

ABSTRACT
Biofilms are thought to play an important role during colonization of the nasopharynx by Streptococcus pneumoniae, yet how they form in vivo and the determinants responsible remain unknown. Using scanning electron microscopy, we show that biofilm aggregates of increasing complexity form on murine nasal septa following intranasal inoculation. These biofilms were highly distinct from in vitro biofilms, as they were discontiguous and appeared to incorporate nonbacterial components such as intact host cells. Biofilms initially formed on the surface of ciliated epithelial cells and, as cells were sloughed off, were found on the basement membrane. The size and number of biofilm aggregates within nasal lavage fluid were digitally quantitated and revealed strain-specific capabilities that loosely correlated with the ability to form robust in vitro biofilms. We tested the ability of isogenic mutants deficient in CbpA, pneumolysin, hydrogen peroxide, LytA, LuxS, CiaR/H, and PsrP to form biofilms within the nasopharynx. This analysis revealed that CiaR/H was absolutely required for colonization, that PsrP and SpxB strongly impacted aggregate formation, and that other determinants affected aggregate morphology in a modest fashion. We determined that mice colonized with ΔpsrP mutants had greater levels of the proinflammatory cytokines tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), IL-1β, and KC in nasal lavage fluid than did mice colonized with wild-type controls. This phenotype correlated with a diminished capacity of biofilm pneumococci to invade host cells in vitro despite enhanced attachment. Our results show that biofilms form during colonization and suggest that they may contribute to persistence through a hyperadhesive, noninvasive state that elicits a dampened cytokine response.
IMPORTANCE
This work demonstrates the first temporal characterization of Streptococcus pneumoniae biofilm formation in vivo. Our results show that the morphology of biofilms formed by both invasive and noninvasive clinical isolates in vivo is distinct from that of formed biofilms in vitro, yet propensity to form biofilms in vivo loosely correlates with the degree of in vitro biofilm formation on a microtiter plate. We show that host components, including intact host cells, influence the formation of in vivo structures. We also found that efficient biofilm formation in vivo requires multiple bacterial determinants. While some factors are essential for in vivo biofilm formation (CiaRH, PsrP, and SpxB), other factors are less critical (CbpA, LytA, LuxS, and pneumolysin). In comparison to their planktonic counterparts, biofilm pneumococci are hyperadhesive but less invasive and elicit a weaker proinflammatory cytokine response. These findings give insight into the requirements for and potential role of biofilms during prolonged asymptomatic colonization.
Introduction
Streptococcus pneumoniae (the pneumococcus) is a Gram-positive bacterium that colonizes the human nasopharynx. Although colonization is typically asymptomatic, S. pneumoniae is an opportunistic pathogen capable of a myriad of infections, including sinusitis, otitis media, pneumonia, bacteremia, and meningitis (1). In general, the attack rate for the pneumococcus is very low, yet such vast numbers of individuals are colonized that the overall disease burden is tremendous. For example, in children of <5 years of age >14.5 million episodes of invasive pneumococcal disease (IPD) are recorded annually, accounting for ~800,000 deaths worldwide (3).
Colonization with the pneumococcus typically occurs without overt inflammation and can last up to several months. It is estimated that 25 to 40% of healthy children in daycare settings and 10 to 15% of adults are colonized at any given time (4). Although carriage is known to be a prerequisite for disease development, our understanding of the host-pathogen interactions that allow S. pneumoniae persistence within the nasopharynx is incomplete. Importantly, considerable evidence now suggests that biofilms, microbial communities attached to a surface and encased within an extracellular matrix (5), play an important role during colonization. In support of this notion, mixed biofilms containing S. pneumoniae have been observed on adenoid tissue and mucosal epithelial cells isolated from patients with chronic rhinosinusitis (6–9). Pneumococcal biofilms have been detected on nasal septa of experimentally colonized mice and can form on cultured respiratory epithelial cells in vitro (10, 11). Pneumococcal aggregates composed of hundreds to thousands of individual pneumococci were observed in nasal lavage elutes taken from asymptomatically colonized mice (12).
The formation of a biofilm presumably confers several advantages to the pneumococcus. Bacteria within biofilms are more resistant to environmental stressors, including host defenses such as defensins and phagocytic cells, than their planktonic counterparts (5, 13–15). Biofilm pneumococci are desiccation resistant and hyperadhesive (16, 17). Pneumococci in biofilms have been reported to reduce the expression of pneumolysin (17–19), a pore-forming toxin that is detected by Toll-like receptor 4 and activates the NLRP-3 inflammasome (20, 21). This, along with other modifications that occur during the biofilm lifestyle (17, 22), potentially promotes asymptomatic colonization. Importantly, the vast majority of studies on S. pneumoniae biofilm formation have been performed in vitro and the requirements or impact of in vivo biofilm formation on colonization remains untested.
In this study, we characterized the temporal formation of biofilms within the nasopharynx of colonized mice and determined the contribution of pneumococcal determinants previously implicated in either in vitro biofilm formation or nasopharyngeal colonization. We included mutants deficient in (i) the adhesin choline binding protein A (CbpA), which mediates bacterial attachment to laminin receptor and polymeric immunoglobulin receptor on mucosal epithelial cells (23, 24); (ii) the pore-forming toxin pneumolysin (Ply); (iii) the enzyme pyruvate oxidase, encoded by the gene spxB, which is responsible for the production of and pneumococcal protection from hydrogen peroxide (25); (iv) autolysin (LytA), the major cell wall hydrolase responsible for pneumococcal DNA release (26); (v) S-ribosylhomocysteine lyase (LuxS), the enzyme responsible for production of the quorum sensing molecule homoserine lactone autoinducer 2 (AI-2), known to be required for in vitro biofilm formation (27); (vi) CiaR/H, the oxygen-sensitive two-component signal transduction system which mediates the stress response (28); and (vii) pneumococcal serine-rich repeat protein (PsrP), a lung cell and intraspecies adhesin previously shown to be required for robust in vitro and in vivo biofilm formation (12). Our results highlight the key role of some of these factors, as well as the compensatory ability of S. pneumoniae biofilm-related mechanisms. In addition to this, we also show that biofilm pneumococci provoke a weaker inflammatory response than their planktonic counterparts and, while hyperadhesive, are less invasive. These latter findings help to elucidate the role that biofilms play during prolonged asymptomatic colonization with S. pneumoniae.
RESULTS
Robust in vitro biofilm formation is strain dependent and not correlated with successful colonization of mice.
We first examined the ability of our noninvasive nasopharyngeal isolates 6A10 and 6B8, as well as the invasive isolates TIGR4, 6A16, 6A18, and 6B21, to form biofilms in vitro using a 6-well microtiter plate 18-h model of biofilm formation (Fig. 1A). 6A10 and 6B8 formed dense biofilms characterized by a thick confluent monolayer of bacteria on the bottom of the well. In contrast, TIGR4, 6A16, and 6A18 formed thin biofilms, with the underlying polystyrene surface of the plate remaining visible. The invasive isolate 6B21 formed robust biofilms equivalent to those of noninvasive 6A10 and 6B8. We subsequently assayed the same strains for their ability to colonize the nasopharynx of BALB/c mice over a 2-week period. All strains showed indiscernible colonizing capabilities as measured by CFU/ml of nasal lavage fluid (NALF) (Fig. 1B). Of note, recoverable CFU peaked at 3 to 5 days postinfection (dpi) and dropped approximately 100-fold by 14 dpi. Enumeration of intimately attached bacteria (i.e., those not dislodged following lavage with saline) in homogenized nasal septum samples by both quantitative reverse transcription-PCR (qRT-PCR) and plating also showed no difference between the strains at day 7 (see Fig. S1 in the supplemental material). Importantly, enumeration of bacterial load by qRT-PCR seemed to be more sensitive, with an ~10-fold increase in extrapolated CFU load. In summary, and consistent with previously published studies (29), we saw no correlation between robust in vitro biofilm formation by distinct clinical isolates and the ability to colonize the nasopharynx of mice. Nonetheless, in vitro biofilm formation was loosely correlated with the anatomical site from which clinical isolates were collected, consistent with the report by Trappetti et al. (30).
