

Abstract
Cardiovascular disease (CVD) is a leading determinant of mortality and morbidity in the world. Epidemiologic studies suggest that flavonoid intake plays a role in the prevention of CVD. Consumption of cocoa products rich in flavonoids lowers blood pressure and improves endothelial function in healthy subjects as well as in subjects with vascular dysfunction such as smokers and diabetics. The vascular actions of cocoa follow the stimulation of nitric oxide (NO). These actions can be reproduced by the administration of the cocoa flavanol (−)-epicatechin (EPI). Previously, using human endothelial cells cultured in calcium-free media, we documented EPI effects on eNOS independently of its translocation from the plasmalemma. To further define the mechanisms behind EPI-eNOS activation in Ca2+ -deprived endothelial cells, we evaluated the effects of EPI on the eNOS/AKT/HSP90 signaling pathway. Results document an EPI-induced phosphorylation/activation of eNOS, AKT, and HSP90. We also demonstrate that EPI induces a partial AKT/HSP90 migration from the cytoplasm to the caveolar membrane fraction. Immunoprecipitation assays of caveolar fractions demonstrate a physical association between HSP90, AKT, and eNOS. Thus, under Ca2+-free conditions, EPI stimulates NO synthesis via the formation of an active complex between eNOS, AKT, and HSP90.
Keywords: eNOS, Epicatechin, Cocoa flavanols, Endothelial cells
Introduction
Cardiovascular diseases (CVD) are a leading cause of morbidity and mortality, and the pathogenesis of these diseases is frequently linked to endothelial cell dysfunction [1]. The consumption of cacao-derived products, particularly from dark chocolate (i.e., cocoa) appears to provide beneficial cardiovascular effects [2, 3] The vascular actions of cocoa on the vasculature are related to its ability to activate endothelial nitric oxide synthase (eNOS), and as a consequence nitric oxide (NO) production [3, 4]. These effects can be reproduced by (−)-epicatechin (EPI), which is the most abundant flavanol present in cacao [4].
Recently, we investigated the effects of Ca2+ depletion on eNOS activation/phosphorylation and translocation. Human coronary artery endothelial cells (HCAEC) were treated with EPI or with bradykinin (BK), a well known Ca2+-dependent eNOS activator. Results demonstrate that both EPI and BK induce increases in intracellular calcium and NO levels. However, under Ca2+-free conditions, EPI (but not BK) is still capable of inducing NO production through eNOS phosphorylation at Serine 615, 633, and 1177. Interestingly, EPI-induced translocation of eNOS from the plasmalemma was abolished upon Ca2+ depletion. Thus, under Ca2+-free conditions, EPI stimulates NO synthesis independent of calmodulin binding to eNOS and of its translocation into the cytoplasm. We also examined the effect of EPI on the NO/cGMP/vasodilator-stimulated phosphoprotein pathway activation in isolated Ca2+-deprived canine mesenteric arteries [5]. Results demonstrated that under these conditions, EPI induces the activation of this vasorelaxation related pathway and that this effect can be inhibited by pretreatment with the eNOS inhibitor L-NG-nitroarginine methyl ester (L-NAME). However, the molecular mechanisms underlying these effects have not been explored.
In this study, we demostrate that in HCAEC under calcium-free conditions, EPI induces the physical association and activity of HSP90/AKT/eNOS at caveolae providing a mechanistic explanation for the ability of EPI to stimulate eNOS under calcium-free conditions.
Methods
Materials
HCAEC and HCAEC growth medium (MesoEndo) were purchased from Cell Applications, Inc. Calcium and phenol red free Dulbecco’s Modified Eagle’s Media (DMEM) and Hank’s solution were from Gibco BRL. EPI, protease and phosphatase inhibitor cocktails, caffeine, EGTA, cholera toxin subunit B peroxidase conjugate (CTB-HRP), phenyl-methyl sulphonyl fluoride (PMSF) were obtained from Sigma-Aldrich Chemicals. Halt Protease and phosphatase inhibitor cocktail EDTA-free was from Thermo Scientific. BAPTA-AM was from Enzo-Life Sciences. SH-5 was from Alexis Biochemicals, AKT Inhibitor IV was from Calbiochem. Geldanamycin (GA) was from EMD-Biosciences and NVP-AUY922 was from Selleckchem. Phospho eNOS-Ser615 primary antibody was from Upstate Co., phospho eNOS-Ser1177, eNOS, HSP90, phospho HSP90, AKT, phospho-AKT-Ser473 primary antibodies, normal rabbit IgG control, and HRP-conjugated secondary antibodies were from Cell Signaling Technology. Protein G-agarose was obtained from Santa Cruz Biotechnologies. Ultracentrifuge tubes (catalog No. 347356), from Beckman Coulter, Polyvidinil fluoride (PVDF) transfer membrane was from Millipore. ECL Plus Western blot detection kit from Amersham-GE. The Nitrite/Nitrate Fluorometric Assay Kit was from Cayman Chemical, Co.
Cell culture
HCAEC from 14-, 40-, and 60-year-old healthy males were maintained in a humidified atmosphere at 37 °C with 5 % CO2 and 95 % O2 in HCAEC growth medium. HCAEC between passages 12–14 were used for all experiments. Treatments were applied to confluent cell cultures.
