

Abstract
Although oligodendrocytes (OLGs) are thought to be vulnerable to hypoxia and ischemia, little is known about the detailed mechanism by which these insults induce OLG death. From the clinical viewpoint, it is imperative to protect OLGs as well as neurons against ischemic injury (stroke), because they are the only myelin-forming cells of the central nervous system. Using the Cre/loxP system, we have established a transgenic mouse line that selectively expresses p35, a broad-spectrum caspase inhibitor, in OLGs. After hypoxia, cultured OLGs derived from wild-type mice exhibited significant upregulation of caspase-11 and substantial activation of caspase-3, which led to cell loss. Expression of p35 or elimination of caspase-11 suppressed the caspase-3 activation and conferred significant protection against hypoxic injury. Expression of p35 in OLGs in vivo resulted in significant protection from ischemia-induced cell injury, thus indicating that caspases are involved in the ischemia-induced cell death of OLGs. Furthermore, the induction of caspase-11 was evident in the ischemic brains of wild-type mice, and OLGs exhibited resistance to brain ischemia in mice deficient in caspase-11, suggesting that caspase-11 is critically implicated in the mechanism(s) underlying ischemia-induced OLG death. Caspases may therefore offer a good therapeutic target for reducing ischemia-induced damage to OLGs.
Introduction
In most cases of acute ischemic stroke, the cerebral white matter is affected. With the advent of sensitive neuroimaging technologies, white matter lesions, termed leukoaraiosis, have been detected in many elderly people (1, 2). Leukoaraiosis is thought to be the result of chronic ischemic injury to the brain and to contribute sometimes to the development of cognitive dysfunction (1, 2). Oligodendrocytes (OLGs), a major cellular component of white matter, are the only central nervous system (CNS) myelin–forming cells and thus play a pivotal role in the proper execution of neural functions. In vitro and in vivo evidence has shown that OLGs are very vulnerable to hypoxic and ischemic insults (3–7). In an in vitro model of ischemia, more than 80% of cultured OLGs died after 6 hours of hypoxia combined with glucose deprivation (7). In a rodent focal ischemia model, OLGs showed conspicuous cellular swelling as early as 30 minutes after ischemia (3). Thus, therapeutic interventions need to be designed to protect OLGs as well as neurons, to reduce neural dysfunction stemming from ischemic brain damage. For this purpose, it is necessary to elucidate the precise mechanisms underlying the ischemia-induced death of OLGs, which are currently largely unknown.
Caspases are evolutionarily conserved executioners of programmed cell death in normal development and are also implicated in a variety of pathological conditions including cerebral ischemia (8). Caspases have been shown to be activated in OLGs subsequent to a variety of stimuli, including γ-irradiation and application of TNF-α (9, 10). Nevertheless, to our knowledge, there is no compelling evidence demonstrating that caspases are involved in hypoxia- or ischemia-induced OLG death. To clarify this point, it would be helpful to use transgenic mice in which the functions of caspases are inactivated selectively in OLGs, for the following reasons. First, as already stated, the actions of most, if not all, caspases are essential in the programmed cell death that is required for normal development (8). Therefore, simply “knocking out” some caspases results in embryonic or perinatal lethality, thus prohibiting the analysis of disease conditions induced in adults (11–13). Second, the pathophysiology of cerebral ischemia is quite complex. In addition to energy failure owing to the lack of oxygen and glucose, excitotoxicity (14, 15), and free radical formation (16), proinflammatory cytokines released from microglia and invading inflammatory cells (17–19) are known to contribute to the development of ischemic damage. In particular, IL-1β, a major proinflammatory cytokine implicated in ischemic brain injury, is cleaved from its inactive precursor by caspase-1 (IL-1β–converting enzyme [ICE]) and secreted (20, 21). Thus, it is evident that inactivation of caspases in cells other than OLGs may render the interpretation of outcomes difficult by modifying the disease conditions associated with cerebral ischemia. The Cre/loxP site-specific recombination system combined with transgenic technologies has enabled us to direct the expression of transgenes in a spatially and/or temporally restricted manner (22, 23). p35, a baculovirus-derived protein, is a broad-spectrum caspase inhibitor that has been demonstrated to function effectively in mammals (24–29). We established a transgenic mouse line bearing a transgene consisting of a p35 gene whose ORF was disrupted by the insertion of a DNA segment flanked by loxP sites (27). By crossing these transgenic mice with those expressing the cre recombinase transgene under the control of the myelin basic protein (MBP) promoter, we achieved the selective expression of the antiapoptotic protein, p35, in OLGs in the CNS (27).
Here, we provide in vitro and in vivo evidence that caspase-related pathways play an important role in hypoxia- and ischemia-induced cell death of OLGs, thus raising the possibility that therapeutic interventions targeted at caspase inhibition may be effective in preventing the ischemia-induced death of OLGs as well as of neurons.
Methods
Transgenic and knockout mice.
Transgenic mice carrying the p35 transgene, whose open reading frame (ORF) was disrupted by the insertion of a DNA segment consisting of a neo gene flanked by loxP sites, downstream of the CAG promoter, were described in detail previously (27). Briefly, a fragment containing the CAG promoter, loxP-neo-loxP, p35 coding sequence, and a polyadenylation signal was cloned into pBluescript and named pA23. This fragment was injected into fertilized C57BL/6 eggs to generate p35 transgenic mice. Transgenic mice expressing the cre recombinase transgene under the MBP promoter were also characterized previously (27). The transgene containing 6.3 kb of the MBP promoter, nuclear localizing signal (NLS) followed by cre recombinase coding region, and a polyadenylation signal was constructed. This transgene was injected into fertilized C57BL/6 eggs to generate transgenic mice. The caspase-11–deficient mice have been described in detail previously (30). Briefly, mutant J1 embryonic stem (ES) cell clones carrying a null mutant caspase-11 allele were injected into C57BL/6 blastocysts. The resulting chimeric males were then mated with C57BL/6 × DBA2 F1 females to obtain germline transmission of the mutant allele. Chimera of clone 444 produced germ-line–transmitted mutant mice. The offsprings from clone 444 were backcrossed to C57BL/6 eight times to ensure the genetic background homogeneity. Heterozygous littermates of this F8 were mated, and the resulting offsprings were used in this study. We used C57BL/6 mice as wild-type control in the present study.
PCR and RT-PCR.
