), synteny between the two genomes in this region is lacking. Other possible routes for the fermentation and regeneration of NAD+ by the RAAC3 TM7 organism include two dehydrogenases related to d-lactate dehydrogenase, which are conserved in other TM7 genomes (12). Malate/lactate dehydrogenase, which has been reported in another TM7 genome (12), was not identified in RAAC3. As an alternative means of producing ATP, RAAC3 encodes the arginine deiminase pathway (Fig. 4C). We identified genes in this pathway in other TM7 genome sequences (12, 19), suggesting that it may be common to members of the TM7 phylum.
The TM7 (RAAC3) genome has several genes involved in complex carbon degradation, including beta-glucosidase, a predicted secreted endo-1,3(4)β-glucanase, α-amylase, and a glycogen phosphorylase-like protein. The presence of a bifunctional trehalose synthase/phosphatase indicates the use of trehalose, which is synthesized from glucose-6-phosphate and UDP-glucose. Alpha,alpha-trehalase is also present, providing for the subsequent degradation of trehalose and release of glucose for cellular consumption.
The RAAC3 TM7 genome contains a complete ubiquinol oxidase (cytochrome bo) operon, with intact functional residues, as well as residues known to distinguish cytochrome O ubiquinol oxidases from their closely related counterparts, cytochrome c oxidases (38). This complex could be used for oxygen scavenging, as all other information points to a fermentation-based metabolism and we did not find the complex in other TM7 genomes. The electron source for ubiquinol oxidase may be a single-subunit form of NADH: ubiquinone dehydrogenase (NDH) most similar to type II NDH in structural searches (39, 40). This NDH is located adjacent to the ubiquinol oxidase operon in the RAAC3 genome and has homologs in other TM7 genomes (12, 19).
RAAC4 (OD1).
OD1 is a very diverse CP, so it is unsurprising that variation in metabolic capabilities exists. Like TM7, the RAAC4 OD1 genome contains genes for the pentose phosphate pathway, as does the recently sequenced AAA011-A08 genome (22). However, the AAA255-P19 OD1 genome from the same work does not contain this pathway and two other partial OD1 genomes examined support only the latter half of the pathway (see Fig. S5 in the supplemental material). RAAC4 possesses a modified EMP pathway from mannose-6-phosphate isomerase to pyruvate (Fig. 4D). Also similar to TM7, and consistent with other OD1 members analyzed here, it does not contain any identifiable PFL, PFOR, or pyruvate dehydrogenase genes or genes for the utilization of acetyl-CoA. Like the WWE3 organism, the OD1 organism may be capable of fermentation to lactate, as indicated by a 2-hydroxy-acid dehydrogenase family gene found adjacent to enolase and pyruvate kinase in the genome. The synteny of the genes in this cluster is not conserved in the other OD1 genomes examined. A predicted lactate transporter is encoded elsewhere in the RAAC4 genome. The presence of a putative cellulosome-related gene cluster, two glycosyl hydrolases, and an end-specific cellobiohydrolase suggests that OD1 is capable of complex carbon degradation. The RAAC4 OD1 genome also contains a gene cluster involved in alternative polyamine biosynthesis, as described by Lee et al. (41), which may be important in biofilm formation.
The RAAC4 OD1 genome may use a membrane-bound sodium/proton-pumping pyrophosphatase to generate a proton motive force. Electron transport proteins include a rubredoxin and flavoprotein of unknown function, perhaps involved in oxygenic stress tolerance. While other, previously described, OD1 genomes were found to contain putative nickel-iron hydrogenases involved in uptake and hydrogen production, no hydrogenases could be identified in RAAC4.
Essential biosynthetic pathways.
While the RAAC genomes have easily identifiable enzymes for the interconversion of amino acids and nucleotides (e.g., serine hydroxymethyltransferase [with the exception of WWE3] and dCTP- or dCMP-deaminase), complete biosynthesis pathways for nucleotides (see Table S2 and Fig. S6 in the supplemental material), lipids, and most amino acids (42) could not be identified in any of the four genomes on the basis of annotations or by using KAAS (33). On the basis of this preliminary analysis, we concluded that these organisms may be auxotrophic for many essential metabolites or may contain novel biosynthetic pathways.
Further analysis was performed to assess the completeness of nucleotide biosynthesis by using reference sets of genes from diverse genomes to query RAAC genomes (see Materials and Methods). The members of SR1 appear to be missing most of this pathway. If novel genes present in this phylum could form parts of this pathway, we might expect them to be conserved and possibly located near identifiable genes involved in nucleotide biosynthesis. However, few biosynthesis genes are present in all three of the SR1 genomes and the regions containing these genes lack synteny, offering no clues to novel conserved genes that may perform the missing functions. The possibility remains that such genes are found elsewhere in the genomes. Similarly, the WWE3 and TM7 genomes show few genes involved in nucleotide biosynthesis and lack synteny surrounding these genes.
Some sequenced representatives of OD1 may have functional pathways for nucleotide biosynthesis (e.g., single-cell genome AAA255-P19) (22), but RAAC4 (OD1) does not (see Fig. S6). Gene loss or horizontal gene transfer could explain these differences in metabolic potential between the members of OD1. Whereas the AAA255-P19 genome contains pyrimidine and purine biosynthesis genes in distinct operons, genes that correspond to these pathways in the other CP genomes are not found in clusters (see Table S2).
Given the lack of complete pathways for the biosynthesis of some essential metabolites, we examined the possibility that the RAAC CP organisms could scavenge these compounds from their surroundings. The RAAC genomes contain numerous nucleases and proteases, as well as several transporters, whose substrates are unknown.
Cell surface and environmental interactions.