HCAEC [Ca2+]i depletion
HCAEC were deprived of Ca2+ by washing them (3 × 1 min) with Epilife media (pH 7.4) without Ca2+ or phenol red supplemented with 0.1 mmol/L EGTA, 25 μmol/L BAPTA (Ca2+ chelators), and 0.1 mmol/L caffeine to deplete [Ca2+]i deposits.
Indirect NOx measurements
NO subproducts (i.e., nitrates and nitrites) levels were measured using a fluorescent kit and a fluorometer (FLx800 Bio-Tek Instruments INC). EPI was diluted in water (water was used as vehicle for control cells). For these experiments, cells were treated with EPI [1 μmol/L], and culture media samples were collected at 10 min as an end point to measure extracellular NO indirectly [6].
Immunoprecipitation
Immunoprecipitation assays were performed as previously described [7]. In brief, cells were lysed with 50 μl of non-denaturing extraction buffer (0.5 %, Triton X-100, 50 mmol/L Tris–HCl, pH 7.4, 0.15 mol/L NaCl, and 0.1 mmol/L EDTA) and supplemented with protease and phosphatase inhibitor cocktail, plus 0.15 mmol/L PMSF, 5 mmol/L Na3VO4, and 1 mmol/L NaF. Homogenates were incubated on ice for 10 min and passed through an insulin syringe five times. The homogenate was incubated on ice with shaking for 10 min and centrifuged (10 min) at 12,000×g at 4 °C. A total of 0.5 mg protein was diluted in binding buffer (10 mM Tris–HCl, pH 7.9, 2 mM MgCl2, 0.15 mM NaCl, 10 % glycerol, 0.15 mM PMSF, supplemented with protease and phosphatase inhibitor cocktails, plus 0.15 mmol/L PMSF, 5 mmol/L Na3VO4 and 1 mmol/L NaF) to a final concentration of 1 μg/μL. Subsequently, samples were pre-cleared by adding 1 μg of normal rabbit IgG control and 20 μL prot-G-agarose with mixing for 30 min (4 °C) and centrifuged at 12,000×g for 10 min at 4 °C. The supernatant was recovered and incubated for 3 h at 4 °C under mild agitation with 3 μg of immunoprecipitating anti-eNOS antibody. Twenty microliters of protein G-Sepharose were added, and the mixture was incubated over night at 4 °C with shaking. The immunoprecipitation mixture was centrifuged at 3,500×g for 4 min at 4 °C, and the supernatant was recovered and stored at 4 °C. The pellet was washed with binding buffer for 15 min at 4 °C with shaking and centrifuged at 3,500×g for 4 min at 4 °C. Washes were repeated 3×. The immunoprecipitated proteins in the pellet and those remaining in the supernatant were applied to a 4–15 % gradient SDS-PAGE for immunoblotting. Co-immunoprecipitation was also performed with anti-HSP90 or anti-AKT antibodies to confirm results. The assay was carried out at least three times with each immunoprecipitating antibody.
Immunoblotting
Cells grown on 10-cm dishes were homogenized in 50 μl lysis buffer (1 % triton X-100, 20 mmol/L Tris–HCl pH 4, 140 mmol/L NaCl, 2 mmol/L EDTA, and 0.1 % SDS) with protease and phosphatase inhibitor cocktails supplemented with 0.15 mmol/L PMSF, 5 mmol/L Na3VO4, and 5 mmol/L NaF. Homogenates were passed through an insulin syringe five times, sonicated for 15 min at 4 °C and centrifuged (12,000×g) for 10 min. The total protein content was measured in the supernatant. A total of 40 μg of protein was loaded onto a 4–15 % SDS-PAGE, electrotransferred, incubated for 1 h in blocking solution (5 % non-fat dry milk in TBS plus 0.1 % Tween 20 [TBS-T]), and followed by either 3-h incubation at room temperature or overnight incubation at 4 °C with primary antibodies. Primary antibodies were typically diluted 1:1,000 or 1:2,000 in TBS-T. Membranes were washed (3× for 5 min) in TBS-T and incubated 1 h at room temperature in the presence of HRP-conjugated secondary antibodies diluted 1:10,000 in blocking solution. Membranes were again washed 3 times in TBS-T, and the immunoblots developed using the ECL detection kit. Band intensities were digitally quantified.
Detergent-resistant membrane (DRM) isolation
Detergent-resistant membrane (lipid rafts and caveolae) isolation was performed as previously described [8]. In brief, approximately 4.5 × 106 cells were lysed with 300 μL of cold TNE buffer (20 mmol/L Tris, 140 mmol/L NaCl, 2 mmol/L EDTA) containing 0.05 % Triton X-100 plus protease and phosphatase inhibitors. Lysates were mixed with 375 μL of 80 % sucrose in TNE-Triton X-100 buffer and transferred to ultracentrifuge tubes. Cell lysates, placed in 45 % sucrose, were gently overlaid with 1 ml of 35 % sucrose in TNE-Triton X-100 buffer, while the latter fraction was overlaid with 400 μL of 5 % sucrose in TNE-Triton X-100 buffer. Samples were centrifuged at 4 °C for 16 h at 170,000×g in an Optima TLX ultracentrifuge using the TLS 55 rotor (Beckman Coulter) to form a ~45–5 % sucrose gradient. After centrifugation, eight fractions were collected. Five μL of each sucrose gradient fraction was placed onto a PVDF membrane. The drop was allowed to dry, and the PVDF membrane was incubated 1 h at room temperature (RT) in blocking solution. The PVDF membrane was subsequently incubated with 1:2,000 CT-B-HRP (used as GM1 marker) dilution in blocking solution and developed using the ECL detection kit.