For genotype identification of the mice used in the experiments, we carried out PCR to amplify the p35 and cre transgenes using genomic DNA samples as described previously (27). To confirm the expression of the p35 transgene in cultured OLGs and Cre/p35 tg mouse brains, we performed RT-PCR. Total RNA was isolated using Trizol (Life Technologies Inc., Rockville, Maryland, USA) according to the manufacturer’s instructions, and the samples were used for RT-PCR as described previously (27).
OLG primary cultures.
OLG cultures were prepared as described previously either from embryonic days 17–19 pups of wild-type female mice bred with male mice hemizygous for the p35 transgene or from caspase-11 knockout mouse (caspase-11–/–) pups harvested at the same embryonic stages (27). We used cultures enriched in mature OLGs characterized by extensive arborization of processes, myelin formation, and positive immunoreactivity for MBP, a well-established marker for mature OLGs. We confirmed that more than 95% of the cells were immunoreactive for MBP. In all the experiments related to p35 transgenic mice, OLGs derived from wild-type littermates were used for comparison with p35 transgene-bearing OLGs (p35-OLGs).
Expression of p35 in OLG cultures.
To achieve p35 expression in cultured OLGs harvested from embryos hemizygous for the p35 transgene, we infected the cells with a recombinant adenovirus bearing a cre expression cassette (Adex-cre) at a concentration of 2 × 105 pfu/mL 24 hours before the induction of hypoxia. We validated the use of this titer for adequate infection efficiency by adding the identical concentration of a lacZ expression adenovirus vector to a different culture for 24 hours, which was then fixed with 1% glutaraldehyde for 10 minutes and incubated in the β-galactosidase staining solution (20) at 37°C overnight. The expression of the p35 transgene was confirmed by RT-PCR and immunostaining. For immunostaining, OLG cultures treated with Adex-cre for 24 hours were fixed with 4% paraformaldehyde/0.1 M PBS for 10 minutes, permeabilized with 0.1% Triton X-100/1% H2O2/PBS for 5 minutes, and blocked with 4% normal goat serum/PBS (NGS/PBS) for 60 minutes. Anti-p35 antibody was applied at a dilution of 1:50 at 4°C overnight and horseradish peroxidase–conjugated (HRP-conjugated) anti-mouse IgG (Chemicon International, Temecula, California, USA) was applied at a dilution of 1:500 for 2 hours at room temperature. Immunoreactivity was visualized using a Tyramide Signal Amplification (TSA-Direct) kit (NEN Life Science Products, Boston, Massachusetts, USA) according to the manufacturer’s instructions. For identification of cells immunoreactive for p35, we performed double immunostaining using a rabbit polyclonal antibody raised against pi-GST, a specific marker of the cell body of mature OLGs (31) (MBL, 1:200). The antibody raised against p35 was produced as described previously (27).
Hypoxia experiments in OLG cultures.
OLGs grown on coverslips were subjected to hypoxia for 6 hours in a Gas-Pak anaerobic chamber (Becton Dickinson Microbiology Systems, Cockeysville, Maryland, USA) at 37°C. In parallel, sister cultures were grown for the same duration under normoxic conditions and served as controls. After hypoxia, cells were immunostained with anti-MBP rabbit polyclonal antibody (IBL) at a dilution of 1:2,000. FITC-conjugated anti-rabbit IgG (1:500; Life Technologies Inc.) was used as the secondary antibody. For the assessment of cell viability, we counted the numbers of MBP-positive cells with intact processes at a magnification of ×100 using a Carl Zeiss (Jena, Germany) Axioplan-2 fluorescence microscope. To evaluate the occurrence of DNA fragmentation in the cells that remained after hypoxia, we performed the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay according to the manufacturer’s instructions (Apoptag; Intergen Co., Purchase, New York, USA). Nuclear staining was performed using Hoechst 33342 (10 μM). To examine the occurrence of DNA fragmentation, TUNEL-positive cells were counted from six independent random samplings of approximately 150 cells and expressed as a percentage of the total cell number. A comparison was made between wild-type mice (WT-OLGs) and p35-OLGs in a blind manner. For the surviving cell assay and the TUNEL assay, statistical analysis was performed using ANOVA followed by Scheffé’s post hoc test and a nonpaired t test, respectively, and a P value of < 0.01 was considered statistically significant. All the data in the present study were expressed as means ± SEM.
Cytochrome c immunostaining in OLG cultures.
Before fixing the cells, MitoTracker Red (Molecular Probes Inc., Eugene, Oregon, USA) was added to the medium at a concentration of 0.1 μM for 20 minutes. Pretreatment for immunostaining was performed as already described here. Mouse monoclonal anti-cytochrome c (7H8.2C12; 1:200; PharMingen, San Diego, California, USA) and FITC-conjugated mouse IgG (1:200; Jackson ImmunoResearch Laboratories Inc., West Grove, Pennsylvania, USA) were used. The OLGs were observed using a Carl-Zeiss Axioplan-2 fluorescence microscope connected to a Zeiss LSM 510. Immunostaining patterns were classified into punctate and diffuse patterns. To assess the translocation of cytochrome c from mitochondria, the number of OLGs exhibiting a diffuse staining pattern was determined from ten independent random samplings of approximately 100 cells and expressed as a percentage of the total cell number. Statistical analysis was performed using ANOVA followed by Scheffé’s post hoc test, and a P value of < 0.01 was considered statistically significant.
Caspase activity assays.
Cultured OLGs were collected in a 1.5-mL Eppendorf tube and spun in a tabletop centrifuge at 1,800 g for 5 minutes at room temperature. For OLGs subjected to hypoxia, cells were collected 6 hours after hypoxia. The pellet was suspended in a lysis buffer consisting of 50 mM TrisCl (pH 7.5), 1 mM EDTA, 10 mM EGTA, and 10 μM digitonin, and incubated at 37°C for 10 minutes. The resultant lysate was spun at 15,800 g for 10 minutes, and the supernatant was used for caspase activity assays. The amount of protein in the samples was determined using a BCA Protein Assay Kit (Pierce Chemical Co., Rockford, Illinois, USA). Cell lysates containing 3 μg of protein were used for the assays, the detailed procedures of which have been described previously (29). The measurements were carried out in triplicate. To evaluate hypoxia-induced activation of caspases, we calculated the ratios of the caspase-like activity in lysates of hypoxia-treated OLGs to that of control OLGs. A comparison was made using a nonpaired t test, and a P value of < 0.01 was considered statistically significant.
Western blot analysis.
