

Abstract
Objective
The objective of the present study is to formulate and characterize the properties of complexed glimepiride nanocrystals (GLP) by various techniques at different stages of its development, and to study the effect of PEG 20000 and P90G on particle size reduction and stability of nanocrystals.
Method
Precipitated (GLP-PEG) and complexed NCs (GLP-PEG-P90G) of glimepiride were characterized for particle size, size distribution, zeta potential and stability assessment using photon correlation spectroscopy (PCS). The crystallinity was analyzed using differential scanning calorimetry (DSC) and X-ray powder diffraction spectroscopy (XRPD). The surface morphology and chemical stability were assessed by means of scanning electron microscopy (SEM) and infrared spectroscopy (FTIR).
Results
A formulation with drug–polymer ratio of 1:1 was most ideal in developing stable NCs as it exhibited smaller particle size and high stability. A high zeta potential was observed in all NCs after complexation indicating improved stability. DSC and XRPD studies showed no change in crystallinity after complexation. SEM analysis of complexed NCs showed presence of spherical shape particles (size below 1 μm) with a lipid coat on the surface. Stability studies on optimized formulation (F1) revealed no change in particle size during 3-month period. FTIR studies prove that the chemical identity of GLP was preserved in the samples and the formulation was stable.
Conclusion
Solid-state characterization studies reveal that complexed GLP NCs are promising carriers for drug delivery and they can be safely and effectively used in design of various formulations. Also, PEG 20000 and P90G are excellent polymer and lipid for particle size reduction (nanonization) and stabilization of nanocrystals.
Keywords: Lipid nanocrystals, Precipitation, Surface properties, Crystallinity, Nanonization
1. Introduction
Solid-state characterization studies are widely used to explore the comprehensive structure–functional relationships between the nanoparticle structure and pharmacological properties of a drug. This could help in uncomplicated development, easy adoption and safe administration of a drug.1, 2 Currently different nanoparticulate systems are in progressive research to improve the bioavailability of a drug.3 The performance of these carriers depends on variety of factors like the surface properties, particle size, shape, composition and its stability both in vitro and in vivo.4 The above factors have to be fully established and their effects on drug pharmacokinetics and pharmacodynamics need to be clearly elucidated to assess the safety and competence of various nanoformulations.5
Compared to all nanoparticulates, nanocrystals (NCs) are considered to be the least complex, as it contain 100% drug with no carriers.6 Due to their nanosize, they offer excellent solubility and are considered to solve the issues of poor solubility.7, 8 NCs possess major limitations like crystal growth (aggregation) on contact with fluids or electrolytes, and loss in its functional properties.9 A strategy to overcome the limitations is to alter the surface properties of NCs by attaching ligands to them or by increasing its stealthiness.10 This approach could decrease particle aggregation and improve in vivo stability.11
During the production process, real time monitoring of immediate NCs and assurance test of final product are necessary. This is critical in product development as it assist in evaluating the performance of the NCs in drug delivery.12 Parameters like particle size, surface charge, size distribution, crystallinity and aggregation need to be controlled precisely as they may affect the ADME (absorption, distribution, metabolism and excretion) and toxicity of nanoformulation.13
The objective of the present study is to characterize the properties of Glimepiride NCs (GLP) by various techniques at different stages of its formulation. NCs were developed by precipitation process using PEG 20000 and stabilized (complexed) by means of Phospholipon 90G (P90G). Glimepiride is a weak acidic, second generation oral hypoglycemic agent (BCS Class II drug), with high permeability, low aqueous solubility (∼3.84–02 mg/ml at 37 °C) and poor dissolution rate.14, 15, 16 It has a log P value of 3.2 and a pKa of 6.2. GLP is a drug of choice for long-term therapy for diabetes mellitus and it requires rapid GI absorption to prevent a sudden increase in the blood glucose level after food intake.17 Particle and solid-state characterization studies facilitate in development of a stable formulation with fewer drugs–excipient interactions and it enables to design a formulation with improved therapeutic efficiency.