None of the RAAC1 to RAAC4 CP organisms appear to make lipid A or lipopolysaccharide, as indicated by the absence of genes for biosynthesis, including lpxC and kdsA (43), and as suggested by previous work on another TM7 genome (12). All of the genomes except the WWE3 genome contain complete, identifiable pathways for peptidoglycan synthesis (see Fig. S7A in the supplemental material). The abundance of glycosyltransferases in all four genomes, particularly the WWE3 genome, suggests that the organisms devote significant energy to the production of polysaccharides, glycoproteins, and/or a glycosylated S-layer. Additionally, the SR1 and WWE3 genomes contain genes for the synthesis of dTDP-rhamnose (see Fig. S7A). The genomes encode proteins containing one or more of the following domains: concanavalins/lectins, pectin lyases, fibronectin III, beta propeller, and polycystic kidney disease, some of which are predicted to be cell surface domains in Bacteria and Archaea (see Fig. S7B) (44). Many of these predicted proteins are large, up to 5,900 amino acids, and some have signal peptides or sortase motifs suggesting possible cell wall localization.
Sortases, which covalently attach surface proteins to the cell wall of Gram-positive bacteria, are present in the SR1, WWE3, and TM7 genomes, and predicted sorted proteins were found in WWE3 and TM7. Each of the four genomes encodes the required components for type IV pilus biosynthesis, including pilT (45), for twitching motility, and for several predicted pilins (see Fig. S7B) (46). The TM7 genome has multiple type II and IV pilus-related gene clusters and additional pilins, totaling 60 genes, a full 6% of the TM7 genome. Importantly, the pili present in these CP genomes are not related to the sortase-associated pili more commonly found in Gram-positive bacteria (47). Rather, they are homologous to type IV pili sometimes involved in the uptake of environmental DNA (48). Consistent with this possible function, each genome contains at least one copy of ComEC, the DNA-specific pore-forming protein required for competence, and DNA protection protein DprA (see Fig. S7B).
Translation and coding.
Codon usage in both the SR1 and OD1 genomes is skewed toward low-GC codons, as expected given the low GC content across these genomes (see Fig. S8 in the supplemental material). Additionally, the SR1 genome uses alternate coding, as reported for another SR1 genome (10). This genome, RAAC1, and the previously reported SR1 genomes (8, 10) contain nearly identical genes for tRNAUCA, suggesting that the corresponding codon, UGA, is not read as termination but rather as an amino acid. Concordantly, open reading frame (ORF) detection with code 11 (bacterial) yielded extremely short sequences and an unreasonably high frequency of split genes (a total of 2,288 predicted ORFs with an average length of 289 bp), while translation with code 4 (UGA read as tryptophan) gave typical ORF sizes and complete genes (Table 1). However, unlike code 4, where UGA encodes tryptophan, conserved positions in protein alignments indicate that UGA most likely encodes glycine, as described for the oral SR1 genotype by Campbell et al. (10). Interestingly, the SR1 genome also harbors duplicated and interrupted tRNA synthetases. Specifically, there is an extra, fragmented copy of both valine- and alanine-tRNA synthetases and an unusual isoleucine-tRNA synthetase that appears to be split into four regions by a nudix hydrolase domain and two phosphoglycerate mutase domains. The same tRNA synthetases were also found in the ACD80 genome.
The OD1 genome has an extremely small number of UGA stop codons (5% of all ORFs, compared to 22% in WWE3 and 15% in TM7), suggesting that this codon may be approaching extinction and possible reassignment, leading to alternate coding. The OD1 genome also contains a predicted suppressor tRNA that reads the codon UAA. However, the majority of the predicted genes in this genome use UAA for termination, which suggests that this may be a pseudo-tRNA or recognize a different codon.
DISCUSSION
Metabolic predictions for all four genomes point to a primarily fermentation-based lifestyle and an inability to synthesize essential metabolites. However, many predicted ORFs were annotated only at the protein domain level or not at all (Table 1), and some unannotated proteins may complete metabolic pathways that appear to be broken in or absent from the CP genomes. As more sequence data from the CP become available, conserved protein families will likely emerge, preparing the way for further exploration at the phylogenetic, biochemical, and structural levels.
The most remarkable feature of all four complete or essentially complete CP genomes is their small size. The first estimate of the genome size of a member of TM7, based on fragmentary single-cell sequencing data, was substantially larger (20). Our result, a genome size of 0.85 Mbp for RAAC3, parallels the recent finding of a 1.01-Mbp genome for another TM7 organism (12). Campbell et al. suggested that the genome of a member of SR1 was <2 Mbp (10), somewhat larger than the 1.17-Mbp genome size of RAAC1. The expectation of small OP11 and OD1 genome sizes was also noted by Wrighton et al. (8). Together, data presented here and recently published results suggest that small genome sizes are common across multiple phyla.
Genomes of the small sizes reported here are found in some free-living marine bacteria and in obligate symbionts (Fig. 3), both of which may be descended from organisms with larger genomes. Mechanisms for genome reduction in free-living bacteria include streamlining (e.g., decreased intergenic distance and loss of nonessential genes and pathways) or metabolic specialization, and in obligate symbionts, directed loss of genes whose functions are provided by the host (49–51). No close relatives with larger genomes have been sequenced to date, and small genome size may indeed be an ancestral trait of these CP.
Examination of pseudogenes can sometimes reveal the evolutionary trajectory of bacteria with reduced genome sizes. Genome erosion and accumulation of pseudogenes are characteristic of the early stages of evolving symbiosis in bacteria (51), whereas an elimination of pseudogenes is suggestive of genomic streamlining (50) or later stages of symbiosis (51). However, until more closely related CP genomes are sequenced, it will be difficult to determine which unannotated genes are truly pseudogenes and which serve novel functions in certain lineages. The coding density of the four CP genomes (around 0.90; Table 1) is lower than that reported for some organisms that have undergone genomic streamlining (e.g., 0.97 in Prochlorococcus marinus and Pelagibacter ubique) (49, 52) but higher than the value for an obligate symbiont, where streamlining was not a mechanism of genome reduction (0.81 in Candidatus Atelocyanobacterium thalassa) (53, 54).