Data analysis
A minimum of three experiments were performed (each in triplicate) unless otherwise noted. Statistical analysis was performed using t test or ANOVA and Tukey post hoc tests for multiple comparisons. Significance is noted at P < 0.05.
Results
EPI-induced activation of HSP90/AKT pathway in a Ca2+ independent manner
We measured the activation of molecules upstream eNOS (HSP90 and AKT) after EPI treatment of cells [1 μmol/L]. Under these conditions an ~70 % increase in HSP90 phosphorylation was evidenced. Treatment with the HSP90 inhibitor geldanamycin (GA) [10 μmol/L] prevented EPI-induced increases in phosphorylation of HSP90 (Fig. 1a). Results also demonstrated an ~65 % increase on AKT phosphorylation. Pre-treatment with the AKT inhibitor SH-5 [20 μmol/L] blocked EPI effects while EPI effects on AKT phosphorylation were not affected by the HSP90 inhibitor GA. (Fig. 1b). We performed a complementary set of experiments using different inhibitors for AKT (inhibitor IV [1 μM]) and HSP90 (NVP-AUY922 [0.1 μM]). In both cases, the inhibition of EPI-induced effects (results not shown) was consistent with the inhibition obtained in the presence of SH5 and GA demonstrating that under Ca2+-free conditions eNOS activation depends on AKT and HSP90 activities.
Fig. 1.
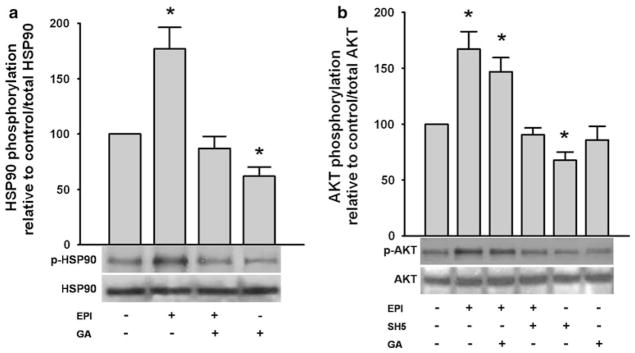
EPI-induced phosphorylation of HSP90 and AKT. Treatment of HCAEC with EPI [1 μmol/L] induces the phosphorylation/activation of HSP90 and AKT. Relative HSP90 phosphorylation increased by ~75 % (a), while AKT phosphorylation increased by ~60 % (b). The use of the HSP90 inhibitor GA, suppresses EPI-induced HSP90 phosphorylation to control levels (a). However, GA does not affect the EPI-induced phosphorylation of AKT (b). Treatment of HCAEC with the AKT inhibitor SH5 abolishes EPI-induced phosphorylation of AKT (b). Treatment with GA or SH5-induced lower protein phosphorylation levels than controls. Data are expressed as mean ± SD (n = 3, * P < 0.05 versus control by t test)
EPI induces phosphorylation of eNOS and NO production under Ca2+-free conditions
We evaluated the phosphorylation of eNOS Ser1177 and Ser615 (stimulation sites) and its dependence on HSP90/AKT in EPI-treated cells under Ca2+-free conditions. Changes in the phosphorylation status were observed in both activation residues. EPI-induced increases in phosphorylation of Ser1177 was ~120 %, while phosphorylation increases on Ser615 was ~65 % compared to unstimulated cells (Fig. 2a). Pre-treatment with the AKT inhibitor SH5 decreased EPI-induced phosphorylation of Ser1177 (only ~25 % over the control), whereas increases on phosphorylation of Ser615 were completely blocked. HCAEC pretreatment with HSP90 inhibitor GA, prevented Ser1177 phosphorylation to ~50 % and Ser615 to ~35 % as compared with EPI. HCAEC treatment with both SH5 and GA inhibitors decreased Ser1177 phosphorylation to ~20 % while Ser615 phosphorylation was abolished (Fig. 2a).
Fig. 2.
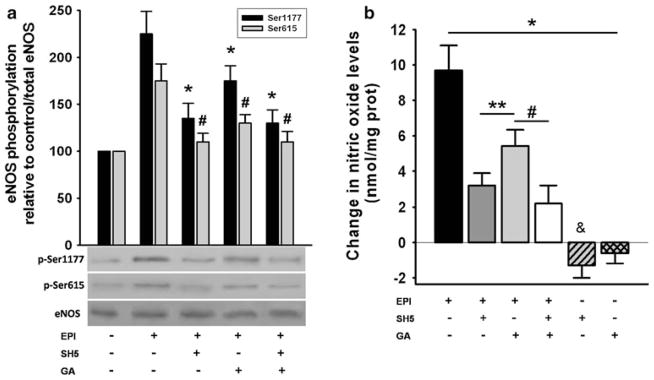
EPI-induced eNOS phosphorylation and stimulation of NO production. In HCAEC EPI-[1 μmol/L] induced the activation of eNOS through Ser-1177 and 615 phosphorylation (a). eNOS phosphorylation on Ser-1177 increased by ~115 % and on Ser-615 by 75 % as compared with untreated cells (control). Treatment with SH5 or GA decreased the EPI-induced phosphorylation to 25 and 50 % on Ser-1177, respectively, while the increase in phosphorylation of Ser-615 was abolished with SH5 and decreased to 20 % with GA. Treatment of the cells with both inhibitors decreased the phosphorylation of Ser-1177 to 25 %, and abolished the phosphorylation of Ser-615. Cell incubation with the inhibitors alone, induced decreases in residue phosphorylation to levels lower than control (data not shown). Data are expressed as mean ± SD (n = 3, and analyzed by t test, * P < 0.05 Ser1177 and # P < 0.05 Ser615 versus EPI treatment). b EPI-induced increases in NO production. EPI effects were decreased with inhibitor treatment to 30 % with SH5, to ~60 % with GA and to ~20 % with both. Cells treated with the inhibitors alone induced decreases in NO levels lower than controls. Control NO values = 0. Data are expressed as mean ± SD (n = 3), * P < 0.05 versus EPI treatment, # P < 0.05 versus control by ANOVA
With AKT and/or HSP90 inhibition, NO production follows a similar pattern as that of eNOS Ser1177 and Ser615 phosphorylation. Results indicate that EPI-induced NO production under Ca2+-free conditions is modulated by AKT-induced changes in the phosphorylation status of eNOS Ser1177 and Ser615 (Fig. 2b).