FIG 1 .
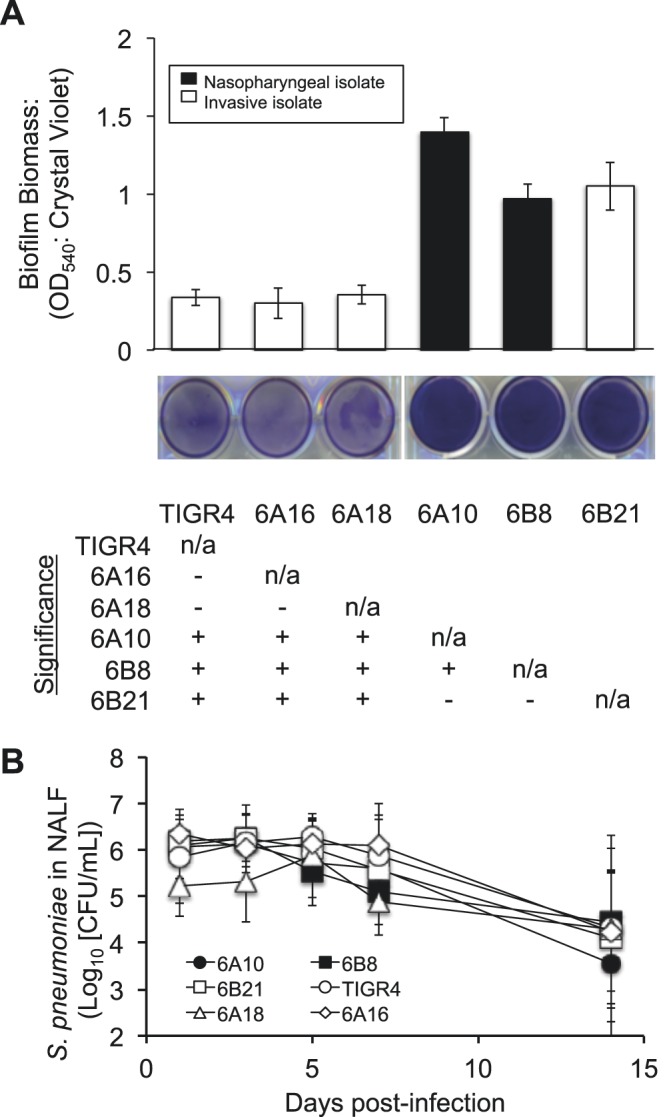
In vitro biofilm formation by S. pneumoniae clinical isolates and corresponding rates of colonization. (A) Mean biomass of biofilms formed by clinical isolates on 6-well polystyrene plates after 18 h of growth, as measured by crystal violet staining (n = 4/strain). Accompanying representative images of stained wells are provided beneath the graph. The table represents statistical significance between strains (+, significance; −, no significance) grown on untreated plates as determined by one-way ANOVA. n/a, not applicable; OD540, optical density at 540 nm. (B) CFU determination of S. pneumoniae in NALF collected from colonized mice at days 1, 3, 5, 7, and 14 postinfection. No significant differences were observed between strains on any given day as tested by one-way ANOVA (n = 9 to 12/cohort).
Biofilm aggregates form on the nasal septa of colonized mice.
To discern if differences between a high- and a low-in-vitro-biofilm-forming strain occurred in vivo, we examined isolated nasal septa from naive mice (control) and mice colonized with 6A10 or TIGR4 at 3, 7, and 14 dpi by using scanning electron microscopy (SEM). Importantly, control mice displayed an undisturbed septal epithelium with intact cilia and a few scattered immune cells (Fig. 2A and B). On day 3, the septa of mice colonized with 6A10 showed the presence of small bacterial aggregates resting on top of the ciliated epithelial cells (Fig. 2C). In some areas, there seemed to be an irregular loss of epithelial cells with what appeared to be the basement membrane exposed and biofilm aggregates attached in their place. Thus, epithelial cells seem to have been sloughed off. At 7 dpi, the formation of larger 6A10 bacterial aggregates became obvious, accompanied by greater areas of exposed basement membrane (Fig. 2E and I). Finally, septa recovered from 6A10-colonized mice at 14 dpi were characterized by an overwhelming number of large bacterial aggregates encased within a matrix structure and attached to the completely bare basement membrane. Unlike earlier time points, in the areas where aggregates were observed, no ciliated epithelial cells were seen in any mice collected at day 14 (Fig. 2G). Septa collected from mice colonized with TIGR4 showed a similar pattern of aggregate formation and epithelial sloughing, although aggregates, in general, seemed to be considerably smaller than those for 6A10 (Fig. 2D, F, H, and J). Interestingly, what appeared to be host cells were frequently seen as part of the biofilm aggregates of both strains (Fig. 2G, I, and J). Similar observations were noted for nasal septa colonized by the remaining clinical isolates (data not shown).
FIG 2 .

Pneumococcal biofilms are present on mucosal epithelial cells in the septa of experimentally infected mice. Scanning electron microscopy of isolated nasal septa from naive or colonized mice. (A and B) Low (A)- and high (B)-magnification views of healthy septal epithelia. (C) 6A10-colonized nasal septa at 3 dpi. Both matrix-associated bacteria and free diplococci are present on top of ciliated epithelial cells. (D) TIGR4-colonized septa at 3 dpi. Small aggregates of bacteria are present. (E and I) 6A10-colonized septa at 7 dpi. Bacteria are encased within a matrix, and aggregates contain a number of visible host components. Large aggregates are associated with an absence of cilia. (F and J) TIGR4-colonized septa at 7 dpi. Clusters of aggregated bacteria rest on the epithelial surface and are associated with a number of host cells. (G and H) 6A10 (G)- and TIGR4 (H)-colonized respiratory epithelium at 14 dpi. Cilia are absent, and bacterial aggregates are seen on the exposed cells. Bacteria are encased within a thick, visible matrix, and individual cells are difficult to distinguish. 6A10 aggregates are large and highly structured, whereas TIGR4 aggregates were mottled and noticeably smaller. (I and J) Aggregates formed by both 6A10 (I) and TIGR4 (J) contain a number of host components, including inflammatory cells. Arrows indicate common features of biofilm architecture: exposed basement membrane, bacterial cells within matrix material, and incorporated ciliated cells and leukocytes.
In vivo pneumococcal aggregate formation over time loosely correlates with in vitro ability.