Genome reduction has been suggested as a driving factor in the switch to alternate coding in some symbiotic alphaproteobacteria and mitochondria (55, 56). In these groups, which use code 4, a single tRNATrp has mutated to accommodate both UGG and UGA via wobble pairing, allowing the elimination of tRNA and termination factor genes (55, 56). While there is currently no consistent bioinformatic method for defining tRNA-to-amino-acid specificity (57), protein alignments and biochemical evidence (10) are convincing arguments for the recoding of UGA to glycine in all of the SR1 genomes reported thus far, and this coding in RAAC1 supports the suggestion by Campbell et al. that this may be a phylum level trait (10). In contrast to the code 4 organisms mentioned above, the SR1 genomes appear to use not wobble pairing but rather an additional tRNAGlyUCA to achieve alternate coding (10). The use of UGA for glycine could be a mechanism for reducing genomic GC content, as all other glycine codons are more GC rich (see Fig. S8 in the supplemental material).
The CP genomes appear smaller than those of typical free-living bacteria, at least in part because of missing metabolic functions. Recently, McLean et al. noted a similarly small genome size for a member of TM6 (23) and suggested that it may be a symbiont. Our analysis suggests that this may also be true for some or all of the organisms reported here. If several core biosynthetic pathways are, in fact, absent from the CP bacteria described here, the organisms likely rely heavily on one (possibly a member of the phylum Tenericutes in the case of the members of SR1) or more community members in a manner similar to symbiosis. Type IV pili, encoded by all four genomes, may aid the cells in interacting with the environment and with other organisms via adhesion to extracellular surfaces, DNA uptake, and biofilm formation (46). Other adhesion- or biofilm-related proteins may also be important to the life strategies of these organisms. Transporters, nucleases, and proteases could allow the organisms to make use of metabolites provided by biomass in their environment or by a host. A potential dependence on other organisms may explain why these CP bacteria remain uncultured.
MATERIALS AND METHODS
Field experiment.
The experimental conditions used in this study and sample collection in the field will be described elsewhere in detail (Handley et al., unpublished ). The present study focused on acetate-amended sediment collected between August and November of 2010. Thirteen flowthrough columns packed with sieved (<2 mm) sediment were placed into one of three wells at the U.S. Department of Energy Integrated Field Research Challenge (IFRC) aquifer in Rifle, CO. Columns were equilibrated to conditions in the subsurface for 1 week. Subsequently, groundwater amended with 10 mM sodium acetate was pumped upward through the columns at an approximate rate of 52 ml day−1. Individual columns were sacrificed for sampling between 13 and 63 days of amendment such that a range of geochemical conditions from iron reduction to sulfate reduction was sampled. Unamended sieved sediment was taken as a background sample. Geochemical measurements of filtered column effluent were made on the day of column sacrifice or on the day before. Aqueous ferrous iron and sulfide were quantified immediately following the collection of effluent with 1,10-phenanthroline and methylene blue colorimetric assays (Hach Company, Loveland, CO). Sulfate was measured by ion chromatography (ICS-2100 [Dionex, Sunnyvale, CA] fitted with an AS-18 guard and analytical column).
DNA extraction and sequencing.
DNA was extracted from an average of 25 ± 7 g of acetate-amended sediment (samples 2 to 14) and 42.1 g of sieved background sediment with PowerMax Soil DNA Isolation kits (MoBio Laboratories, Inc., Carlsbad, CA), with the following modifications of the manufacturer’s instructions. Sediment was vortexed for an additional 3 min in SDS at maximum speed and then incubated for 30 min at 65°C in place of bead beating. Extracted DNA was precipitated with cold ethanol, Na-acetate (0.3 M, pH 5.2), and glycogen (50 µg/ml) and re-eluted in 50 µl elution buffer. Illumina sequencing was conducted at the UC Davis DNA Technologies Core Facility (http://dnatech.genomecenter.ucdavis.edu) by using paired-end 101-bp reads with an insert size of 500 bp. Sequencing was distributed across five lanes to produce between 3 and 6 Gbp of sequence for each of the 13 amended samples and 15 Gbp for the background sample.
Metagenomic assembly and curation.
Sequence data sets for each sample were assembled independently by using idba_ud with default parameters (58). This generated 61.0 to 107.1 Mbp of sequence information per sample and 16.8 Mbp for background sediment on scaffolds of >5 kb. Genes on all scaffolds >5 kb in length were predicted by using Prodigal with the metagenome option (59, 60). Scaffolds for which Prodigal chose code 4 (UGA translated as tryptophan instead of termination) were manually curated into a genome identified as belonging to SR1. The genome was retranslated with UGA encoding glycine (see Results). For each scaffold, we determined the GC content, coverage, genetic code, and profile of phylogenetic affiliation based on the best hit for each gene in Uniref90 (61). On the basis of analyses of these data, as well as emergent self-organizing map (ESOM)-based analyses of tetranucleotide frequencies and time series relative abundance (62, 63), draft genomes were generated that included scaffolds from multiple samples. Scaffolds for the same genome found in different samples were aligned to yield longer fragments, leveraging the observation that fragmentation of assemblies is, to some extent, dependent on the context (community composition). Read mapping used Bowtie (64), and paired-read information was used to extend and join contigs and to fill in gaps left by the assembler (63). A few regions, particularly those containing short repeats (a few hundred bases or less), could not be completely resolved, but the connectivity of their two sides was confirmed.
Functional annotation.