Cells were extracted as already described here, and the lysates were mixed with 2× sample buffer and boiled at 100°C for 5–10 minutes. Aliquots containing 5–7 μg of protein were separated on 15% SDS-PAGE gels and transferred to Immobilon-P membranes (Millipore Corp., Bedford, Massachusetts, USA). The membranes were incubated with the corresponding primary antibodies (anti-active form of caspase-3 polyclonal rabbit antibody [1:500; PharMingen]; anti–caspase-11 rat mAb (1:100; ref. 32); anti–β-tubulin mouse mAb [1:2,000; Sigma Chemical Co., St. Louis, Missouri, USA]; anti–caspase-1 rat mAb (1:100; ref. 30); anti–caspase-2 mouse mAb [1:500; Transduction Laboratories, Lexington, Kentucky, USA]; anti–caspase-9 rabbit polyclonal antibody [1:500; MBL]). After three washes, secondary antibodies were applied at room temperature for 2 hours. For caspase-3 and β-tubulin immunoblots, alkaline phosphatase-conjugated anti-rabbit IgG (Chemicon International) and anti-mouse IgG (Jackson ImmunoResearch Laboratories Inc.) were used, respectively, at a 1:500 dilution, and immunoreactive bands were visualized in a solution containing 0.1 M TrisCl (pH 9.5), 0.1 M NaCl, 50 mM MgCl2, 350 μg/mL 4-nitro blue tetrazolium, and 180 μg/mL 5-bromo-4-chloro-3-indolyl phosphate. For caspase-11 immunoblots, HRP-conjugated anti-rat IgG antibody (Chemicon International) was applied, and the blots were developed using an enhanced chemiluminescence system (ECL Plus; Amersham, Buckinghamshire, United Kingdom).
Caspase-3 immunostaining.
To demonstrate the activation of caspase-3 in situ, immunostaining for the activated form of caspase-3 was performed. We used a polyclonal rabbit antibody raised against the active form of caspase-3 (67341A; PharMingen) at a dilution of 1:1,000. Rhodamine-conjugated anti-rabbit IgG antibody (Chemicon International) was used as secondary antibody at 1:500. Otherwise, the procedures for immunostaining were as described earlier here. Counterstaining with Hoechst 33342 (10 μM) was carried out to visualize nuclear morphology.
Focal cerebral ischemia model.
The Animal Welfare Guidelines of Osaka University were followed for all the studies and experiments with mice. Male mice weighing 18–25 g were subjected to permanent focal cerebral ischemia as described elsewhere, with some modifications (17, 18). Mice were anesthetized with 1.5–2.0% isoflurane/30% O2/70% N2O using a facemask. Core temperature was continuously monitored and maintained at 37°C throughout surgery. After a midline neck incision, the left common carotid artery was carefully isolated from the vagal nerve and ligated. The external carotid artery was also ligated, and the internal carotid artery was carefully isolated. An 8-0 nylon filament (Ethilon; Ethicon Inc., Sommerville, New Jersey, USA), whose tip was coated with silicon resin (Xantopren; Heraeus, Dormagen, Germany), was inserted into the common carotid artery through a small incision made in the proximity of the carotid bifurcation and advanced to the proximal part of the anterior cerebral artery to compromise the middle cerebral artery (MCA) flow. The filament was fixed in position by ligature. In sham-operated animals, these procedures, except for the insertion of an intraluminal filament, were performed. To ensure that the MCA was successfully occluded, several animals, anesthetized by an intraperitoneal injection of excess pentobarbital sodium (100 mg/kg), were perfused transcardially with India ink diluted with heparinized PBS 10 minutes after the surgery followed by perfusion fixation with 4% paraformaldehyde/PBS. The brain was removed and cut coronally into 1-mm–thick sections for subsequent inspection. Brain areas receiving sufficient perfusion were stained by the India ink, but those devoid of blood flow were demarcated as “white areas.” We also stained brain coronal sections obtained from several animals subjected to 24 hours of MCA occlusion with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma Chemical Co.) in PBS to confirm the formation of infarct in the MCA area. For all the animals used for histological examinations described later here, neurological deficits were scored as follows: grade 0, no observable neurological deficits; grade 1, failure to extend the right forepaw; grade 2, circling to the contralateral side; and grade 3, loss of walking or righting reflex (18). We evaluated the neurological deficits 60 minutes after MCA occlusion (MCAO), at which time the effect of the anesthesia had completely worn off. For histological examinations, mice were deeply anesthetized at 6 or 24 hours after MCAO with excess pentobarbital sodium (100 mg/kg) and perfused transcardially with heparinized PBS followed by 4% paraformaldehyde/PBS for tissue fixation. Brains were postfixed in 4% paraformaldehyde/PBS at 4°C overnight.
Histological examinations.
The overall histology of the brains was assessed by hematoxylin and eosin (H&E) staining. For immunohistochemistry, frozen sections were thawed and washed in PBS three times. The sections were permeabilized with 0.1% Triton X-100/PBS at room temperature for 10 minutes and then blocked in 4% NGS/PBS for 60 minutes. Subsequently, they were incubated with primary antibodies at 4°C overnight. Primary antibodies and their dilutions were as follows: anti-p35 mouse polyclonal antibody, 1:1,000; anti–pi-GST rabbit polyclonal antibody (MBL), 1:200; and anti–caspase-11 rat mAb, 1:100. For pi-GST immunostaining, rhodamine-conjugated anti-rabbit IgG antibody (Chemicon International) at 1:200 was used as the secondary antibody. For p35 and caspase-11 immunostaining, immunoreactivity was visualized using TSA-Direct according to the manufacturer’s instructions. Double-labeling with TUNEL and pi-GST immunohistochemistry was performed to investigate whether DNA fragmentation was induced in OLGs located in the infarcts that resulted from MCAO, as described previously (33). To explore the survival of OLGs after ischemic insult, the numbers of remaining cortical pi-GST–positive cells that had morphological features consistent with those of OLGs were counted 6 hours and 24 hours after ischemia by a naive observer at a magnification of ×100 using a Carl-Zeiss Axioplan-2 fluorescence microscope. The region of interest (ROI) was set at the granular insular cortex at the level of 1.10 mm anterior to bregma, according to Franklin and Paxinos (34). As controls, the numbers of pi-GST–positive cells in the same ROI from sham-operated animals were counted. Statistical analysis was performed using ANOVA followed by Scheffé’s post hoc test within each genotype (wild-type littermates, p35 tg, Cre/p35 tg, and caspase-11 KO). Statistical significance was set at P < 0.01.