To provide a quantitative analysis of the observed biofilm aggregates, we stained and digitally quantitated the size and number of bacterial aggregates within NALF using ImageJ software (Fig. 3). Consistent with our SEM results, these analyses revealed the more frequent occurrence of large bacterial aggregates from 3 to 14 dpi for strain 6A10. This was also observed for 6A16, 6B8, and 6B21. In some instances, extremely large aggregates were observed at 14 dpi that occupied the majority of our visual field (see Fig. S2 in the supplemental material). In contrast, aggregates detected from the NALF of mice colonized with TIGR4 and 6A18 were much smaller and remained consistent in size between 3 and 14 dpi. Thus, the high-biofilm-forming strains all formed large aggregates in vivo, and the majority of low-in-vitro-biofilm-forming strains did not. The exception was 6A16, which formed large aggregates in vivo. We conclude that there is a general, but incomplete, correlation between in vitro biofilm formation and in vivo aggregate formation. Importantly, these results provide the first temporal and quantitative evidence for the changes in pneumococcal biofilm aggregate size seen during colonization. Recovered aggregates were confirmed to be S. pneumoniae by visual confirmation of diplococci within Gram-stained elutes using a microscope and testing for optochin sensitivity on blood agar plates following culture.
FIG 3 .

In vivo aggregates detected in NALF of mice colonized with S. pneumoniae. Following processing with ImageJ software (see Materials and Methods), individual biofilm aggregates were grouped into classes based on pixel size, from small aggregates (1 to 10/11 to 1,000) to medium (1,001 to 10,000/10,001 to 100,000) to large (>100,000). Shown is the contribution of each size class to the total aggregate area as determined by a percentage of the total pixel area. Asterisks denote a significant difference between size areas compared to day 3. Statistical analysis was done using Student’s t test.
Assessment of the contribution of individual virulence determinants to biofilm formation.
To determine the individual contribution of established virulence determinants to in vivo biofilm formation, we tested a large panel of 6A10 isogenic mutants. We chose to use 6A10 as our reference strain since it formed the most robust in vivo biofilms. The mutants selected for examination were chosen since previous studies had shown the corresponding gene products to be required during in vitro biofilm formation (10, 12, 27, 31–34) or nasopharyngeal colonization (31, 35–39). We first assayed these mutants in vitro using the 18-h, 6-well microtiter plate biofilm model; consistent with previous publications, no significant differences from the wild type were observed for the majority of the mutants (29, 31). The two exceptions were the spxB mutant, which had an increase in biofilm formation, and the ciaR/H mutant, which had a significant decrease in biofilm formation (see Fig. S3 in the supplemental material). A strong trend for reduced biofilm formation was observed for the mutant lacking psrP-secY2A2 (P = 0.062). In vivo, loss of CiaR/H resulted in the most obvious defect, with no recoverable bacteria detected from the nasopharynx of mice at any time point, indicating that loss of CiaR/H resulted in a major fitness defect (see Fig. S4A). For this reason, 6A10ΔciaR/H was not included in further experiments. None of the remaining mutants exhibited a significant difference from the wild type in their ability to colonize when using CFU/ml of nasal lavage fluid to examine colonization over time or when homogenized nasal septa from mice at day 7 were tested for levels of intimately attached bacteria by qRT-PCR and colony counts (see Fig. S4B).
Compared to the wild-type controls, SEM of septa collected from mice at day 7 revealed that the loss of individual determinants resulted in various degrees of attenuation (Fig. 4). Mice colonized with 6A10ΔcbpA had what appeared to be a normal number of biofilm aggregates, although the morphology of these biofilms appeared smaller and seemed to be less structurally complex. While not enumerated, visual inspection suggested that 6A10ΔcbpA aggregates contained fewer host cell fragments than did wild-type formed biofilms. 6A10ΔcbpA aggregates also seemed to be flatter and scattered. Mice colonized with 6A10ΔlytA also contained smaller aggregates, although these aggregates retained the same morphological characteristics as wild-type pneumococci. Mutants lacking LuxS were also morphologically similar to wild-type bacteria but also appeared to be greatly reduced in size. Of note, 6A10ΔlytA- and 6A10ΔluxS-colonized epithelia did not seem to display the same level of basement membrane exposure as mice colonized with 6A10. Loss of pneumolysin seemed to enhance the size of the aggregates observed in SEM images, albeit the presence of host cells in the biofilm aggregates remained constant and exposure of the basement membrane continued to occur. Analysis of septa from mice colonized with 6A10ΔspxB revealed that this mutant was severely attenuated for aggregate formation in vivo. Formed aggregates were barely detectable and appeared to lack the matrix structure seen in wild-type aggregates. Lastly, the septal epithelia of mice colonized with the 6A10ΩpsrP-secY2A2 deficient mutant displayed a complete absence of aggregates and undisturbed epithelia. This is consistent with our earlier finding that TIGR4 required PsrP for in vivo biofilm formation (12). Importantly, individual diplococci were not readily discernible in any of the SEM images despite being present in NALF samples (Fig. 3 and 5). This may be due to their small size or their loss during the collection and fixation process.
FIG 4 .

Biofilm aggregates on septal epithelia of mice colonized with isogenic mutants. Nasal septa from 6A10-colonized mice collected at 7 dpi at low and high magnification. Aggregate formation in vivo was minimally decreased or equal to that of the wild type in strains lacking CbpA or pneumolysin, as structures were still fairly large in size and contained host components. Mutants lacking LytA, LuxS, and SpxB exhibited a decreased ability to form large aggregates, and observed aggregates rested on top of ciliated epithelial cells, in contrast to the exposed basement membrane observed in wild-type-colonized samples. The PsrP mutant strain was completely unable to form biofilm aggregates, and colonized mice displayed a healthy ciliated epithelia. The presence of scattered immune cells could be seen.
FIG 5 .

Mutant strains are able to colonize efficiently but exhibit a decreased ability to form aggregates in vivo. ImageJ quantitation of aggregate size distribution over time by 6A10 or 6A10 isogenic mutants. Statistical significance was determined by 2-tailed Student’s t test versus the 6A10 control at the corresponding time point and is represented with an asterisk.
To quantitate the differences described above, digitalized images of crystal violet (CV)-stained bacteria in NALF of colonized mice were again analyzed (Fig. 5). At day 3, aggregates detected in nasal lavage fluid showed no differences in size distribution, with the exception of 6A10ΔlytA and 6A10ΩpsrP-secY2A2 samples, which showed a modest, but significant, increase in the prevalence of smaller aggregates and planktonic bacteria. At day 7 postinfection, other mutants began to show a similar phenotype, with 6A10ΔcbpA, 6A10ΔlytA, 6A10ΔluxS, and, unexpectedly, 6A10Δpln showing an increased proportion of small aggregates compared to wild-type 6A10. 6A10ΩpsrP-secY2A2 NALF contained an increased amount of small aggregates and an associated decrease in larger aggregates. By 14 days postinfection, all of the mutant strains assayed displayed a decreased ability to form the larger aggregates, with 6A10ΔspxB- and 6A10ΩpsrP-secY2A2-colonized mice exhibiting a near-complete absence of the large aggregates. Importantly, the amount of smaller aggregates found within the NALF of mice colonized with 6A10ΔcbpA, 6A10ΔlytA, 6A10ΔluxS, 6A10Δpln, and 6A10ΔspxB was significantly larger than those found in the NALF of 6A10-colonized mice, indicating that while large aggregates failed to form, smaller aggregates and individual pneumococci remained present. These results indicate that there is, in fact, a significant difference in the aggregates formed by mutant strains in vivo.