Predicted ORFs were run through a multidatabase search pipeline for functional prediction as previously described (8). Briefly, sequence similarity searches were performed with USEARCH (65) against UniRef90 (61) and the KEGG database (Kyoto University Bioinformatics Center). Domain level functional annotation was done with InterProScan (66). RNA was predicted by a combination of database searching and tRNAScan-SE (67) for tRNAs. Cellular localization was predicted with PSORTb (version 3.0) (68) and by detection of the sortase cleavage motif, LPXTG (69), with in-house scripts.
Phylogenetic analysis.
Genomes were placed into phylogenetic context based on analysis of the 16S rRNA gene sequences. Sequences were aligned with the SILVA database by using the SINA alignment service (http://www.arb-silva.de/aligner) (70). Representative sequences from TM7, PER, BD1-5, SR1, OD1, WS6, WWE3, and OP11 were obtained from SILVA in aligned form (see the accession numbers provided in Fig. S1). Conserved gaps were removed from compiled aligned sequences with GapStreeze v.2.1.0 with the gap tolerance set to 99% (http://www.hiv.lanl.gov/content/sequence/GAPSTREEZE/gap.html). The alignment was further trimmed to remove uninformative regions. A maximum-likelihood tree was constructed with RAxML by using the GTRCAT model with 1,000 bootstraps. For WWE3, which is not recognized as a phylum by SILVA and is called “otu_4443” in GreenGenes (accessed June 2013), the SINA aligner was used to find sequences with >80% identity to the genomic 16S rRNA gene sequence. These sequences were included on a 16S rRNA gene tree containing multiple CP and formed a phylum level branch.
Overall community composition.
Percent relative abundances of genomes in each of the 13 samples and the background sediment sample were calculated on the basis of mapping of unpaired reads from each sample against each genome with Bowtie2 (version 2.0.4) with the settings --phred33 and --fast with default specificity (71). Community composition by sample was determined with EMIRGE (72) by using 80 iterations and SILVA 108 clustered at 97% identity as the reference database. Chimeras were detected with Uchime (65). Reconstructed 16S rRNA gene sequences were analyzed with RDP-classifier (http://rdp.cme.msu.edu/classifier/classifier.jsp), and relative abundances were calculated by EMIRGE on the basis of read mapping normalized for sequence length. operational taxonomic unit abundances were summed within each phylogenetic order represented (members of the class Proteobacteria were summed at the class level). All orders with abundances of <5% in all samples were included in the category “other” (Fig. 3).
Novel protein analysis.
An original database and an updated database of HMMs representing sifting families (32) were obtained from http://edhar.genomecenter.ucdavis.edu/sifting_families. These were compiled into a searchable form with hmmpress (Janelia Farm, 2010, h3.0). Amino acid sequences from each genome were searched against the database by using hmmsearch with a reporting cutoff of 1E-5 and parsed with an alignment coverage threshold of 80% for both the HMM and the query gene.
Metabolic pathway analysis.
The initial analysis relied upon gene annotations from an in-house pipeline (described above) with functional residues confirmed in proteins of interest. Subsequently, amino acid sequences were submitted to KAAS (http://www.genome.jp/kaas-bin/kaas_main?mode=partial) (33) by using a customized search list of diverse members of the domains Bacteria and Archaea (KAAS identification codes pfa, eco, son, cje, gme, sme, rsp, mtu, bsu, cac, ctr, bfr, fjo, emi, cau, tma, mja, afu, pho, tac, ape, sso, pai, tne, tko, pab, pfu, mma, aae, dra, det, cte, pma, syw, fnu, fsu, cao, sru, lil, and fra). Searches were run independently in both bidirectional and single-directional best-hit modes. Additional searches for specific genes (Fig. 4; see Table S1 in the supplemental material) were conducted by generating a diverse reference set from 75 bacterial and archaeal genomes in the IMG database (73) and using these as queries for BLAST (74) to search for potential homologs within the CP genomes.
Nucleotide and amino acid sequence accession numbers.
All of the sequences and annotations determined in this study can be accessed at http://genegrabber.berkeley.edu/aac. Sequences are also available at NCBI through BioProject numbers PRJNA217185 (RAAC1), PRJNA217183 (RAAC2), PRJNA217186 (RAAC3), and PRJNA216121 (RAAC4).