EPI-treatment induces eNOS, AKT, and HSP90 association
Agonist stimulation of endothelial cells, triggers the formation of a complex between HSP90, AKT, and eNOS which leads to its phosphorylation and activation [9]. We thus, explored if in calcium depleted cells EPI stimulates the association of eNOS to AKT/HSP90. We used specific antibodies to immunoprecipitate eNOS, AKT or HSP90 in control, EPI, EPI plus SH5 or EPI plus GA-treated HCAEC. The immunoprecipitated (IP) phase obtained from the eNOS antibody was used for immunoblot analysis of total eNOS, HSP90, phosphorylated HSP90, AKT, and phosphorylated AKT (Fig. 3a). In unstimulated cells, there was a very low amount of complexed proteins, as shown by Western blots. In contrast, in EPI-treated HCAEC, eNOS is attached to the phosphorylated (i.e., active) forms of AKT and HSP90 (Fig. 3a, b), indicating that EPI in the absence of Ca2+ promotes the binding of eNOS/AKT/HSP90 proteins to form a functional protein complex. On the other hand, in Ca2+-deprived HCAEC-treated with SH5 the association of AKT with eNOS is decreased, whereas GA treatment only abolishes the association of HSP90 to eNOS. The incubation with both inhibitors, blocked the association of all three proteins. Western blots of the supernatant (SN) phase from the IP did not identify total eNOS, which indicates that the enzyme was still bound to caveolae after treatment (data not shown).
Fig. 3.
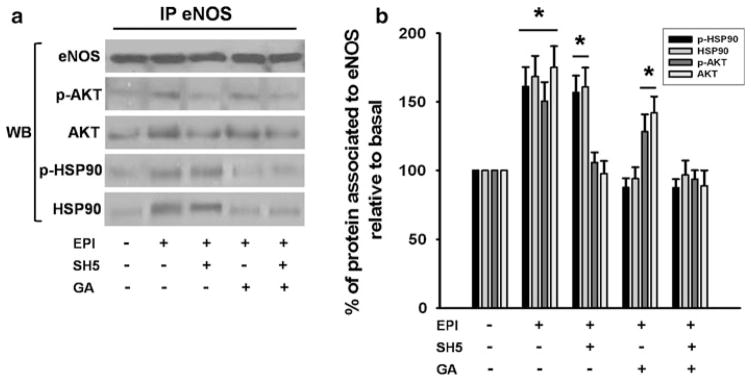
EPI induces eNOS, AKT, HSP90 association in HCAEC. (a) Western blots were performed on immunoprecipitates (IP) to detect total eNOS, phospho-AKT, total AKT, phospho HSP90, and total HSP90. In control HCAEC, eNOS was not associated with HSP90 and mildly associated with AKT. EPI treatment induced the association of eNOS with AKT, HSP90 and their active forms (a, b). SH5 inhibits EPI-induced association of AKT to eNOS, while GA inhibits EPI-induced association of eNOS/HSP90 (a, b). Treatment with both inhibitors blocks EPI-induced association of AKT and HSP90 with eNOS (a, b). Data are expressed as mean ± SD (n = 3), * P < 0.05 by ANOVA
EPI induces the mobilization of AKT and HSP90 to detergent-resistant membranes (DRM) cell fractions
Using sucrose gradient based subcellular fractionation with concentrations ranging from ~45–5 % we previously determined that in EPI-treated HCAEC under Ca2+-free conditions, the activation of eNOS takes place without translocation of the enzyme from caveolae (low-density fractions of membrane lipids DMR) [5, 10]. Again, we utilized this approach to establish the migration pattern of proteins enriched in DRM and to identify the molecules involved in eNOS activation/phosphorylation under Ca2+-free conditions at the caveolar domain. Each of the subcellular fractions were used to measure eNOS, AKT, and HSP90 by Western blot of control and EPI-treated samples. An antibody to CT-B-HRP-conjugated (which binds specifically to ganglioside GM1) was used as marker for DRM (lipid rafts/caveolae) enriched in low-density fractions. In non-stimulated cells, results demonstrate that in Ca2+-free HCAEC, eNOS was detected in the low-density sucrose fractions, along with the DRM marker GM1 (Fig. 4a, c), while most of the AKT and HSP90 were mainly contained within the 45–35 % sucrose fraction (high density fractions) (Fig. 4a, d and e, respectively). Western blot from the sucrose gradient fractions of EPI-treated HCAEC demonstrate that eNOS was present mostly in the interphase (IF) and 5 % sucrose fraction, similar to the localization pattern found in control experiments, suggesting no translocation from low-density membrane lipids (corresponding to lipid rafts or caveolae domains) to high density fractions (Fig. 4b, c). Furthermore, in EPI-treated cells, AKT and HSP90 were localized mostly in low-density fractions (sucrose fractions IF and 5 %) along with lipid raft/caveolae marker GM1 which indicates the mobilization of those proteins from the high density fractions to the low- density fractions suggesting an interaction with eNOS (Fig. 4b, d and e). Altogether, these results suggest that under Ca2+-free conditions, the activation of eNOS requires recruitment of AKT and HSP90 to the low-density region of membrane DRM (lipid raft/caveolar fraction).