Biofilm-deficient pneumococci elicit a stronger proinflammatory response.
Since the absence of overt inflammation would presumably serve to prolong colonization, we tested if the biofilm phenotype affected the host response. Consistent with this notion, we observed a muted interleukin-6 (IL-6) and IL-8 response to biofilm pneumococci by Detroit-562 nasopharyngeal cells (Fig. 6A). This phenomenon was recapitulated in J774A.1 macrophages that showed decreased IL-1β, IL-6, and tumor necrosis factor alpha (TNF-α), in response to biofilm pneumococci (Fig. 6A). We next sought to test this in vivo. Since naturally occurring biofilm-deficient mutants do not exist, we first compared cytokine levels in forced nasal washes from mice colonized for 7 days with the low-biofilm-forming strain TIGR4 and the high-biofilm-forming strain 6A10 (Fig. 6B). In both instances, minimal levels of TNF-α and IL-1β were observed, no differences were detected for KC, and a significantly lower level of IL-6 was observed for 6A10 than for TIGR4. Since PsrP mutants do not form biofilms in vivo (Fig. 4 and 6C), we also tested the cytokine response to TIGR4 and 6A10 mutants lacking PsrP. Mice colonized with T4ΔpsrP had higher levels of TNF-α and IL-6 in pooled nasal lavage samples at days 1, 3, and 5 (Fig. 6D) and higher IL-6 and KC levels in the forced lavage samples at day 7 than did the wild type (Fig. 6B). Importantly, the latter findings are tempered by the fact that, for TIGR4, more CFU were present in the nasopharynx of mice colonized with the PsrP mutant than in mice colonized with the wild type [TIGR4 recovered log10(CFU/ml) = 5.375, T4ΔpsrP log10(CFU/ml) = 6.133, P = 0.006; TIGR4 log10(copy number by qRT-PCR) = 5.999, T4ΔpsrP log10(copy number by qRT-PCR) = 7.123, P = 0.023]. The latter has previously been attributed to the high metabolic cost of producing PsrP (40). For 6A10, where no differences between the psrP-deficient mutant and the wild type in colonization levels were detectable (see Fig. S3 in the supplemental material), 6A10ΩpsrP-secY2A2 elicited a strong trend toward greater IL-6 and IL-1β levels in forced lavage samples (Fig. 6B).
FIG 6 .

S. pneumoniae that is unable to form biofilms elicits an enhanced cytokine response during colonization. (A) Assorted cytokine levels in Detroit-562 pharyngeal epithelial cell or J774A.1 macrophage supernatants 24 h following a 4-h exposure to 106 CFU of either planktonic or biofilm-derived TIGR4 pneumococci. (B) Levels of TNF-α, IL-6, IL-1β, and KC in nasal lavage samples collected at 7 days postinfection from mice colonized with wild-type or PsrP-deficient isogenic mutants of strains TIGR4 and 6A10 (n = 5/cohort). Cytokine levels were measured using ELISA. Significance was tested by Student’s t test (n = 5/cohort). Asterisks denote a statistically significant difference (P < 0.05) for the mutant versus its respective wild type. Hash tag denotes statistically significant difference versus TIGR4. (C) Representative scanning electron micrograph of septa isolated from mouse colonized with T4ΔpsrP. Note that T4ΔpsrP does not form the biofilm aggregates observed in mice infected with TIGR4 or cause the sloughing of mucosal epithelial cells. (D) Levels of TNF-α and IL-6 in pooled nasal lavage samples collected from mice colonized with wild-type or PsrP-deficient isogenic mutants of strain TIGR4 on days 1, 3, and 5 postinfection (n = 6/cohort). Cytokine levels were measured using ELISA.
Biofilm bacteria may fail to evoke a strong proinflammatory response due to a number of reasons, and this result seemed paradoxical given our prior observation of cell sloughing on the nasal septa of colonized mice by SEM. For this reason, we examined the ability of biofilm pneumococci to invade host cells. Biofilm pneumococci were found to be hyperadhesive compared to their planktonic counterparts. In contrast, their capacity for cellular invasion was dramatically decreased. Overall, the invasive index (invasive events measured per adherence event) was 33-fold lower for biofilm pneumococci than the planktonic value (Fig. 7). Given these results, we surmise that biofilm bacteria may not be invading the cells to which they are attached in vivo, whereas planktonic pneumococci are capable of invasion. Importantly, histological analysis of nasal septa from 6A10-colonized mice confirmed the disruption of ciliated epithelial cells (see Fig. S5 in the supplemental material), indicating that cell sloughing was not an artifact of our SEM and that, despite a reduction in the invasive potential and a weaker inflammatory host response to biofilm pneumococci, the host was indeed responding to the infection.
FIG 7 .
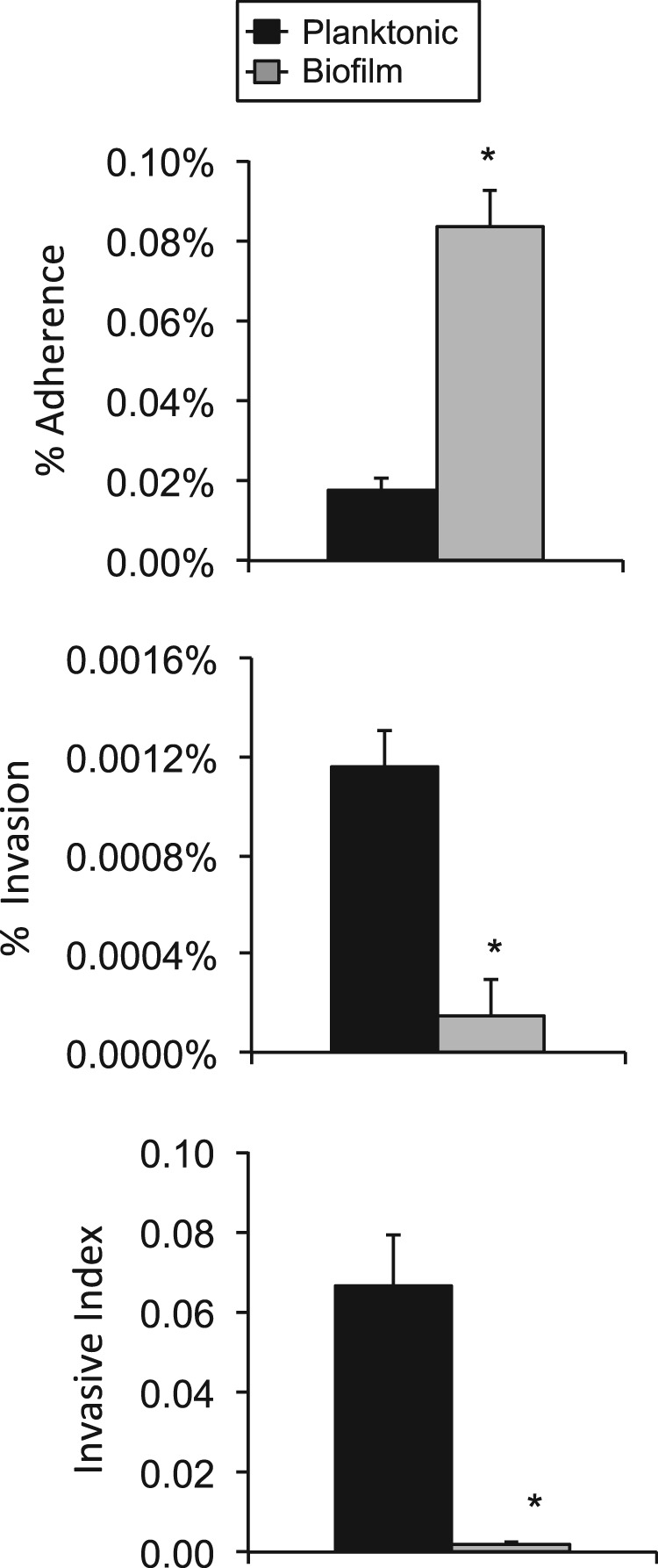
Biofilm bacteria are hyperadhesive but less invasive. Percentage of TIGR4 cells from the total inoculum that attached and invaded Detroit-562 pharyngeal epithelial cells. The ratio of invasive pneumococci to those that are attached is also shown. Experiments were performed in triplicate. Statistical analysis was performed using a two-tailed Student t test. Asterisks denote a significant difference (P < 0.05).