Results
For in vitro experiments, we prepared OLG cultures from transgenic mice bearing the p35 transgene (p35 tg). We first ensured the successful expression of the p35 transgene in our OLG cultures using adenovirus-mediated delivery of the cre recombinase transgene. We added an adenovirus vector carrying a cre transgene expression cassette at the concentration of 2.0 × 105 pfu/mL. The majority of OLGs in cultures infected with a lacZ-expression adenovirus vector at the same concentration were positive for β-galactosidase activity (Figure 1a), confirming that this titer was sufficient for effective transfection efficiency in our culture system. The expression of the p35 transgene was verified by RT-PCR and double immunostaining using antibodies against p35 and pi-glutathione S-transferase (GST), a marker for the cell body of OLGs (31) (Figure 1, b–d).
Figure 1.

Expression of the p35 transgene in OLG cultures. (a) The majority of OLGs transfected with a lacZ expression adenovirus vector were positive for β-galactosidase activity. Original magnification, ×200. (b) The expression of the p35 transgene was confirmed by RT-PCR. Lane 1, brain sample from a transgenic mouse bearing the cre recombinase transgene (Cre tg); lane 2, brain sample from a transgenic mouse bearing both the cre recombinase and p35 transgenes (Cre/p35 tg); lane 3, sample obtained from cultured p35 transgene–bearing OLGs infected with an adenovirus vector carrying a cre recombinase expression cassette. In each sample, ice cDNA was amplified as an internal control of cDNA synthesis. (c and d) Double immunostaining for p35 (c) and pi-GST (d).
To investigate the effect of p35 expression in OLGs on hypoxia-induced cell toxicity, we subjected OLG cultures to hypoxia for 6 hours. Several lines of evidence have shown that OLGs are vulnerable to hypoxic insult (5, 7). In our system, the oxygen concentration in the anaerobic chamber was demonstrated to decrease to 0.4% within 100 minutes (35). The hypoxic insult resulted in a substantial loss of OLGs that had been harvested from WT-OLGs (112.7 ± 8.5 to 52.2 ± 2.7 per visual field; P < 0.01, ANOVA; Figure 2a). Cell toxicity was morphologically characterized by cell lysis, destruction of processes, and formation of cell debris (Figure 2, b and c). On the other hand, p35-expressing OLGs (p35-OLGs) were extremely resistant to the hypoxic insult (Figure 2, d and e). After hypoxia, the number of p35-OLGs with intact processes exhibited only a slight decrease that was not statistically significant (113.9 ± 5.5 to 102 ± 3.2 per visual field; Figure 2a). To evaluate the viability of the remaining cells, we performed the TUNEL assay. The proportion of TUNEL-positive cells was significantly lower in the p35-OLGs (23.4 ± 1.8% vs. 42.4 ± 1.8%; P < 0.01, t test; Figure 2f).
Figure 2.

Hypoxic injury to OLG cultures. OLGs were examined 6 hours after hypoxia. We counted the numbers of MBP-positive cells with morphological features consistent with mature oligodendrocytes. (a) The numbers of mature OLGs with intact arborizations counted at a magnification of ×100. The quantification was carried out in 18 independent visual fields from three distinct cultures for each group. Statistical analysis was performed using ANOVA followed by Scheffé’s post hoc test. AP < 0.01. (b and d) OLGs from wild-type mice (wild-type, b) and p35 tg (Cre/p35, d), both of which were treated with the cre recombinase expression adenovirus vector, immunostained with an MBP-specific antibody. OLGs were characterized morphologically by extensive arborization of processes and myelin formation. (c) In cultures of wild-type OLGs subjected to 6 hours of hypoxia, cell lysis, destruction of processes, and scattered cell debris were seen. (e) The hypoxia-induced morphological changes were attenuated in Cre/p35. Original magnification, ×200. (f) The proportions of TUNEL-positive cells after hypoxia in wild-type and Cre/p35 OLGs. The quantification was performed in six independent random samplings of approximately 150 cells each, and the data were expressed as a percentage of the total cell number (means ± SEM). Statistical analysis was performed using a nonpaired t test. AP < 0.01.
It has been shown that p35 prevents cell death by inhibiting caspases (26), implying that the hypoxia-induced cell death in our cultures was mediated by the activation of caspases. Therefore, we investigated the involvement of caspase-3, a representative effector caspase, in the hypoxic cell injury. In Western blot analysis using an antibody raised against the active form of caspase-3, we detected a 20-kDa band corresponding to a cleaved product of caspase-3 that is produced upon its activation, but only in cell lysates prepared from WT-OLGs subjected to hypoxia (Figure 3a). Furthermore, this antibody immunolabeled the cytoplasm of numerous hypoxia-treated WT-OLGs, whose nuclei exhibited a condensed morphology that is consistent with apoptotic demise (Figure 3, d and e). There was no immunoreactivity in control cells or in cells that retained normal nuclear morphology after hypoxia (Figure 3, b–e). In line with these findings, the hypoxic insult resulted in a significant increase in caspase-3-like (DEVDase) activity in WT-OLGs (340.0 ± 25.0%; Figure 3f). As expected, there was no significant change in DEVDase activity in p35-OLGs after hypoxia (96.1 ± 13.3%; Figure 3f). There was no significant change in caspase-1-like (YVADase) activity after hypoxia in WT-OLGs or p35-OLGs (93.3 ± 14.4% and 86.9 ± 12.4%, respectively; Figure 3g).
Figure 3.

Activation of caspase-3 in OLGs after hypoxia. All the samples of hypoxia-treated OLGs were prepared 6 hours after hypoxia. (a) Western blot analysis showing that the cleavage of procaspase-3 occurred in wild-type, but not in Cre/p35, OLGs after hypoxia. (b and c) Under normal conditions, WT-OLGs did not exhibit immunoreactivity for the active form of caspase-3. (d and e) WT-OLGs showing immunoreactivity for the active form of caspase-3 and a condensed nuclear morphology. Note that a neighboring OLG that did not display immunoreactivity for the active form of caspase-3 had a normal nuclear morphology even after hypoxia (arrows). Original magnification, ×200. (f) Hypoxia-induced caspase-3–like protease (DEVDase) activation in wild-type and Cre/p35 cells. The measurements were carried out in triplicate for two different samples. The changes in DEVDase activity after hypoxia were expressed as a percentage. Statistical analysis was performed with a nonpaired t test. AP < 0.01. (g) Hypoxia-induced caspase-1–like protease (YVADase) activation in wild-type and Cre/p35 OLGs. The changes in YVADase activity after hypoxia were expressed as a percentage (means ± SEM). The measurements were carried out in triplicate for two different samples.