Fig. 4.

EPI treatment induces the mobilization of AKT and HSP90 to caveolae/lipid rafts. Total protein extracts from control and EPI-treated HCAEC were loaded onto a sucrose gradient. Sucrose density gradients (~45, 35 and 5 %) were used for the detection of eNOS, AKT, HSP90, and ganglioside M1 (GM1) in the various fractions (Fx). Non-stimulated HCAEC shows eNOS localization to the low-sucrose density fraction along with GM1, while AKT and HSP90 were predominantly located at high density fractions (a, c). In EPI-treated cells AKT and HSP90 located at low-sucrose density fractions as evidenced by the presence of eNOS and GM1 (b, d and e). Data are expressed as mean ± SD (n = 3, * P < 0.05 by t test)
EPI-treatment induces eNOS, AKT, and HSP90 association to caveolae
In order to determine if EPI is able to induce the eNOS/AKT/HSP90 complex formation at caveolae (DRM), we performed IP assays using Cav-1 antiboby on the mixture of fractions 1–4 and 5–8 from non-stimulated as well as EPI-treated cells. As demonstrated in Fig. 5a and b, Cav-1 and eNOS co-precipitate in the mixture of low-density fractions (5–8) corresponding to DRM/caveolar fraction of both control and EPI-treated cells, which indicates that the enzyme is still bound to caveolae after treatment. Meanwhile, AKT as well as HSP90 co-precipitate with Cav-1 and eNOS only in mixtures of low-density fractions (5–8) from EPI-treated cells (Fig. 5a, b), which indicates caveolae protein complex formation.
Fig. 5.

EPI treatment induces Cav-1, eNOS, AKT, and HSP90 complex formation in caveolae/lipid rafts. High density sucrose gradient fractions (Fx) 1–4 were mixed as well as low-density fractions (5–8) from control and EPI-treated HCAEC. Fraction mixtures were used to assess protein association by immunoprecipitation. Cav-1 antibody was used to precipitate eNOS, AKT, and HSP90 in the high and low-density fraction mixtures (a, b). In control and EPI-treated cells high density fractions (1–4) do not demonstrate protein association. In low-density fractions (5–8) from non-stimulated cells, an association between Cav-1 andeNOS was detected. In contrast, in EPI-treated cells fractions 5–8 demonstrate a Cav-1, eNOS, AKT, and HSP90 association (a, b). Sucrose gradient mixture fractions from control and EPI-treated cells were used for immunoprecipitation (IP) using an AKT antibody (c). Western blots (WB) were used to detect eNOS, phospo-eNOS (Ser-1177 and 615), AKT, phospho-AKT, HSP90, and phospho-HSP90 in control and treated cells segregated by fractions mixtures. Data are expressed as mean ± SD (n = 3, * P < 0.05 by t test)
In order to further verify whether EPI is able to induce the activation/phosphorylation of eNOS via its binding to the AKT/HSP90 protein complex on lipid raft/caveolae, we performed IP assays using specific antibodies for AKT (Fig. 5c) and HSP90 (data not shown). As before, we used mixtures of sucrose gradient fractions (Fig. 5a) (1–4) and (5–8) obtained from control and EPI-treated HCAEC. The immunoprecipitated phase with the AKT antibody was then used for immunoblot analysis of total eNOS, eNOS Ser-1177, and Ser-615, as well as AKT, phospho-AKT and HSP90, and phospho HSP90 (Fig. 5c). In fractions 1–4 from control cells, as well as in EPI-treated cells, there is a low amount of eNOS and no detectable phosphorylated form of the enzyme. In contrast, the IP phase of EPI-treated HCAEC from the fraction mixture 5–8 demonstrated enzyme enrichment and enhanced phosphorylation of Ser-1177, and Ser-615, indicating that under calcium-free conditions there is phosphorylation and eNOS activation. AKT was detected in all fractions. However, it was enriched in control 1–4 fractions and in EPI-treated cells in the 5–8 fractions, whereas the phosphorylated form of AKT was only present in EPI-treated cells (predominantly in fraction 5–8). These results can be interpreted as evidencing an association between the active form of AKT with eNOS in caveolar fractions. Meanwhile, HSP90 demonstrated an association with AKT in all the fractions of EPI-treated cells with enrichment in fractions 5–8. These results evidence the conformation of a functional protein complex between the active forms of eNOS, AKT, and HSP90 in DRM (low-density fraction) of EPI-treated HCAEC under Ca2+-free conditions (Fig. 5c).