DISCUSSION
This study is the first temporal analysis of pneumococcal biofilm formation during nasopharyngeal colonization and the first comprehensive determination of the required S. pneumoniae virulence determinants. Analysis of dislodged bacteria in NALF showed that aggregates were able to form within the nasopharynx and increased dramatically in size over time, although this ability was markedly strain dependent. The ability of clinical isolates to form aggregates in vivo was loosely correlated with their ability to form biofilms in vitro and the anatomical site from which the bacteria were isolated, albeit no correlation with the ability to successfully colonize the nasopharynx was found. In vivo biofilms formed by S. pneumoniae were striking in their difference from those formed on the polystyrene microtiter plate surface. Biofilms within the nasopharynx were neither confluent nor contiguous and incorporated host cells. Aggregates of all sizes were observed in vivo, with some most likely exceeding several thousand pneumococci at later stages. For all wild-type strains tested, biofilm aggregates on mucosal epithelial cells transitioned from spotty aggregates on top of ciliated cells to larger clumps and structures attached directly to the basement membrane. Very few planktonic bacteria were observed at any time point, possibly due to our collection techniques; unattached bacteria are not likely to have remained on septal samples throughout the washing steps. The hyperadhesive state of biofilm pneumococci may have enabled their visualization by SEM. Two recent studies using in vitro systems have shifted more closely toward the in vivo phenotype by growing bacterial biofilms either on fixed epithelial cells or in a continuous reactor that exchanges the medium (10, 11). In these instances, the formed biofilms are more similar to what we observed within the nasopharynx.
Using this in vivo model, we determined that a number of pneumococcal factors contribute to in vivo biofilm formation. Importantly, all of these factors, with the exception of pneumolysin, contributed in some manner to robust in vivo biofilm formation, with a stark requirement observed for CiaR/H, SpxB, and PsrP. We deemed mutants lacking CiaR/H as unfit due to a complete inability to establish infection, thus precluding the mutant’s ability to form in vivo biofilms. CbpA has been shown to be critical for colonization (31, 39), likely due to its ability to bind laminin receptor and pIgR (23, 24). Thus, loss of this protein removes a number of binding opportunities for the pneumococcus that may explain the altered morphology. Prior to this study, CbpA has been shown to be important for biofilm formation only when tested for in an unencapsulated background in vitro (31). Loss of LytA, the major autolysin of S. pneumoniae, has also been shown to result in decreased biofilm formation in vitro (33). Debris from dead pneumococci may contribute to the formation of the biofilm matrix and compose the bulk of the biofilm aggregate (33, 41). Consistent with this, our data show that the lytA mutant formed aggregates of greatly reduced size and complexity in vivo. Hydrogen peroxide has been shown to trigger autolysis; this may explain why the SpxB− mutant also resulted in very small aggregates being formed.
Surprisingly, 6A10ΔluxS displayed only a modest decrease for in vivo biofilm aggregate size. LuxS had been shown by multiple investigators to be important during biofilm formation in vitro, both in standard and on cultured epithelial cell models (11, 27, 34). LuxS is an important factor in the pneumococcal autoinducer 2 (AI-2) quorum sensing system. Inhibition of this system is thought to block the bacterial response to the host environment, thus preventing the signaling necessary for establishment of the biofilm phenotype. Importantly, S. pneumoniae contains a second quorum sensing system, the Com system, which has been shown to play a role in biofilm formation, specifically on cultured respiratory cells (11). Therefore, we speculate that loss of LuxS and AI-2 system components may be compensated for. Deletion of pneumolysin had a modest effect; this was not unexpected, as studies have shown that the expression of pln is reduced during in vitro biofilm formation (17). Finally, PsrP had a dramatic effect on biofilm formation in vivo. PsrP is now understood to be an intraspecies adhesin that promotes biofilm formation through the homodimerization of its BR domain to PsrP on other pneumococci (12). It also binds to cytokeratin 10 on host cells (42), although this ligand is thought to be absent on the nasal septa. While TIGR4, 6A10, 6A16, and 6B8 all carry PsrP, strains 6A18 and 6B21 do not and still formed in vivo biofilms (43), suggesting that other factors are capable of compensating for the absence of PsrP.
We determined that biofilm pneumococci elicit a significantly weaker immune response from nasopharyngeal and macrophage cell lines in vitro than do their planktonic counterparts. The notion that this occurs in vivo is supported by our observation of a greater proinflammatory response to TIGR4 than to 6A10, as well as the greater response to the PsrP-deficient mutants than to their wild-type controls. One caveat is that, for TIGR4, there are more CFU present for the mutant, which most likely also contributes to the higher inflammatory response. Importantly, this decrease in the immune response is in contrast to the highly reproducible observation that formation of biofilm aggregates is associated with a loss of ciliated epithelial cells. At this time, we cannot explain these seemingly contradictory results, but one possible explanation is that sloughing of epithelial cells is not an inflammatory event and that biofilm bacteria in contact with underlying cells fail to elicit a robust immune response. This may be due to decreased invasion of tissues or, alternatively, the previously reported reduction in pneumolysin production.
One highly important consideration is that, regardless of differences in biofilm-forming ability and differences in the detected inflammatory response to the colonizing strain, we did not observe any significant differences in colonization levels as measured by bacterial titers between clinical isolates or the majority of mutants. While this may be due to the technical limitations of our approach, it fails to provide answers regarding the advantage of forming biofilms in vivo. One obvious and yet untested possibility is for transmission. The biofilm aggregates are presumably more resistant to desiccation than individual pneumococci, and their hyperadhesive state makes them highly suitable vehicles for transmission on fomites. Alternatively, differences in biofilm-forming ability may be an important factor in disease development versus asymptomatic colonization in response to additional stimulation. This is evidenced by a recent paper by Marks et al., showing that external factors affect biofilm formation and that dispersed bacteria are more virulent than their biofilm counterpart (44).
Despite considerable differences in aggregate size, no significant differences in recoverable CFU between wild-type strains or the majority of mutants tested were observed in vivo. This may be indicative of a technical failure for the traditional recovery methods in the enumeration of colonizing bacteria. Large aggregates containing thousands of bacteria are presumably counted as a single CFU during plating, thus leading to a lower reading of overall burden, particularly at later time points. Additionally, collection of NALF alone does not account for bacteria that may remain more tightly attached to the epithelial surface or bacteria that have been internalized. PCR-based approaches would presumably count both live and dead pneumococci that are part of in vivo aggregates. In vitro, a large amount of the biofilm is dead bacteria (17). Interestingly, we detected ~10-fold-more S. pneumoniae bacteria in homogenized nasal septum samples by qRT-PCR than by standard CFU counts from paired samples. Thus, the enumeration of bacteria within the nasopharynx remains problematic, with analysis of burden affected by the particular strain’s ability to form biofilm aggregates and the presence of dead bacteria within aggregates.