Next, we explored the effect of hypoxia on the subcellular distribution of cytochrome c, a well-known apoptogenic factor that acts upstream of caspase-3 activation (36, 37). To this end, before fixing the cells we treated them with a mitochondrial marker, MitoTracker Red, and then fixed the cells and immunolabeled them with an anti–cytochrome c antibody. Under normal conditions, the distribution of cytochrome c immunoreactivity showed a punctate pattern that coincided with the localization of mitochondria as evidenced by the MitoTracker Red staining (Figure 4, a–c). After hypoxic insult, a fraction of the cells exhibited a diffuse pattern of immunostaining, which most likely indicated that cytochrome c had translocated from the mitochondria into the cytosol (Figure 4, d–i). We evaluated the translocation of cytochrome c from the mitochondria by comparing the change in the proportion of cells manifesting the diffuse staining pattern after hypoxia. In both WT-OLGs and p35-OLGs, hypoxia induced a similar degree of change in the proportion of cells with diffuse staining (4.7 ± 0.1% to 21.6 ± 2.3% and 6.3 ± 0.7% to 16.9 ± 1.4%, respectively; P < 0.01, ANOVA; Figure 4j). These results suggest that p35 prevents OLG cell death after cytochrome c release.
Figure 4.

Translocation of cytochrome c from mitochondria in OLGs after hypoxia. (a) A WT-OLG under normal conditions showing a punctate immunostaining pattern for cytochrome c that coincided with the localization of mitochondria, as revealed by the mitochondrial marker MitoTracker Red (b). (c) A merged image of a and b. (d–f) A WT-OLG subjected to 6 hours of hypoxia exhibiting a diffuse pattern of immunostaining for cytochrome c. The cell assumed a round morphology without processes, a morphological sign indicative of an unhealthy state. (g–i) A p35-expressing OLG showing a diffuse staining pattern for cytochrome c but maintaining processes. Original magnification, ×400. (j) The proportions of OLGs showing a diffuse pattern of cytochrome c immunostaining. Quantification was performed with ten random samplings of approximately 100 cells each. The data are expressed as a percentage of the total cell number (means ± SEM). Statistical analysis was performed by ANOVA followed by Scheffé’s post hoc test. AP < 0.01.
We then used Western blotting to examine the expression of caspases in OLGs shortly after 6 hours of hypoxia. We did not observe any significant changes in the expression level of caspases-1, 2, 3, and 9 (data not shown). In contrast, we observed a dramatic increase in the expression of caspase-11 after hypoxic insult, both in WT-OLGs and p35-OLGs (Figure 5a). We therefore investigated the expression of caspase-11 in ischemia-induced death of OLGs in vivo by double immunostaining for caspase-11 and pi-GST. Intriguingly, the induction of caspase-11 was observed as little as 1 hour after the MCAO in OLGs situated in the cerebral cortex in the MCA territory (Figure 6, a–i), suggesting that caspase-11 may play an important role in an upstream event that leads to cell death. The immunoreactivity of caspase-11 in OLGs persisted for at least 24 hours after MCAO (data not shown).
Figure 5.

Caspase-11–dependent caspase-3 activation in OLGs was induced by hypoxia. For Western blot analysis and caspase-like activity assays, all the samples of hypoxia-treated OLGs were prepared 6 hours after hypoxia. (a) Western blot analysis showing that caspase-11 (43 kDa) was upregulated in response to hypoxia in WT-OLGs and p35-OLGs. (b) Hypoxia-induced DEVDase activation in wild type and caspase-11–/–. The measurements were carried out twice in triplicate, and the results were expressed and assessed as described in Figure 3. AP < 0.01. (c) Western blot analysis showing that hypoxia-induced cleavage of caspase-3 was attenuated in caspase-11–/– mice–derived OLGs (KO) in comparison with those from wild-type mice (WT). (d) Hypoxia-induced death of OLGs derived from caspase-11–/– mice. The data were obtained from six independent visual fields for each genotype and expressed and assessed as described in Figure 2.
Figure 6.

Double immunostaining of wild-type mouse brain sections (cerebral cortex subjected to ischemia) for pi-GST (red) and caspase-11 (green). (a and d) In the sham-operated animal, there was no immunoreactivity for caspase-11. After MCAO, immunoreactivity for caspase-11 became evident in OLGs (arrows) at 1 (b and e) and 6 hours (c and f). Original magnification, ×200. Laser confocal images (g–i) revealed that the immunoreactivity for caspase-11 was recognized in the perikarya of OLGs identified as pi-GST–positive cells (g, pi-GST; h, caspase-11; i, merged). The images were taken from the deep layers of the granular insular cortex on the ischemic side.
There is evidence that there exists a death-executing pathway in which the activation of caspase-11 lies upstream of caspase-3 activation (38). To investigate whether this pathway was operative in the hypoxia-induced death of OLGs in our cultures, we prepared OLG cultures from caspase-11–/– mice. In control cultures, 6 hours of hypoxia resulted in a 4.5-fold increase in DEVDase activity (455.5 ± 29.3%; Figure 5b). In cultures derived from caspase-11–/– mice, there was only a 2.6-fold rise in DEVDase activity (263.5 ± 25.4%; P < 0.01, t test; Figure 5b). Furthermore, only a faint band corresponding to the activated form of caspase-3 (p20) was detected in caspase-11–/– OLG lysate under hypoxic conditions (Figure 5c). This finding clearly showed that a caspase-11–dependent caspase-3 activation machinery was operative in the hypoxia-induced toxicity of our cultures, although it appeared not to be the exclusive mechanism. Correspondingly, hypoxic insult led to only a slight rise in cell death in OLGs from caspase-11–/– mice (95.2 ± 10.2 to 73.0 ± 7.2 per visual field; Figure 5d). Furthermore, OLGs of caspase-11–/– mice exhibited resistance to ischemic insult in vivo (see Figure 8g). These results strongly indicated that the inactivation of caspase-11 conferred protection to the OLGs against the hypoxia- and ischemia-induced cell death.
Figure 8.