Discussion
We recently reported that EPI is capable of stimulating NO production via eNOS activation in a calcium dependent and in a calcium and translocation-independent manner [5, 6]. These most recent findings, prompted us to examine the mechanisms through which EPI induces eNOS activation in calcium-deprived HCAEC.
The molecular mechanisms that regulate eNOS activity in endothelial cells have been extensively studied. eNOS activity can be regulated by (1) its subcellular localization (association with caveolar microdomains), (2) phosphorylation status, and (3) through its interaction with specific proteins [11]. Under basal conditions, the majority of eNOS is present at caveolae, where it is bound to Cav-1. The association between these proteins is known to maintain eNOS in an inactive state. Such inhibition can be removed when eNOS associates to Ca2+/calmodulin (Ca2+-CaM) and it translocates from the caveolae into the cytoplasm [12, 13]. In response to physiological stimuli, eNOS activity is modulated through phosphorylation of key residues [14–16]. For instance, the phosphorylation of eNOS-Ser1177, Ser633, and Ser615 (human sequence) is associated with increases in enzymatic activity [17, 18], while the phosphorylation of Thr495 is related with eNOS inactivation [19, 20]. An increased association of eNOS to AKT and HSP90 is observed with its activation [11, 21]. Recently, we demonstrated that EPI induces eNOS activation and NO production via the PI3K/AKT/PKA and Ca2+-CaM/CaMKII pathways in HCAEC under standard Ca2+ containing conditions [5]. However, it is well established that eNOS activation can occur both under Ca2+-dependent and independent manners [22]. Most ligands, including bradykinin and acetylcholine, stimulate eNOS by raising the intracellular calcium concentration ([Ca2+]i), which in turn yields a Ca2+-CaM complex that binds to and activates eNOS [23, 24]. It has been reported that in the absence of calcium, AKT, and the protein chaperonin HSP90 play important roles in the activation of eNOS [25]. It also have been demonstrated that calcium and receptor-independent eNOS phosphorylation at Ser1177 is a process mediated by AKT and HSP90 [25]. Shear stress can also activate eNOS independently of large and sustained increases in [Ca2+]i, but this stimulation results in rather low NO production compared with agonist-evoked effects, where large increases in [Ca2+]i are induced [26]. However, Ca2+-independent eNOS phosphorylation has not been previously reported to occur as a consequence of the stimulation of receptors by commonly known agonists. It has been reported that a number of dihydropyridine Ca2+ antagonists, including nifedipine [27] and amlodipine [28] enhance NO production in endothelial cells independently of increases in [Ca2+]i inducing the relaxation of coronary arteries by a mechanism dependent on eNOS-Ser1177 phosphorylation [10]. In this study, we demonstrate that EPI induces increases in NO production through eNOS Ser615 and Ser1177 phosphorylation in the absence of cytosolic Ca2+ and that this phenomenon is mediated by AKT and HSP90 activities. Lack of changes secondary to the inhibition of PKA (H89 [10 μM]) and AMPK (Dorsomorphin dihydrochloride [0.1 μM]) (results not shown) further indicate that under Ca2+- free conditions PKA and AMPK do not participate in EPI-induced effects.
With non-receptor mediated stimuli such as shear stress [29] or ceramides [30], the activation of eNOS can take place independently of CaM binding and enzyme translocation while requiring small increases in [Ca2+]i. In a previous study using calcium-deprived cells, we demonstrated that EPI-induced NO production, even when eNOS translocation into the cytoplasm was abolished [5]. In the present study, we demonstrate via subcellular fractionation and immunoprecipitation assays the lack of decoupling of eNOS from caveolae. AKT and HSP90 also migrated from the non-lipid raft/caveolar fraction to DRM low-density fractions (where caveolar fractions are located), demonstrating that these proteins migrate and associate with eNOS at caveolae, inducing its activity (Figs. 4, 5 and 6).
Fig. 6.
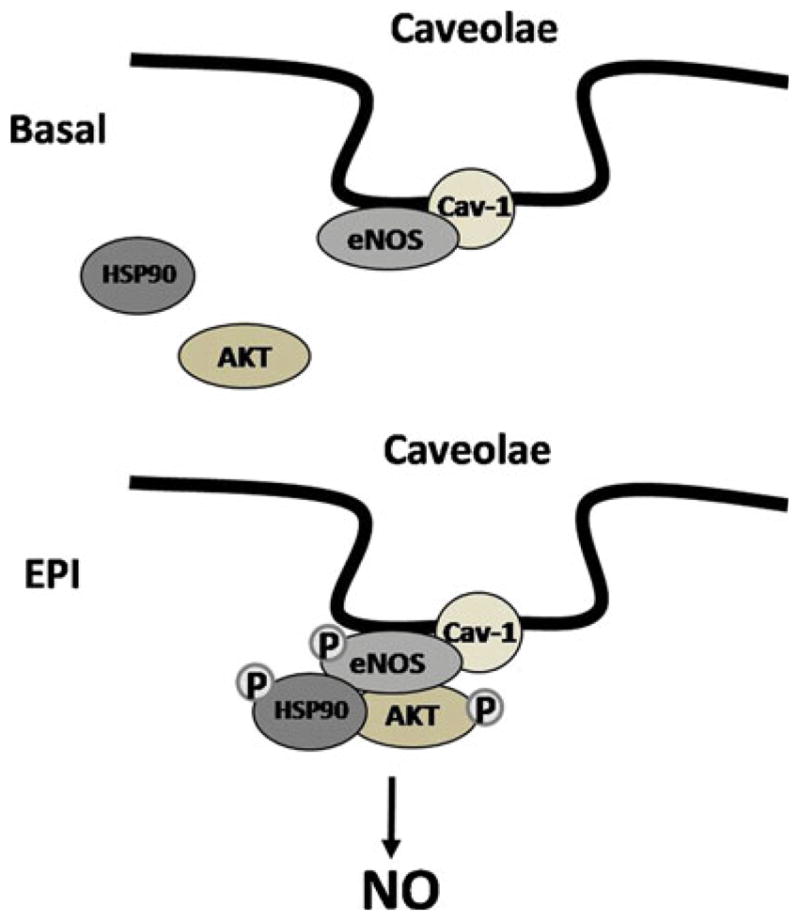
Schematic representation of the mechanism involved in HCAEC eNOS activation as observed under Ca2+-free conditions. AKT and HSP90 play an important role in phosphorylation/activation of eNOS in caveolae with EPI-treatment
It has also been demonstrated in unstimulated cells, that AKT resides in the cytoplasm with its kinase domain masked by C-terminal hydrophobic domain. In response to agonist stimulation, AKT is recruited to the plasma membrane (probably by HSP90) via its N-terminal PH domain. Membrane associated AKT is sequentially phosphorylated at two activation domains threonine 308 and serine 473 [9]. Other proteins that participate in the regulation of eNOS activity are also sequestered in these plasmalemmal microdomains (including G protein-coupled receptors), thus anchoring eNOS to caveolae may facilitate its coupling to and activation by these signaling molecules [31].