In summary, here we have shown that biofilms develop over time in vivo and that their morphology is highly distinct from that seen in a microtiter plate model. We have identified key virulence determinants in this process and shown that biofilm pneumococci are less invasive and elicit a weaker inflammatory response; this may facilitate long-term colonization, although direct proof remains lacking. Importantly, how biofilm contributes to successful asymptomatic colonization remains an open question. Thus, future studies continue to be warranted to help understand the role that pneumococcal biofilms play in vivo.
MATERIALS AND METHODS
Bacterial strains.
S. pneumoniae strains used in this study include TIGR4 (T4), a virulent serotype 4 clinical isolate (45), as well as serotype 6A and 6B isolates previously obtained from the nasopharynx of healthy children (strains 6A10 and 6B8) and those with IPD (6A16, 6A18, and 6B21) (29). The TIGR4 mutant deficient in psrP (T4ΔpsrP) has been previously described (12). Isogenic deletion mutants in the 6A10 background were created by allelic exchange using mutagenic PCR constructs consisting of fragments of the flanking genes around an erythromycin resistance cassette (46). Mutants created for this study included those deficient in cbpA (6A10ΔcbpA), nanA (6A10ΔnanA), pln (6A10Δpln), spxB (6A10ΔspxB), ciaR/H (6A10ΔciaR/H), lytA (6A10ΔlytA), luxS (6A10ΔluxS), and the pathogenicity island encoding PsrP (6A10ΩpsrP-secY2A2). Mutants were kept under antibiotic selection with 1 µg/ml erythromycin at all times with the exception of outgrowth for experiments.
In vitro biofilm formation.
For experiments examining early biofilm formation, in vitro biofilms were grown as previously described using the microtiter plate model with biofilm biomass quantitated by spectrophotometry for attached crystal violet (CV) (47). Specifically, bacteria were diluted from frozen stocks to 105 CFU/ml in Todd-Hewitt broth (THB) and 2 ml of the suspension was added to each well of a 6-well polystyrene plate. Plates were incubated for 18 h at 37°C in 5% CO2 prior to washing and staining. For experiments examining the host cell response to biofilms, mature biofilms were grown using a continuous flow biofilm line reactor as previously described (12). Biofilms were grown in THB for 2 days at 37°C in 5% CO2. Planktonic cultures used to seed the line reactor and their extracted biofilms were frozen and used in a paired fashion.
Imaging and quantitative analysis of bacterial aggregates in nasal lavage samples.
All animal experiments were reviewed and approved by the University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee. Female 6-week-old BALB/c mice (Jackson Laboratories, Bar Harbor, ME) were infected intranasally with 105 CFU of S. pneumoniae in 10 µl phosphate-buffered saline (PBS). Mice were anesthetized using 2% vaporized isoflurane and laid on their side, at which point bacterial solution was pipetted onto the right nostril, where it was inhaled. On days 1, 3, 5, 7, and 14 postinfection, nasal lavage was performed on anesthetized mice by administering and then aspirating 10 µl of PBS from the nares. A portion of the recovered nasal lavage fluid (NALF) was serially diluted, plated onto blood agar plates, and incubated at 37°C in 5% CO2 overnight to extrapolate CFU/ml from colony counts. The remaining portion of collected NALF was stained using a 1:1 ratio of 1% CV and observed under high magnification with a light microscope. Digital images were captured at random and compiled, and the three median images were chosen for quantitative analysis using ImageJ software. Images were converted to black and white to allow for quantitation of aggregates using the software particle-counting application. Particle count data for each image were then exported to a spreadsheet, sorted, and separated into size classes. The area of aggregates in each size class was then obtained and converted to a percentage of the total aggregate area. Averages were calculated per mouse and then per experimental group.
Cytokine analysis of NALF and enumeration of nasal septum-attached pneumococci.
NALF from infected mice was collected at 1, 3, and 5 days postinfection (dpi). NALF was pooled, frozen, and subsequently used to measure the temporal proinflammatory cytokine response to colonization by enzyme-linked immunosorbent assay (ELISA). In separate experiments, forced NALF was collected from euthanized mice at day 7. This was done by puncturing the trachea with a catheter and pushing 500 µl PBS outward through the nares into a collection chamber. To determine the number of pneumococci intimately attached to nasopharyngeal cells (i.e., resistant to the lavage), nasal septa from these latter mice were excised and homogenized in PBS, with samples serially diluted and plated for extrapolation of bacterial titers from colony counts. Homogenized septa were also processed for enumeration of bacteria by qRT-PCR using probes for the genes pln and lytA (48).
SEM and histology.
Nasal septa were excised according to published protocols (49), with the exception that colonized mice were exsanguinated and perfused with PBS following euthanasia to prevent the accumulation of erythrocytes during the dissection procedures. Tissue samples were processed for scanning electron microscopy (SEM) as previously described, and images were captured with a JEOL-6610 scanning electron microscope (JEOL, Peabody, MA) (12). For histology experiments, intact nares were collected from euthanized mice. Samples were immediately placed in 10% formalin and then decalcified using EDTA. Sections were cut from paraffin-embedded blocks, stained with hematoxylin and eosin (H&E), and visualized by microscopy.
Cell adhesin and invasion assays and in vitro cytokine analyses.
Detroit-562 human pharyngeal cells were cultured at 37°C in 5% CO2. Cell monolayers were grown to ~95% confluence in 24-well plates (~106 cells/well) prior to their use. Adhesion and invasion assays were performed as previously described (50). For in vitro cytokine analysis, Detroit-562 cells and J774A.1 mouse macrophages were exposed to equal CFU of planktonic and biofilm pneumococci for 4 h. Supernatants were collected and examined by ELISA.
Statistical analyses.
Statistical analysis of in vitro biofilm formation data, viable CFU counts recovered from the nasopharynx, and qRT-PCR values was performed by one-way analysis of variance (ANOVA) using SigmaStat 3.1 software (Systat Software Inc., Point Richmond, CA). Analysis of aggregate size class data and cytokine values from recovered NALF was performed using a two-tailed Student t test.