Temporal profiles of ischemia-induced death of OLGs. In wild-type mice, focal cerebral ischemia resulted in a time-dependent decrease in the density of OLGs in the cerebral cortex under ischemia (a–c). In Cre/p35 tg, ischemia-induced death of OLGs was attenuated (d–f). Original magnification, ×100. (g) OLG survival after MCAO. The numbers of pi-GST–positive cells in the granular insular cortex under ischemia were counted at a magnification of ×100 on 12- to 14-μm-thick frozen sections obtained from five to seven different animals from each genotype: wild-type littermates, p35 tg, Cre/p35 tg, and caspase-11–/– mice (caspase-11 KO). The data were expressed as means ± SEM per visual field. Statistical analysis was performed using ANOVA followed by Scheffé’s post hoc test. AP < 0.01, compared with sham-operated animals in each group.
To test directly the involvement of caspases in ischemia-induced OLG cell death, we crossed p35 tg with mice expressing the cre gene in their OLGs (Cre tg). As depicted in Figure 7, the selective expression of p35 in OLGs in the brain was confirmed by immunostaining. To test whether p35-expressing OLGs in vivo were resistant to ischemic insult, we subjected mice hemizygous for both the p35 and cre transgenes (Cre/p35 tg) to permanent focal cerebral ischemia for 6 or 24 hours by MCAO with an intraluminal filament and compared the histopathological outcomes with those obtained from wild-type mice and p35 tg. We verified that cerebral blood flow in the MCA area was compromised and that the formation of infarct was achieved in our ischemia model using wild-type mice (data not shown). In both wild-type (sham-operated) and p35 tg mice, cerebral ischemia caused time-dependent decreases in the density of OLGs in the cortex (wild-type mice, 88.2 ± 6.2 per visual field in sham-operated animals, 74.0 ± 6.0 per visual field at 6 hours, and 45.3 ± 2.1 per visual field at 24 hours; p35 transgenic mice, 91.2 ± 2.5 per visual field in sham-operated animals, 66.0 ± 4.7 per visual field at 6 hours, and 45.2 ± 3.3 per visual field at 24 hours; Figure 8, a–c, and g). In contrast, OLGs in Cre/p35 tg exhibited resistance to the ischemic insult (89.8 ± 6.4 per visual field in sham-operated animals, 96.5 ± 4.1 per visual field at 6 hours, and 79.3 ± 6.5 per visual field at 24 hours; Figure 8, d–f and g). As stated earlier here, OLGs of caspase-11–/– mice were tolerant of the ischemic insult (96.3 ± 3.4 per visual field in sham-operated animals, 90.4 ± 3.6 per visual field at 6 hours, 76.0 ± 4.9 per visual field at 24 hours; Figure 8g). Histological analysis by H&E staining showed that cerebral ischemia resulted in neuronal loss accompanied by overt ischemic changes in the remaining neurons and in tissue edema (Figure 9, a–d). These ischemia-induced histopathological changes were of approximately the same magnitude in wild-type mice and in Cre/p35 tg mice (Figure 9, b and c). The TUNEL staining demonstrated the robust occurrence of DNA fragmentation in ischemic brains (Figure 9e). As expected, TUNEL-positive OLGs were found in wild-type mice (Figure 9e, arrows). In Cre/p35 tg, few TUNEL-positive OLGs were seen against a background of numerous non-OLG, TUNEL-positive cells, most likely representing neurons (Figure 9f). In conjunction with the observation that ischemia-induced loss of OLGs was significantly attenuated in Cre/p35 tg and caspase-11–/– mice, these findings strongly suggested that ischemia-induced death of OLGs is largely dependent on caspase activation, in which caspase-11 might play an important role.
Figure 7.
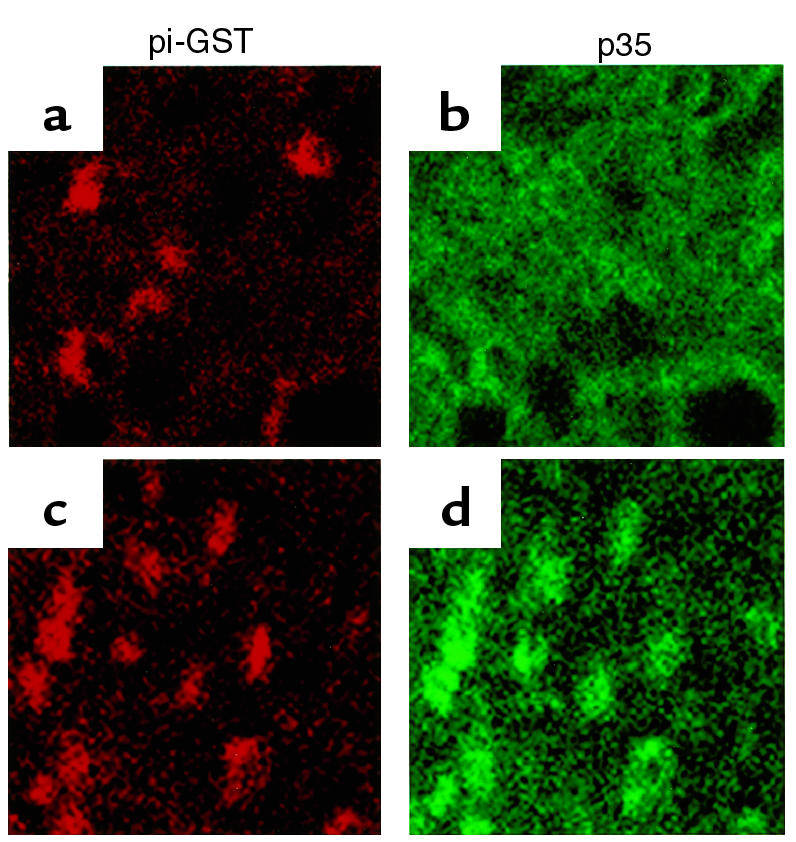
Expression of p35 in the cerebral cortex of Cre/p35 tg was verified by double immunostaining for p35 and pi-GST. (a and b) OLGs identified by immunoreactivity for pi-GST were scattered in the cerebral cortex of a wild-type mouse, and they did not exhibit immunoreactivity for p35. (c and d) OLGs were immunoreactive for p35 in Cre/p35 tg. Original magnification, ×400.
Figure 9.