Results evidencing EPI-induced NO production in calcium depleted HCAEC in the absence of dissociation from caveolae and with the recruitment and/or association of AKT and HSP90 represents a novel mechanism for eNOS activation (Fig. 6). The “physiological” relevance of the EPI-induced Ca2+-independent eNOS activation in endothelial cells needs to be evaluated. However, it may be related to EPI-induced protective effects of cells as seen under stressful conditions such as with ischemia, where calcium homeostasis is altered and/or lost. The results presented here may also help to understand the mechanisms by which EPI may act to maintain and/or recover vascular function in diseases where NO production is limited or endothelial dysfunction is present. However, caution must be exercised when comparing in vitro EPI-induced effects versus those obtained after an oral intake of cocoa products or its flavanols, since EPI undergoes extensive metabolism (GI, liver) and the bioavailability of free EPI is low and some of its metabolites are vasoactive [15].
In conclusion, results demonstrate that EPI can induce the activation of eNOS in calcium depleted HCAE cells independently of its translocation into the cytoplasm in a AKT and HSP90 dependent manner. These actions appear unique and continue to suggest the presence of a specific plasmalemmal target that mediates such effects (i.e. receptor) which warrants further investigation.
Acknowledgments
This study was supported by NIH AT4277, HL43617, P60-MD000220 grants to Dr. Villarreal.
Contributor Information
Israel Ramirez-Sanchez, Department of Medicine, University of California, San Diego, San Diego, CA, USA. Escuela Superior de Medicina, Instituto Politecnico Nacional, Mexico, DF, Mexico.
Hugo Aguilar, Department of Medicine, University of California, San Diego, San Diego, CA, USA.
Guillermo Ceballos, Department of Medicine, University of California, San Diego, San Diego, CA, USA. Escuela Superior de Medicina, Instituto Politecnico Nacional, Mexico, DF, Mexico.
Francisco Villarreal, Email: fvillarr@ucsd.edu, Department of Medicine, University of California, San Diego, San Diego, CA, USA. UCSD Cardiology, 9500 Gilman Dr. 0613J, BSB 4028, La Jolla, CA 92093, USA.
References
- 1.Shimbo D, Graham-Clarke C, Miyake Y, Rodriguez C, Sciacca R, Di Tullio M, Boden-Albala B, Sacco R, Homma Shunichi. The association between endothelial dysfunction and cardiovascular outcomes in a population-based multi-ethnic cohort. Atherosclerosis. 2007;192:197–203. doi: 10.1016/j.atherosclerosis.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Buitrago-Lopez A, Sanderson J, Johnson L, Warnakula S, Wood A, Di Angelantonio E, Franco OH. Chocolate consumption and cardiometabolic disorders: systematic review and meta-analysis. BMJ. 2011;343:1–8. doi: 10.1136/bmj.d4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corti R, Flammer AJ, Hollenberg NK, Lüscher TF. Cocoa and cardiovascular health. Circulation. 2009;119:1433–1441. doi: 10.1161/CIRCULATIONAHA.108.827022. [DOI] [PubMed] [Google Scholar]
- 4.Schroeter H, Heiss C, Balzer J, Kleinbongard P, Keen CL, Hollenberg NK, Sies H, Kwik-Uribe C, Schmitz HH, Kelm M. (−)-Epicatechin mediates beneficial effects of flavanol-rich cocoa on vascular function in humans. Proc Natl Acad Sci USA. 2006;103:1024–1029. doi: 10.1073/pnas.0510168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramirez-Sanchez I, Maya L, Ceballos G, Villarreal F. (−)-Epicatechin induces calcium and translocation independent eNOS activation in arterial endothelial cells. Am L Physiol Cell Physiol. 2011;300:C880–C887. doi: 10.1152/ajpcell.00406.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramirez-Sanchez I, Maya L, Ceballos G, Villarreal F. Epicatechin activation of endothelial cell eNOS, NO and related signaling pathways. Hypertension. 2010;55:1398–1405. doi: 10.1161/HYPERTENSIONAHA.109.147892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramirez-Sanchez I, Ceballos-Reyes G, Rosas-Vargas H, Cerecedo-Mercado D, Zentella-Dehesa A, Salamanca F, Coral-Vazquez RM. Expression and function of utrophin associated protein complex in stretched endothelial cells: dissociation and activation of eNOS. Front Biosci. 2007;12:1956–1962. doi: 10.2741/2201. [DOI] [PubMed] [Google Scholar]
- 8.Melkonian KA, Ostermeyer AG, Chen JZ, Roth MG, Brown DA. Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J Biol Chem. 1999;274:3910–3917. doi: 10.1074/jbc.274.6.3910. [DOI] [PubMed] [Google Scholar]