SUPPLEMENTAL MATERIAL
Bacterial load for intimately attached pneumococci. Quantitation of bacterial load of different colonizing strains of S. pneumoniae attached to septal epithelium at 7 dpi via qRT-PCR, expressed as copies/g tissue (black bars), compared to CFU/g (white bars). The reference gene sequence used was ply. No significant differences were observed between strains on any given day as tested by one-way ANOVA (n = 5/cohort). Download
Representative images of CV-stained bacterial aggregates recovered from the nasopharynx of mice. Samples were collected at 3, 7, and 14 dpi (n = 6 mice/time point and strain). Download
In vitro biofilm formation for 6A10 isogenic mutants. Mean biomass of biofilms formed by isogenic mutants of strain 6A10 on 6-well polystyrene plates after 18 h of growth, as measured by crystal violet staining relative to wild type (n = 3/mutant). Download
Bacterial load for mice colonized with isogenic mutants. (A) Colonization of nasopharynx by strain 6A10 and mutants deficient in CbpA, LytA, LuxS, pneumolysin, SpxB, or PsrP-SecY2A2, as determined by CFU/ml of nasal lavage elute collected at days 3, 7, and 14. (B) Quantitation of bacterial load of isogenic mutants of S. pneumoniae strain 6A10 attached to septal epithelium at 7 dpi via qRT-PCR, expressed as copies/g tissue (black bars), compared to CFU/g (white bars). Significance was tested by one-way ANOVA (n = 5/cohort). Download
H&E-stained cross sections of the septal epithelium collected from a single mouse infected with 6A10. Seven days after challenge into the right nares, the infected right naris showed disruption of the mucosal epithelium and greater disorganization (A). In contrast, the left naris was largely undisturbed and showed intact mucosal epithelial cells with cilia (B). Download
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI078972 to C.J.O. K.B.C. received support through T32DE14318-10 and T32AI7271-24.
We thank Barbara Hunter and Lauren Chestnut at The University of Texas Health Science Center Electron Microscopy Core for their invaluable assistance in the imaging of colonized nasal septa.
Footnotes
Citation Blanchette-Cain K, Hinojosa CA, Akula Suresh Babu R, Lizcano A, Gonzalez-Juarbe N, Munoz-Almagro C, Sanchez CJ, Bergman MA, Orihuela CJ. 2013. Streptococcus pneumoniae biofilm formation is strain dependent, multifactorial, and associated with reduced invasiveness and immunoreactivity during colonization. mBio 4(5):e00745-13. doi:10.1128/mBio.00745-13.
REFERENCES
- 1. Musher D. 2010. Streptococcus pneumoniae, p 2623–2642 In Mandell G, Bennett J, Dolin R. (ed), Mandell, Douglas, and Bennett’s principles and practice of infectious disease, 7th ed, vol 2 Churchill: Livingstone Elsevier, Philadelphia, PA [Google Scholar]
- 2. Reference deleted. [Google Scholar]
- 3. O’Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T, Hib, Pneumococcal Global Burden of Disease Study Team 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902 [DOI] [PubMed] [Google Scholar]
- 4. Crook D, Brueggmann A, Sleeman K, Peto T. 2004. Pneumococcal carriage. ASM Press, Washington, DC [Google Scholar]
- 5. Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322 [DOI] [PubMed] [Google Scholar]
- 6. Sanclement JA, Webster P, Thomas J, Ramadan HH. 2005. Bacterial biofilms in surgical specimens of patients with chronic rhinosinusitis. Laryngoscope 115:578–582 [DOI] [PubMed] [Google Scholar]
- 7. Kania RE, Lamers GE, Vonk MJ, Dorpmans E, Struik J, Tran Ba Huy P, Hiemstra P, Bloemberg GV, Grote JJ. 2008. Characterization of mucosal biofilms on human adenoid tissues. Laryngoscope 118:128–134 [DOI] [PubMed] [Google Scholar]
- 8. Hoa M, Tomovic S, Nistico L, Hall-Stoodley L, Stoodley P, Sachdeva L, Berk R, Coticchia JM. 2009. Identification of adenoid biofilms with middle ear pathogens in otitis-prone children utilizing SEM and FISH. Int. J. Pediatr. Otorhinolaryngol. 73:1242–1248 [DOI] [PubMed] [Google Scholar]
- 9. Psaltis AJ, Ha KR, Beule AG, Tan LW, Wormald PJ. 2007. Confocal scanning laser microscopy evidence of biofilms in patients with chronic rhinosinusitis. Laryngoscope 117:1302–1306 [DOI] [PubMed] [Google Scholar]
- 10. Marks LR, Parameswaran GI, Hakansson AP. 2012. Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect. Immun. 80:2744–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vidal JE, Howery KE, Ludewick HP, Nava P, Klugman KP. 2013. Quorum-sensing systems LuxS/autoinducer 2 and Com regulate Streptococcus pneumoniae biofilms in a bioreactor with living cultures of human respiratory cells. Infect. Immun. 81:1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanchez CJ, Shivshankar P, Stol K, Trakhtenbroit S, Sullam PM, Sauer K, Hermans PW, Orihuela CJ. 2010. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog. 6:e1001044. 10.1371/journal.ppat.1001044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Otto M. 2006. Bacterial evasion of antimicrobial peptides by biofilm formation. Curr. Top. Microbiol. Immunol. 306:251–258 [DOI] [PubMed] [Google Scholar]
- 14. Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, Engebretsen IL, Bayles KW, Horswill AR, Kielian T. 2011. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 186:6585–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stewart PS, Costerton JW. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–138 [DOI] [PubMed] [Google Scholar]
- 16. Walsh RL, Camilli A. 2011. Streptococcus pneumoniae is desiccation tolerant and infectious upon rehydration. mBio 2(3):e00092-11. 10.1128/mBio.00092-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanchez CJ, Kumar N, Lizcano A, Shivshankar P, Dunning Hotopp J, Jorgensen JH, Tettelin H, Orihuela CJ. 2011. Streptococcus pneumoniae in biofilms are unable to cause invasive disease due to altered virulence determinant production. PLoS One 6:e28738. 10.1371/journal.pone.0028738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oggioni MR, Trappetti C, Kadioglu A, Cassone M, Iannelli F, Ricci S, Andrew PW, Pozzi G. 2006. Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol. Microbiol. 61:1196–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cope EK, Goldstein-Daruech N, Kofonow JM, Christensen L, McDermott B, Monroy F, Palmer JN, Chiu AG, Shirtliff ME, Cohen NA, Leid JG. 2011. Regulation of virulence gene expression resulting from Streptococcus pneumoniae and nontypeable Haemophilus influenzae interactions in chronic disease. PLoS One 6:e28523. 10.1371/journal.pone.0028523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U. S. A. 100:1966–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McNeela EA, Burke Á, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Pétrilli V, Andrew PW, Kadioglu A, Lavelle EC. 2010. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 6:e1001191. 10.1371/journal.ppat.1001191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanchez CJ, Hurtgen BJ, Lizcano A, Shivshankar P, Cole GT, Orihuela CJ. 2011. Biofilm and planktonic pneumococci demonstrate disparate immunoreactivity to human convalescent sera. BMC Microbiol. 11:245. 10.1186/1471-2180-11-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, Tuomanen E. 2000. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell 102:827–837 [DOI] [PubMed] [Google Scholar]
- 24. Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala’Aldeen DA, Tuomanen EI. 2009. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Invest. 119:1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pericone CD, Park S, Imlay JA, Weiser JN. 2003. Factors contributing to hydrogen peroxide resistance in Streptococcus pneumoniae include pyruvate oxidase (SpxB) and avoidance of the toxic effects of the Fenton reaction. J. Bacteriol. 185:6815–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eldholm V, Johnsborg O, Haugen K, Ohanstand HS, Håvarstein LS. 2009. Fratricide in Streptococcus pneumoniae: contributions and role of the cell wall hydrolases CbpD, LytA and LytC. Microbiology 155:2223–2234 [DOI] [PubMed] [Google Scholar]