H&E staining and double-labeling with TUNEL and pi-GST immunohistochemistry of brain sections. (a) Normal structure of the cerebral cortex from a sham-operated animal. (b and c) Brain sections obtained at 24 hours after MCAO from a wild-type mouse and Cre/p35 tg, respectively, showing tissue edema and overt ischemic changes in the remaining neurons. Original magnification, ×100. (d) High-powered view (×1,000) of a neuron exhibiting typical ischemic changes, which consisted of a pyknotic nucleus, cytoplasmic eosinophilia, triangular cellular morphology, and perineuronal vacuolation (51); paraffin-embedded sections (7 μm thick). (e) Double-labeling with TUNEL (green) and pi-GST immunohistochemistry (red) showed a robust occurrence of DNA fragmentation in numerous cells including OLGs (arrows). (f) In Cre/p35 tg, few OLGs were positive for TUNEL viewed against a background of numerous TUNEL-positive cells, most likely representing neurons. Original magnification, ×100.
Discussion
In the present study, we demonstrated that hypoxia- and ischemia-induced death of OLGs is accompanied by the activation of caspases. Inactivation of caspases by a broad-spectrum caspase inhibitor, p35, conferred on the OLGs significant protection from hypoxic and ischemic insults, clearly showing that a family of caspases is implicated in these cell-death paradigms. Furthermore, we have shown that caspase-11–dependent caspase-3 activation is involved in the hypoxia-induced death of OLGs. These findings have important clinical implications because they shed light on the intracellular mechanisms of ischemia-induced OLG death and provide several informative clues about how novel therapeutic targets might be used in the treatment of cerebral ischemia.
In our OLG cultures, 6 hours of hypoxia resulted in approximately 50% cell loss, accompanied by DNA fragmentation in about 40% of the remaining cells. This finding is consistent with previous studies reporting the vulnerability of OLGs to hypoxia (5, 7). OLG primary cultures derived from mouse forebrain exhibited approximately 60% cell loss 24 hours after a 2-hour exposure to oxygen/glucose deprivation (5), and only 24% of cultured rat OLGs survived after 6 hours of hypoxia (7). Caspases, central intracellular executioners of cell death, are activated in OLGs after a variety of stimuli, including γ-irradiation and TNF-α treatment (9, 10, 39). We confirmed the activation of caspase-3 in our OLG cultures after hypoxia using Western blot analysis and an activity assay using a fluorogenic substrate. The expression of p35 suppressed the hypoxia-induced caspase-3 activation, as expected from its inhibitory effects on caspases, and significantly attenuated the cell death. These results strongly imply that the hypoxia-induced demise of OLGs is mainly mediated by caspase-3 activation. Caspase-3 is activated by several distinct pathways (40). Cytochrome c released from mitochondria activates caspase-9 with the assistance of Apaf-1 in the presence of (d)ATP, after which pro-caspase-3 is cleaved into its active form by the activated caspase-9 (36, 37). Caspase-3 activation is also accomplished by caspase-8, which is activated downstream or independent of the “death factors” (41). We found that the translocation of cytochrome c from the mitochondria took place after hypoxia in both WT-OLGs and p35-OLGs to a similar degree. This observation indicates that the apoptogenic molecule, cytochrome c, is likely to be involved in the hypoxia-induced activation of caspase-3 and that the release of cytochrome c occurs independent of caspase activity. Recent evidence has shown that a proapoptotic Bcl-2 family member, Bid, upon cleavage by caspase-8, translocates into the mitochondrial outer membrane to liberate cytochrome c (42, 43). However, such a caspase-dependent mechanism of cytochrome c release is not likely to play a significant role in our hypoxia experiment, as the expression of p35 did not attenuate the cytochrome c release in OLGs treated with hypoxia.
Recently, a third caspase-3 activation paradigm was described wherein caspase-11 directly processes and activates caspase-3 (38). In our study, caspase-11 was upregulated and contributed to cell death via caspase-3 activation after hypoxia. We confirmed that OLGs prepared from caspase-11–/– mice were resistant to the hypoxic insult with the significantly attenuated activation of caspase-3 as revealed by the DEVDase activity assay and Western blotting. Because the contribution of caspase-11 to hypoxia-induced death and caspase-3 activation in OLGs is significant, inactivation of caspase-11 is a potential therapeutic target in the treatment of cerebral ischemia. This notion is supported by our in vivo finding that OLGs in caspase-11–/– mice were resistant to ischemia. Furthermore, the early activation of caspase-11 in wild-type mouse brains implies that this initiator caspase may be involved in some upstream event leading to the ischemia-associated demise of OLGs.
We have demonstrated that p35-expressing OLGs are significantly resistant to ischemic injury in vivo, showing unequivocally that a family of caspases plays a pivotal role in the execution of ischemia-induced OLG death. Permanent focal cerebral ischemia produced by MCAO, a commonly used ischemia model, resulted in a time-dependent loss of OLGs in infarct areas. This finding is consistent with previous studies reporting that OLGs are vulnerable to ischemia (3–7). In a transient global ischemia model that has been used to analyze selective vulnerability to ischemia, DNA fragmentation in OLGs preceded that in neurons (6). In focal cerebral ischemia models, OLGs exhibited morphological changes such as cell swelling (3) and biochemical changes exemplified by the accumulation of tau in the early phase of ischemia (44). Additionally, a recent paper reported that caspase-3 activation in OLGs is induced by hypoxia/ischemia in neonatal rats (45). What triggers the death of OLGs in cerebral ischemia remains to be elucidated. There is no doubt that hypoxia is an important component of ischemia-associated pathophysiological events. However, cerebral ischemia is known to cause a variety of other aberrant changes such as excessive glutamate release (14, 15), formation of free radicals that include nitric oxide (16, 46, 47), and TNF-α production (19), all of which are toxic to OLGs (5, 9, 39, 48). Our result, indicating that the expression of p35 conferred substantial protection against ischemic insult, strongly suggests that these toxic alterations may converge to activate caspases in the execution of OLG death. Accumulating evidence has shown that a family of caspases plays an important role in ischemic neuronal death (33, 49, 50). Therefore, the inactivation of caspases is a potential therapeutic target capable of conferring significant protection from ischemic injury on both OLGs and neurons. At present, we have no data regarding the long-term survival of OLGs beyond 24 hours after ischemia. Further experiments are required to clarify this point.
Irrespective of the clinical disease subtypes (atherothrombotic stroke, cardioembolic stroke, and lacunar stroke), acute ischemic stroke affects cerebral white matter in the majority of cases. As OLGs are the only myelin-forming cells in the CNS, it is obvious that therapeutic interventions designed to protect OLGs as well as neurons are required to attenuate the neural dysfunction that results from ischemic brain damage. The present study provides the first in vitro and in vivo evidence to our knowledge that a family of caspases plays a pivotal role in the hypoxia- and ischemia-induced death of OLGs. These results underscore the importance of caspase inactivation as a therapeutic target for the treatment of ischemic stroke.