- 9.Shiojima Ichiro, Walsh Kenneth. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signalin pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 10.Lenasi H, Kohlstedt K, Fichtlscherer B, Mülsch A, Busse R, Fleming I. Amlodipine activates the endothelial nitric oxide synthase by altering phosphorylation on Ser 1177 and Thr495. Cardiovasc Res. 2003;59:844–853. doi: 10.1016/s0008-6363(03)00505-4. [DOI] [PubMed] [Google Scholar]
- 11.Gratton J-P, Fontana J, O’Connor DS, Garcıa-Cardeña G, McC-abe TJ, Sessa WC. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. J Biol Chem. 2000;275:22268–22272. doi: 10.1074/jbc.M001644200. [DOI] [PubMed] [Google Scholar]
- 12.Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. 1997;272:18522–18525. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- 13.Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J Biol Chem. 1997;272:15583–15586. doi: 10.1074/jbc.272.25.15583. [DOI] [PubMed] [Google Scholar]
- 14.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J Biol Chem. 2002;277:3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 15.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 16.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCabe TJ, Fulton D, Roman LJ, Sessa WC. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J Biol Chem. 2000;275:6123–6128. doi: 10.1074/jbc.275.9.6123. [DOI] [PubMed] [Google Scholar]
- 18.Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, Corson MA. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- 19.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- 20.Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J Biol Chem. 2001;276:16587–16591. doi: 10.1074/jbc.M100229200. [DOI] [PubMed] [Google Scholar]
- 21.Joy S, Siow RCM, Rowlands DJ, Becker M, Wyatt AW, Aaronson PI, Coen CW, Kallo I, Jacob R, Mann GE. The isoflavone equol mediates rapid vascular relaxation. Ca2+-independent activation of endothelial nitric-oxide synthase/Hsp90 involving ERK1/2 and Akt phosphorylation in human endothelial cells. J Biol Chem. 2006;281:27335–27345. doi: 10.1074/jbc.M602803200. [DOI] [PubMed] [Google Scholar]
- 22.Omura M, Kobayashi S, Mizukami Y, Mogami K, Todoroki-Ikeda N, Miyake T, Matsuzaki M. Eicosapentaenoic acid (EPA) induces Ca(2+)-independent activation and translocation of endothelial nitric oxide synthase and endothelium-dependent vasorelaxation. FEBS Lett. 2001;487:361–366. doi: 10.1016/s0014-5793(00)02351-6. [DOI] [PubMed] [Google Scholar]
- 23.Michel T, Feron O. Nitric oxide synthases: which, where, how, and why? J Clin Invest. 1997;100:2146–2152. doi: 10.1172/JCI119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010;459:793–806. doi: 10.1007/s00424-009-0767-7. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi S, Mendelsohn ME. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt: calcium-independent eNOS activation involves formation of an HSP90-Akt-CaM-bound eNOS complex. J Biol Chem. 2003;278:30821–30827. doi: 10.1074/jbc.M304471200. [DOI] [PubMed] [Google Scholar]
- 26.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–E75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 27.Kitakaze M, Asamuma H, Takashima S, Minamino T, Ueda Y, Sakata Y, Asakura M, Sanada S, Kuzuya T, Hori M. Nifedipine-induced coronary vasodilation in ischemic hearts is attribuible to bradykinin- and NO- dependent mechanisms in dogs. Circulation. 2000;101:311–317. doi: 10.1161/01.cir.101.3.311. [DOI] [PubMed] [Google Scholar]
- 28.Zhang XP, Hintze TH. Amlodipine releases nitric oxide from canine coronary microvessels: an unexpected mechanism of action of a calcium channel-blocking agent. Circulation. 1998;97:580–876. doi: 10.1161/01.cir.97.6.576. [DOI] [PubMed] [Google Scholar]
- 29.Fleming I, Bauersachs J, Fisslthaler B, Busse R. Ca2+-independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibitors and fluid shear stress. Circ Res. 1998;82:686–695. doi: 10.1161/01.res.82.6.686. [DOI] [PubMed] [Google Scholar]
- 30.Igarashi J, Thatte HS, Prabhakar P, Golan DE, Michel T. Calcium-independent activation of endothelial nitric oxide synthase by ceramide. Proc Natl Acad Sci USA. 1999;96:12583–12588. doi: 10.1073/pnas.96.22.12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goetz Regina M, Thatte Hemant S, Prakash Prabhakar, Cho Michael R, Thomas Michel, Golan David E. Estradiol induces the calcium-dependent translocation of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1999;96:2788–2793. doi: 10.1073/pnas.96.6.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]