SUPPLEMENTAL MATERIAL
Phylogeny of selected CP based on 16S rRNA genes including NCBI accession numbers for the tree shown in Fig. 1. Bipartition values from 1,000 bootstraps are expressed as percentages for all bootstraps >50%. The 16S rRNA genes from genomes in this study and other genomic sequences are in red. Previously reported TM7 single-cell genomic sequences are represented by TM7 genomosp. GTL1 (accession no. AAXS01000094 (20) single cell TM7a (accession no. NZ_ABBV01000356) (19), and Candidatus Saccharimonas aalborgensis (accession no. NC_021219) (12) and are 81 and 85% identical to RAAC3 at the 16S rRNA gene level, respectively. The SR1 sequences ACD80 and SR1-OR1 are 87 and 89% identical to RAAC1, respectively. The OD1 sequences AAA011-A08 and AAA255-P19 represent partial genomes described in reference 22 and accessed through IMG (73). RAAC4 shares 75% sequence identity with AAA011-A08, its nearest relative with a genomic sequence. Download
Phylogenetic tree of the DNA-directed RNA polymerase subunit beta protein of selected organisms, including CP with partial or complete genome sequences. The tree was constructed by using RAxML with 1,000 bootstrap replicates from an alignment of 99 taxa with 900 aligned positions. Sequences from genomes in this study are red, while sequences from genomes used for comparison are blue. Reference and ACD sequences were taken from a report by Wrighton et al. (8); other CP sequences in blue are from public databases or the IMG database (10, 12, 22). Download
Patterns of SR1 relative abundance in relation to sulfide and the relative abundance of other organisms. (A) Log relative abundances of both RAAC1 (SR1) and a member of the phylum Tenericutes correlate with the log sulfide concentration (µM) in each sample until peak sulfate reduction. The two outliers, samples 13 and 14, were taken after peak sulfate reduction, when SR1 abundance was highest, and were not included in the regression. (B) The log relative abundances of the SR1 and Tenericutes species are correlated when all samples are included (adjusted R2 = 0.9). For all regressions, P < 0.0005. Download
Summary of hmmsearch results against the Sfam database (29). CP genomes from this study are RAAC1 to RAAC4. For comparison, there are three phyla for which only one sequenced genome exists (Gemmatimonas aurantiaca, Caldisericum exile, and Desulfurispirillum indicum), two small genomes (approximately comparable in size to the CP genomes) with well-sequenced relatives are represented by Prochlorococcus marinus and Pelagibacter ubique, and a representative of a well-sequenced group with a larger genome, Geobacter metallireducens GS15. For each genome, the percentage of predicted proteins with no hits in the SFam database is shown above the column). Genomes not from this study were obtained from the NCBI Genome (http://www.ncbi.nlm.nih.gov/genome/) and Joint Genome Institute IMG (http://img.jgi.doe.gov/cgi-bin/w/main.cgi) databases. Download
Comparison of the EMP (blue), pentose phosphate (purple), and fermentation (green) pathways across multiple complete and partial CP genomes and the complete RAAC1 to RAAC4 genomes. We note that all SR1 genomes appear to be missing the upper EMP pathway and do not contain the pentose phosphate pathway. The TM7 genomes lack identifiable genes for enolase. The OD1 genomes lack clear means for making acetyl-CoA from pyruvate. Download
Comparison of pyrimidine biosynthesis (yellow) and purine biosynthesis (red) across multiple partial CP genomes and the complete RAAC1 to RAAC4 genomes. Only the AAA255-P19 genome (22) appears to have nearly complete pathways for nucleotide biosynthesis. The presence or absence of this metabolism appears to vary across the CP. The large number of nucleases present in the CP genomes could provide an alternate source of nucleotides. Note that ACD22, ACD24, and ACD25 may contain multiple copies of genes because of multiple strains or species within a bin, although these genomes are incomplete. Download
Counts by genome of proteins involved in cell wall and cell surface biosynthesis (A) and cell-environment interactions (B). Yellow lists in panel B are not mutually exclusive, as a given protein can have more than one function or domain and was counted in each appropriate category. Download
Codon usage shown for selected amino acids reflects coding bias in SR1 and OD1 genomes but not WWE3 and TM7 genomes. Codon usage was calculated as a percentage of the total, including the termination codon. Dashed lines group triplets that typically encode the same amino acid. SR1 uses UGA to encode glycine instead of termination, and hence it is found much more frequently in that genome. The OD1 genome uses UGA as a termination codon for only 34 out of 687 predicted ORFs, representing 0.02% of the codons in the genome. Download
Gene identifiers for enzymes numbered in Fig. 4. Green, identified; yellow, homology and function unclear; red, not identified by BLAST with a diverse reference set, KAAS, or in-house annotation pipeline; NA, not detected by in-house annotation pipeline. The relevant DNA and protein sequences are accessible at http://genegrabber.berkeley.edu/aac/organisms.
Complete nucleotide biosynthesis pathways were not identified in any of the four CP genomes. Gene identifiers are listed when detected. Colors are the same as in Table S1. All genomes were further searched by using a curated reference set of archaeal purine biosynthesis genes collected on the basis of the work of Brown et al. (A. M. Brown, S. L. Hoopes, R. H. White, and C·A. Sarisky, Biol. Direct 6:63, 2011, doi:10.1186/1745-6150-6-63), and no complete pathway was identified in any of the four genomes. The relevant DNA and protein sequences are accessible at http://genegrabber.berkeley.edu/aac/organisms.
ACKNOWLEDGMENTS
We thank Kyle Frischkorn (University of California, Berkeley [UCB]) and Sean Mullin (UCB) for DNA extraction and PCR experiments, Per Nielsen (Aalborg University, Aalborg, Denmark) and Marc Strous (Max Planck Institute, Bremen, Germany) for helpful comments on the manuscript, Cameron Thrash (Oregon State University) for useful discussions, and Thomas Sharpton (University of California, San Francisco) for assistance with SFam. Additionally, we thank Andrea Singh (UCB) for bioinformatic support, Kenneth Williams (Rifle site management), H. O’Geen (DNA Technologies Core Facility) for sequencing, and Stephanie Tabb for graphic design assistance.
Funding was provided by in part by NSF-GRFP and the Berkeley Fellowship (to R.S.K.) and by the IFRC Subsurface Biogeochemical Research Program, Office of Science, Biological and Environmental Research, U.S. Department of Energy (to J.F.B.).
Footnotes
Citation Kantor RS, Wrighton KC, Handley KM, Sharon I, Hug LA, Castelle CJ, Thomas BC, Banfield JF. 2013. Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio 4(5):e00708-13. doi:10.1128/mBio.00708-13.