Acknowledgments
We are grateful to I. Saito at University of Tokyo for providing us recombinant adenovirus vectors bearing the cre recombinase or lacZ expression cassette. This work was supported in part by grants from the Japanese Ministry of Education, Sports and Culture (to H. Okano and M. Miura) and the Human Frontier Science Program (to H. Okano). This work was also supported in part by research grants from Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Corporation to (H. Okano). S. Hisahara is a Research Fellow of the Japan Society for the Promotion of Science.
References
- 1.Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28:652–659. doi: 10.1161/01.str.28.3.652. [DOI] [PubMed] [Google Scholar]
- 2.Pantoni L, et al. Role of white matter lesions in cognitive impairment of vascular origin. Alzheimer Dis Assoc Disord. 1999;13(Suppl. 3):S49–S54. [PubMed] [Google Scholar]
- 3.Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27:1641–1646; discussion 1647. doi: 10.1161/01.str.27.9.1641. [DOI] [PubMed] [Google Scholar]
- 4.Mandai K, et al. Ischemic damage and subsequent proliferation of oligodendrocytes in focal cerebral ischemia. Neuroscience. 1997;77:849–861. [PubMed] [Google Scholar]
- 5.McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- 6.Petito CK, Olarte JP, Roberts B, Nowak TS, Jr, Pulsinelli WA. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol. 1998;57:231–238. doi: 10.1097/00005072-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Lyons SA, Kettenmann H. Oligodendrocytes and microglia are selectively vulnerable to combined hypoxia and hypoglycemia injury in vitro. J Cereb Blood Flow Metab. 1998;18:521–530. doi: 10.1097/00004647-199805000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 9.Hisahara S, Shoji S, Okano H, Miura M. ICE/CED-3 family executes oligodendrocyte apoptosis by tumor necrosis factor. J Neurochem. 1997;69:10–20. doi: 10.1046/j.1471-4159.1997.69010010.x. [DOI] [PubMed] [Google Scholar]
- 10.Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV. Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci. 1999;19:3043–3049. doi: 10.1523/JNEUROSCI.19-08-03043.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuida K, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 12.Hakem R, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 13.Kuida K, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 14.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 15.Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- 16.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 17.Friedlander RM, et al. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hara H, et al. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab. 1997;17:370–375. doi: 10.1097/00004647-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2000;20:53–65. doi: 10.1097/00004647-200001000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 21.Kuida K, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 22.Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods. 1998;14:381–392. doi: 10.1006/meth.1998.0593. [DOI] [PubMed] [Google Scholar]
- 23.Lobe CG, et al. Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- 24.Rabizadeh S, LaCount DJ, Friesen PD, Bredesen DE. Expression of the baculovirus p35 gene inhibits mammalian neural cell death. J Neurochem. 1993;61:2318–2321. doi: 10.1111/j.1471-4159.1993.tb07477.x. [DOI] [PubMed] [Google Scholar]
- 25.Martinou I, et al. Viral proteins E1B19K and p35 protect sympathetic neurons from cell death induced by NGF deprivation. J Cell Biol. 1995;128:201–208. doi: 10.1083/jcb.128.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villa P, Kaufmann SH, Earnshaw WC. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- 27.Hisahara S, et al. Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. EMBO J. 2000;19:341–348. doi: 10.1093/emboj/19.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viswanath V, et al. Transgenic mice neuronally expressing baculoviral p35 are resistant to diverse types of induced apoptosis, including seizure-associated neurodegeneration. Proc Natl Acad Sci USA. 2000;97:2270–2275. doi: 10.1073/pnas.030365297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Araki T, et al. Conditional expression of anti-apoptotic protein p35 by Cre-mediated DNA recombination in cardiomyocytes from lox-P-p35-transgenic mice. Cell Death Differ. 2000;7:485–492. doi: 10.1038/sj.cdd.4400674. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 31.Tansey FA, Cammer W. A pi form of glutathione-S-transferase is a myelin- and oligodendrocyte-associated enzyme in mouse brain. J Neurochem. 1991;57:95–102. doi: 10.1111/j.1471-4159.1991.tb02104.x. [DOI] [PubMed] [Google Scholar]
- 32.Wang S, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem. 1996;271:20580–20587. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- 33.Namura S, et al. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franklin, K.J.B., and Paxinos, G. 1997. The mouse brain streotaxic coordinates. Academic Press. San Diego, California, USA. ix–xiv.
- 35.Kendler A, Dawson G. Progressive hypoxia inhibits the de novo synthesis of galactosylceramide in cultured oligodendrocytes. J Biol Chem. 1990;265:12259–12266. [PubMed] [Google Scholar]
- 36.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 37.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 38.Kang SJ, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol. 2000;149:613–622. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casaccia-Bonnefil P. Cell death in the oligodendrocyte lineage: a molecular perspective of life/death decisions in development and disease. Glia. 2000;29:124–135. doi: 10.1002/(sici)1098-1136(20000115)29:2<124::aid-glia5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S. Mechanisms mediating caspase activation in cell death. Cell Death Differ. 1999;6:1060–1066. doi: 10.1038/sj.cdd.4400600. [DOI] [PubMed] [Google Scholar]
- 41.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 43.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 44.Irving EA, Yatsushiro K, McCulloch J, Dewar D. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: involvement of free radicals. J Cereb Blood Flow Metab. 1997;17:612–622. doi: 10.1097/00004647-199706000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Han BH, et al. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000;7:38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida T, Waeber C, Huang Z, Moskowitz MA. Induction of nitric oxide synthase activity in rodent brain following middle cerebral artery occlusion. Neurosci Lett. 1995;194:214–218. doi: 10.1016/0304-3940(95)11752-i. [DOI] [PubMed] [Google Scholar]
- 47.Eliasson MJ, et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitrovic B, et al. Nitric oxide induces necrotic but not apoptotic cell death in oligodendrocytes. Neuroscience. 1995;65:531–539. doi: 10.1016/0306-4522(94)00491-m. [DOI] [PubMed] [Google Scholar]
- 49.Schulz JB, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 50.Velier JJ, et al. Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J Neurosci. 1999;19:5932–5941. doi: 10.1523/JNEUROSCI.19-14-05932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia JH, et al. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol. 1993;142:623–635. [PMC free article] [PubMed] [Google Scholar]