- 27. Trappetti C, Potter AJ, Paton AW, Oggioni MR, Paton JC. 2011. LuxS mediates iron-dependent biofilm formation, competence, and fratricide in Streptococcus pneumoniae. Infect. Immun. 79:4550–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mascher T, Heintz M, Zähner D, Merai M, Hakenbeck R. 2006. The CiaRH system of Streptococcus pneumoniae prevents lysis during stress induced by treatment with cell wall inhibitors and by mutations in pbp2x involved in β-lactam resistance. J. Bacteriol. 188:1959–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lizcano A, Chin T, Sauer K, Tuomanen EI, Orihuela CJ. 2010. Early biofilm formation on microtiter plates is not correlated with the invasive disease potential of Streptococcus pneumoniae. Microb. Pathog. 48:124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trappetti C, van der Maten E, Amin Z, Potter AJ, Chen AY, Mourik P, van Mourik PM, Lawrence AJ, Paton AW, Paton JC. 2013. Site of isolation determines biofilm formation and virulence phenotypes of serotype 3 Streptococcus pneumoniae clinical isolates. Infect. Immun. 81:505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muñoz-Elías EJ, Marcano J, Camilli A. 2008. Isolation of Streptococcus pneumoniae biofilm mutants and their characterization during nasopharyngeal colonization. Infect. Immun. 76:5049–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trappetti C, Kadioglu A, Carter M, Hayre J, Iannelli F, Pozzi G, Andrew PW, Oggioni MR. 2009. Sialic acid: a preventable signal for pneumococcal biofilm formation, colonization, and invasion of the host. J. Infect. Dis. 199:1497–1505 [DOI] [PubMed] [Google Scholar]
- 33. Moscoso M, García E, López R. 2006. Biofilm formation by Streptococcus pneumoniae: role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J. Bacteriol. 188:7785–7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vidal JE, Ludewick HP, Kunkel RM, Zähner D, Klugman KP. 2011. The LuxS-dependent quorum-sensing system regulates early biofilm formation by Streptococcus pneumoniae strain D39. Infect. Immun. 79:4050–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosenow C, Ryan P, Weiser JN, Johnson S, Fontan P, Ortqvist A, Masure HR. 1997. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol. Microbiol. 25:819–829 [DOI] [PubMed] [Google Scholar]
- 36. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301 [DOI] [PubMed] [Google Scholar]
- 37. Regev-Yochay G, Trzcinski K, Thompson CM, Lipsitch M, Malley R. 2007. SpxB is a suicide gene of Streptococcus pneumoniae and confers a selective advantage in an in vivo competitive colonization model. J. Bacteriol. 189:6532–6539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ogunniyi AD, LeMessurier KS, Graham RM, Watt JM, Briles DE, Stroeher UH, Paton JC. 2007. Contributions of pneumolysin, pneumococcal surface protein A (PspA), and PspC to pathogenicity of Streptococcus pneumoniae D39 in a mouse model. Infect. Immun. 75:1843–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Orihuela CJ, Gao G, Francis KP, Yu J, Tuomanen EI. 2004. Tissue-specific contributions of pneumococcal virulence factors to pathogenesis. J. Infect. Dis. 190:1661–1669 [DOI] [PubMed] [Google Scholar]
- 40. Rose L, Shivshankar P, Hinojosa E, Rodriguez A, Sanchez CJ, Orihuela CJ. 2008. Antibodies against PsrP, a novel Streptococcus pneumoniae adhesin, block adhesion and protect mice against pneumococcal challenge. J. Infect. Dis. 198:375–383 [DOI] [PubMed] [Google Scholar]
- 41. Domenech M, García E, Prieto A, Moscoso M. 2013. Insight into the composition of the intercellular matrix of Streptococcus pneumoniae biofilms. Environ. Microbiol. 15:502–516 [DOI] [PubMed] [Google Scholar]
- 42. Shivshankar P, Sanchez C, Rose LF, Orihuela CJ. 2009. The Streptococcus pneumoniae adhesin PsrP binds to keratin 10 on lung cells. Mol. Microbiol. 73:663–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Obert C, Sublett J, Kaushal D, Hinojosa E, Barton T, Tuomanen EI, Orihuela CJ. 2006. Identification of a candidate Streptococcus pneumoniae core genome and regions of diversity correlated with invasive pneumococcal disease. Infect. Immun. 81:4766–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marks LR, Davidson BA, Knight PR, Hakansson AP. 2013. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4(4):e00438-13. 10.1128/mBio.00438-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506 [DOI] [PubMed] [Google Scholar]
- 46. Embry A, Hinojosa E, Orihuela C. 2007. Regions of diversity 8, 9 and 13 contribute to Streptococcus pneumoniae virulence. BMC Microbiol. 7:80. 10.1186/1471-2180-7-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Allegrucci M, Sauer K. 2007. Characterization of colony morphology variants isolated from Streptococcus pneumoniae biofilms. J. Bacteriol. 189:2030–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Sevilla MF, Garcia-Garcia JJ, Esteva C, Moraga F, Hernandez S, Selva L, Coll F, Ciruela P, Planes AM, Codina G, Salleras L, Jordan I, Dominguez A, Munoz-Almagro C. 2012. Clinical presentation of invasive pneumococcal disease in Spain in the era of heptavalent conjugate vaccine. Pediatr. Infect. Dis. J. 31:124–128 [DOI] [PubMed] [Google Scholar]
- 49. Antunes MB, Woodworth BA, Bhargave G, Xiong G, Aguilar JL, Ratner AJ, Kreindler JL, Rubenstein RC, Cohen NA. 2007. Murine nasal septa for respiratory epithelial air-liquid interface cultures. BioTechniques 43:195–204 [DOI] [PubMed] [Google Scholar]
- 50. Orihuela CJ, Tuomanen EI. 2006. Models of pneumococcal disease. Drug Discov. Today Dis. Models 3:69–75 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial load for intimately attached pneumococci. Quantitation of bacterial load of different colonizing strains of S. pneumoniae attached to septal epithelium at 7 dpi via qRT-PCR, expressed as copies/g tissue (black bars), compared to CFU/g (white bars). The reference gene sequence used was ply. No significant differences were observed between strains on any given day as tested by one-way ANOVA (n = 5/cohort). Download
Representative images of CV-stained bacterial aggregates recovered from the nasopharynx of mice. Samples were collected at 3, 7, and 14 dpi (n = 6 mice/time point and strain). Download
In vitro biofilm formation for 6A10 isogenic mutants. Mean biomass of biofilms formed by isogenic mutants of strain 6A10 on 6-well polystyrene plates after 18 h of growth, as measured by crystal violet staining relative to wild type (n = 3/mutant). Download
Bacterial load for mice colonized with isogenic mutants. (A) Colonization of nasopharynx by strain 6A10 and mutants deficient in CbpA, LytA, LuxS, pneumolysin, SpxB, or PsrP-SecY2A2, as determined by CFU/ml of nasal lavage elute collected at days 3, 7, and 14. (B) Quantitation of bacterial load of isogenic mutants of S. pneumoniae strain 6A10 attached to septal epithelium at 7 dpi via qRT-PCR, expressed as copies/g tissue (black bars), compared to CFU/g (white bars). Significance was tested by one-way ANOVA (n = 5/cohort). Download
H&E-stained cross sections of the septal epithelium collected from a single mouse infected with 6A10. Seven days after challenge into the right nares, the infected right naris showed disruption of the mucosal epithelium and greater disorganization (A). In contrast, the left naris was largely undisturbed and showed intact mucosal epithelial cells with cilia (B). Download