REFERENCES
- 1. Harris JK, Caporaso JG, Walker JJ, Spear JR, Gold NJ, Robertson CE, Hugenholtz P, Goodrich J, McDonald D, Knights D, Marshall P, Tufo H, Knight R, Pace NR. 2013. Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J. 7:50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McDonald D, Price MN, Goodrich J, Nawrocki EP, Desantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved GreenGenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6:610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu. Rev. Microbiol. 57:369–394 [DOI] [PubMed] [Google Scholar]
- 4. Pace NR. 2009. Mapping the tree of life: progress and prospects. Microbiol. Mol. Biol. Rev. 73:565–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hugenholtz P, Goebel BM, Pace NR. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180:4765–4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harris JK, Kelley ST, Pace NR. 2004. New perspective on uncultured bacterial phylogenetic division OP11. Appl. Environ. Microbiol. 70:845–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guermazi S, Daegelen P, Dauga C, Rivière D, Bouchez T, Godon JJ, Gyapay G, Sghir A, Pelletier E, Weissenbach J, Le Paslier D. 2008. Discovery and characterization of a new bacterial candidate division by an anaerobic sludge digester metagenomic approach. Environ. Microbiol. 10:2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337:1661–1665 [DOI] [PubMed] [Google Scholar]
- 9. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J. Bacteriol. 192:5002–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campbell JH, O’Donoghue P, Campbell AG, Schwientek P, Sczyrba A, Woyke T, Söll D, Podar M. 2013. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. U. S. A. 110:5540–5545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31:533–538 [DOI] [PubMed] [Google Scholar]
- 13. Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL. 2001. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl. Environ. Microbiol. 67:411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peura S, Eiler A, Bertilsson S, Nykänen H, Tiirola M, Jones RI. 2012. Distinct and diverse anaerobic bacterial communities in boreal lakes dominated by candidate division OD1. ISME J. 6:1640–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Borrel G, Lehours A-C, Bardot C, Bailly X, Fonty G. 2010. Members of candidate divisions OP11, OD1 and SR1 are widespread along the water column of the meromictic Lake Pavin (France). Arch. Microbiol. 192:559–567 [DOI] [PubMed] [Google Scholar]
- 16. Perner M, Seifert R, Weber S, Koschinsky A, Schmidt K, Strauss H, Peters M, Haase K, Imhoff JF. 2007. Microbial CO(2) fixation and sulfur cycling associated with low-temperature emissions at the Lilliput hydrothermal field, southern mid-Atlantic Ridge (9 degrees S). Environ. Microbiol. 9:1186–1201 [DOI] [PubMed] [Google Scholar]
- 17. Davis JP, Youssef NH, Elshahed MS. 2009. Assessment of the diversity, abundance, and ecological distribution of members of candidate division SR1 reveals a high level of phylogenetic diversity but limited morphotypic diversity. Appl. Environ. Microbiol. 75:4139–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dinis JM, Barton DE, Ghadiri J, Surendar D, Reddy K, Velasquez F, Chaffee CL, Lee MC, Gavrilova H, Ozuna H, Smits SA, Ouverney CC. 2011. In search of an uncultured human-associated TM7 bacterium in the environment. PLoS One 6:e21280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA, Quake SR. 2007. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. U. S. A. 104:11889–11894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Podar M, Abulencia CB, Walcher M, Hutchison D, Zengler K, Garcia JA, Holland T, Cotton D, Hauser L, Keller M. 2007. Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl. Environ. Microbiol. 73:3205–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Youssef NH, Blainey PC, Quake SR, Elshahed MS. 2011. Partial genome assembly for a candidate division OP11 single cell from an anoxic spring (Zodletone Spring, Oklahoma). Appl. Environ. Microbiol. 77:7804–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu WT, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437 [DOI] [PubMed] [Google Scholar]
- 23. McLean JS, Lombardo MJ, Badger JH, Edlund A, Novotny M, Yee-Greenbaum J, Vyahhi N, Hall AP, Yang Y, Dupont CL, Ziegler MG, Chitsaz H, Allen AE, Yooseph S, Tesler G, Pevzner PA, Friedman RM, Nealson KH, Venter JC, Lasken RS. 2013. Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc. Natl. Acad. Sci. U. S. A. 110:E2390–E2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lasken RS. 2012. Genomic sequencing of uncultured microorganisms from single cells. Nat. Rev. Microbiol. 10:631–640 [DOI] [PubMed] [Google Scholar]
- 25. Woyke T, Teeling H, Ivanova NN, Huntemann M, Richter M, Gloeckner FO, Boffelli D, Anderson IJ, Barry KW, Shapiro HJ, Szeto E, Kyrpides NC, Mussmann M, Amann R, Bergin C, Ruehland C, Rubin EM, Dubilier N. 2006. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443:950–955 [DOI] [PubMed] [Google Scholar]
- 26. Baker BJ, Comolli LR, Dick GJ, Hauser LJ, Hyatt D, Dill BD, Land ML, VerBerkmoes NC, Hettich RL, Banfield JF. 2010. Enigmatic, ultrasmall, uncultivated Archaea. Proc. Natl. Acad. Sci. U. S. A. 107:8806–8811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. 2012. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335:587–590 [DOI] [PubMed] [Google Scholar]
- 28. Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428:37–43 [DOI] [PubMed] [Google Scholar]
- 29. Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Wu D, Tringe SG, Singer SW, Eisen JA, Banfield JF. 2013. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat. Commun 4:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderson RT, Vrionis HA, Ortiz-Bernad I, Resch CT, Long PE, Dayvault R, Karp K, Marutzky S, Metzler DR, Peacock A, White DC, Lowe M, Lovley DR. 2003. Stimulating the in situ activity of Geobacter species to remove uranium from the groundwater of a uranium-contaminated aquifer. Appl. Environ. Microbiol. 69:5884–5891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams KH, Long PE, Davis JA, Wilkins MJ, N’Guessan AL, Steefel CI, Yang L, Newcomer D, Spane FA, Kerkhof LJ. 2011. Acetate availability and its influence on sustainable bioremediation of uranium-contaminated groundwater. Geomicrobiol. J. 28:519–539 [Google Scholar]
- 32. Sharpton TJ, Jospin G, Wu D, Langille MG, Pollard KS, Eisen JA. 2012. Sifting through genomes with iterative-sequence clustering produces a large, phylogenetically diverse protein-family resource. BMC Bioinformatics 13:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35:W182–W185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pierce E, Xie G, Barabote RD, Saunders E, Han CS, Detter JC, Richardson P, Brettin TS, Das A, Ljungdahl LG, Ragsdale SW. 2008. The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ. Microbiol. 10:2550–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sato T, Atomi H, Imanaka T. 2007. Archaeal type III RubisCOs function in a pathway for AMP metabolism. Science 315:1003–1006 [DOI] [PubMed] [Google Scholar]
- 36. Fauvart M, Braeken K, Daniels R, Vos K, Ndayizeye M, Noben JP, Robben J, Vanderleyden J, Michiels J. 2007. Identification of a novel glyoxylate reductase supports phylogeny-based enzymatic substrate specificity prediction. Biochim. Biophys. Acta 1774:1092–1098 [DOI] [PubMed] [Google Scholar]
- 37. Müller V, Grüber G. 2003. ATP synthases: structure, function and evolution of unique energy converters. Cell. Mol. Life Sci. 60:474–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abramson J, Riistama S, Larsson G, Jasaitis A, Svensson-Ek M, Laakkonen L, Puustinen A, Iwata S, Wikström M. 2000. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Struct. Biol. 7:910–917 [DOI] [PubMed] [Google Scholar]
- 39. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 40. Feng Y, Li W, Li J, Wang J, Ge J, Xu D, Liu Y, Wu K, Zeng Q, Wu JW, Tian C, Zhou B, Yang M. 2012. Structural insight into the type-II mitochondrial NADH dehydrogenases. Nature 491:478–482 [DOI] [PubMed] [Google Scholar]
- 41. Lee J, Sperandio V, Frantz DE, Longgood J, Camilli A, Phillips MA, Michael AJ. 2009. An alternative polyamine biosynthetic pathway is widespread in bacteria and essential for biofilm formation in Vibrio cholerae. J. Biol. Chem. 284:9899–9907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. White D. 2000. The physiology and biochemistry of Prokaryotes (2nd ed.). Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 43. Sutcliffe IC. 2010. A phylum level perspective on bacterial cell envelope architecture. Trends Microbiol. 18:464–470 [DOI] [PubMed] [Google Scholar]
- 44. Jing H, Takagi J, Liu JH, Lindgren S, Zhang RG, Joachimiak A, Wang JH, Springer TA. 2002. Archaeal surface layer proteins contain beta propeller, PKD, and beta helix domains and are related to metazoan cell surface proteins. Structure 10:1453–1464 [DOI] [PubMed] [Google Scholar]
- 45. Aukema KG, Kron EM, Herdendorf TJ, Forest KT. 2005. Functional dissection of a conserved motif within the pilus retraction protein PilT. J. Bacteriol. 187:611–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Proft T, Baker EN. 2009. Pili in gram-negative and Gram-positive bacteria—structure, assembly and their role in disease. Cell. Mol. Life Sci. 66:613–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang HJ, Baker EN. 2012. Structure and assembly of Gram-positive bacterial pili: unique covalent polymers. Curr. Opin. Struct. Biol. 22:200–207 [DOI] [PubMed] [Google Scholar]
- 48. Chen I, Dubnau D. 2004. DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2:241–249 [DOI] [PubMed] [Google Scholar]
- 49. Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappé MS, Short JM, Carrington JC, Mathur EJ. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309:1242–1245 [DOI] [PubMed] [Google Scholar]
- 50. Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ, Rappé MS. 2012. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3:e00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McCutcheon JP, Moran NA. 2012. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10:13–26 [DOI] [PubMed] [Google Scholar]
- 52. Dufresne A, Salanoubat M, Partensky F, Artiguenave F, Axmann IM, Barbe V, Duprat S, Galperin MY, Koonin EV, Le Gall F, Makarova KS, Ostrowski M, Oztas S, Robert C, Rogozin IB, Scanlan DJ, Tandeau de Marsac N, Weissenbach J, Wincker P, Wolf YI, Hess WR. 2003. Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc. Natl. Acad. Sci. U. S. A. 100:10020–10025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tripp HJ, Bench SR, Turk KA, Foster RA, Desany BA, Niazi F, Affourtit JP, Zehr JP. 2010. Metabolic streamlining in an open-ocean nitrogen-fixing cyanobacterium. Nature 464:90–94 [DOI] [PubMed] [Google Scholar]
- 54. Thompson AW, Foster RA, Krupke A, Carter BJ, Musat N, Vaulot D, Kuypers MM, Zehr JP. 2012. Unicellular cyanobacterium symbiotic with a single-celled eukaryotic alga. Science 337:1546–1550 [DOI] [PubMed] [Google Scholar]
- 55. McCutcheon JP, McDonald BR, Moran NA. 2009. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc. Natl. Acad. Sci. U. S. A. 106:15394–15399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Knight RD, Freeland SJ, Landweber LF. 2001. Rewiring the keyboard: evolvability of the genetic code. Nat. Rev. Genet. 2:49–58 [DOI] [PubMed] [Google Scholar]
- 57. Perona JJ, Hadd A. 2012. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry 51:8705–8729 [DOI] [PubMed] [Google Scholar]
- 58. Peng Y, Leung HC, Yiu SM, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428 [DOI] [PubMed] [Google Scholar]
- 59. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hyatt D, Locascio PF, Hauser LJ, Uberbacher EC. 2012. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28:2223–2230 [DOI] [PubMed] [Google Scholar]
- 61. Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. 2007. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23:1282–1288 [DOI] [PubMed] [Google Scholar]
- 62. Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF. 2009. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. 2013. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 23:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461 [DOI] [PubMed] [Google Scholar]
- 66. Zdobnov EM, Apweiler R. 2001. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848 [DOI] [PubMed] [Google Scholar]
- 67. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS. 2010. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26:1608–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Comfort D, Clubb RT. 2004. A comparative genome analysis identifies distinct sorting pathways in Gram-positive bacteria. Infect. Immun. 72:2710–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pruesse E, Peplies J, Glöckner FO. 2012. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Miller CS, Baker BJ, Thomas BC, Singer SW, Banfield JF. 2011. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 12:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40:D115–D122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogeny of selected CP based on 16S rRNA genes including NCBI accession numbers for the tree shown in Fig. 1. Bipartition values from 1,000 bootstraps are expressed as percentages for all bootstraps >50%. The 16S rRNA genes from genomes in this study and other genomic sequences are in red. Previously reported TM7 single-cell genomic sequences are represented by TM7 genomosp. GTL1 (accession no. AAXS01000094 (20) single cell TM7a (accession no. NZ_ABBV01000356) (19), and Candidatus Saccharimonas aalborgensis (accession no. NC_021219) (12) and are 81 and 85% identical to RAAC3 at the 16S rRNA gene level, respectively. The SR1 sequences ACD80 and SR1-OR1 are 87 and 89% identical to RAAC1, respectively. The OD1 sequences AAA011-A08 and AAA255-P19 represent partial genomes described in reference 22 and accessed through IMG (73). RAAC4 shares 75% sequence identity with AAA011-A08, its nearest relative with a genomic sequence. Download
Phylogenetic tree of the DNA-directed RNA polymerase subunit beta protein of selected organisms, including CP with partial or complete genome sequences. The tree was constructed by using RAxML with 1,000 bootstrap replicates from an alignment of 99 taxa with 900 aligned positions. Sequences from genomes in this study are red, while sequences from genomes used for comparison are blue. Reference and ACD sequences were taken from a report by Wrighton et al. (8); other CP sequences in blue are from public databases or the IMG database (10, 12, 22). Download
Patterns of SR1 relative abundance in relation to sulfide and the relative abundance of other organisms. (A) Log relative abundances of both RAAC1 (SR1) and a member of the phylum Tenericutes correlate with the log sulfide concentration (µM) in each sample until peak sulfate reduction. The two outliers, samples 13 and 14, were taken after peak sulfate reduction, when SR1 abundance was highest, and were not included in the regression. (B) The log relative abundances of the SR1 and Tenericutes species are correlated when all samples are included (adjusted R2 = 0.9). For all regressions, P < 0.0005. Download
Summary of hmmsearch results against the Sfam database (29). CP genomes from this study are RAAC1 to RAAC4. For comparison, there are three phyla for which only one sequenced genome exists (Gemmatimonas aurantiaca, Caldisericum exile, and Desulfurispirillum indicum), two small genomes (approximately comparable in size to the CP genomes) with well-sequenced relatives are represented by Prochlorococcus marinus and Pelagibacter ubique, and a representative of a well-sequenced group with a larger genome, Geobacter metallireducens GS15. For each genome, the percentage of predicted proteins with no hits in the SFam database is shown above the column). Genomes not from this study were obtained from the NCBI Genome (http://www.ncbi.nlm.nih.gov/genome/) and Joint Genome Institute IMG (http://img.jgi.doe.gov/cgi-bin/w/main.cgi) databases. Download
Comparison of the EMP (blue), pentose phosphate (purple), and fermentation (green) pathways across multiple complete and partial CP genomes and the complete RAAC1 to RAAC4 genomes. We note that all SR1 genomes appear to be missing the upper EMP pathway and do not contain the pentose phosphate pathway. The TM7 genomes lack identifiable genes for enolase. The OD1 genomes lack clear means for making acetyl-CoA from pyruvate. Download
Comparison of pyrimidine biosynthesis (yellow) and purine biosynthesis (red) across multiple partial CP genomes and the complete RAAC1 to RAAC4 genomes. Only the AAA255-P19 genome (22) appears to have nearly complete pathways for nucleotide biosynthesis. The presence or absence of this metabolism appears to vary across the CP. The large number of nucleases present in the CP genomes could provide an alternate source of nucleotides. Note that ACD22, ACD24, and ACD25 may contain multiple copies of genes because of multiple strains or species within a bin, although these genomes are incomplete. Download
Counts by genome of proteins involved in cell wall and cell surface biosynthesis (A) and cell-environment interactions (B). Yellow lists in panel B are not mutually exclusive, as a given protein can have more than one function or domain and was counted in each appropriate category. Download
Codon usage shown for selected amino acids reflects coding bias in SR1 and OD1 genomes but not WWE3 and TM7 genomes. Codon usage was calculated as a percentage of the total, including the termination codon. Dashed lines group triplets that typically encode the same amino acid. SR1 uses UGA to encode glycine instead of termination, and hence it is found much more frequently in that genome. The OD1 genome uses UGA as a termination codon for only 34 out of 687 predicted ORFs, representing 0.02% of the codons in the genome. Download
Gene identifiers for enzymes numbered in Fig. 4. Green, identified; yellow, homology and function unclear; red, not identified by BLAST with a diverse reference set, KAAS, or in-house annotation pipeline; NA, not detected by in-house annotation pipeline. The relevant DNA and protein sequences are accessible at http://genegrabber.berkeley.edu/aac/organisms.
Complete nucleotide biosynthesis pathways were not identified in any of the four CP genomes. Gene identifiers are listed when detected. Colors are the same as in Table S1. All genomes were further searched by using a curated reference set of archaeal purine biosynthesis genes collected on the basis of the work of Brown et al. (A. M. Brown, S. L. Hoopes, R. H. White, and C·A. Sarisky, Biol. Direct 6:63, 2011, doi:10.1186/1745-6150-6-63), and no complete pathway was identified in any of the four genomes. The relevant DNA and protein sequences are accessible at http://genegrabber.berkeley.edu/aac/organisms.