2. Materials and methods
2.1. Materials
Glimepiride was obtained as a gift sample from S.D Biomed (Malaysia). PEG 20000 and P90G were procured from Sigma Aldrich and GmbH, Germany. Acetone, Tween 80, sodium dodecyl sulfate, polysorbate 80, dichloromethane and methanol were purchased from R & M Chemicals, (Malaysia). Deionized water was obtained from Millipore, MilliQ-Plus. All the other solvents and reagents used were of Anala R grade.
2.2. Methods
GLP was dissolved in dichloromethane (DCM). PEG 20000 was added to the drug solution and stirred (Erla-EMS H7000, Korea) at a temperature not exceeding 60 °C. The drug–polymer solution was injected slowly (1 ml/min) into an aqueous phase containing Tween 80 (2.5% w/v) with mechanical stirring at 450 rpm to precipitate the NCs. The volume (80 ml) of dispersion was adjusted to 100 ml using double distilled water and stirred for 4 h at room temperature. Later, the solution was gently heated (65 °C) with magnetic stirring for 45 min to remove the organic solvent. The contents were centrifuged (Heraeus-Labofuge 200, Germany) at 5000 rpm for 20 min to separate the NCs. The clear supernatant liquid was discarded; the thick viscous dispersion was collected and further redispersed in 15 ml of distilled water and recentrifuged (20,000 rpm) for 10 min to remove the impurities and the residual surfactants. The precipitated NCs were recovered using a vacuum filter (Kontes Ultra ware – 0.2 μm, USA) and dried in a hot air oven (Memmert – UF110, Germany) at 35 °C for 20 min. The procedure was repeated with different drug-carrier ratio to obtain various batches.
2.3. Complexation of GLP-PEG NCs
50 mg of dried NCs were accurately weighed and dispersed in 50 ml of phosphate buffer (pH 7.8) in presence of Tween 80 (0.1% w/v) by gentle stirring for 10 min. P90G (2% w/v) previously solubilized in chloroform was gradually added (3 ml/min) to the dispersion and stirred using a magnetic stirrer at 250 rpm for 30 min at a temperature above its melting point (60 °C). The dispersion was shaken (Daiki Scientific – DK-SI 010, Korea) at 120 rpm for 1 h at 15 °C. Mannitol (5% w/v) was added to the dispersion and shaken for 10 min prior to lyophilization.
2.4. Freeze drying
The milky homogenous dispersion “prepared in 2.3 above” was subjected to freeze drying in a freeze dryer (Thermo scientific, USA), with an inbuilt Pirani 501 microprocessor. The samples were lyophilized at a slow freezing temperature (shelf temperature −40 °C at 6 torr and 10−1 mbar pressure) for 10 h. The lyophilized products were stored in borosilicate glass vials and stored in a dessicator at room temperature until further use.
2.5. Particle characterization
2.5.1. Photon correlation spectroscopy (PCS)
The mean particle size and polydispersity index (PDI) were measured using Malvern Zetasizer Nano ZS (Malvern Instruments, UK). 2 mg of sample was dispersed in 150 ml of deionized water containing 0.25% w/v of Tween 80 and 0.5% w/v of sodium dodecyl sulfate (SDS). The dispersion was sonicated in a bath sonicator (Power sonic 410, Lab Tech, Korea) and left aside for 24 h prior to analysis. 4 μl of each suspension was diluted with 2 ml of deionized water and the samples were pipetted into disposable polystyrene cuvette. The samples were measured for mean particle size and PDI at a fixed angle of 90° at a temperature of 25 °C after 5 runs. A refractive index of 1.590 and 1.300 was used for the drug and solvent respectively.
2.5.2. Zeta potential measurement (ZP)
The zeta potential was measured using Malvern Zetasizer Nano ZS (M3-PALS, Malvern Instruments, UK). Samples were dispersed in deionized water and left for 24 h, and were injected into a clear disposable zeta cell after suitable dilution. The zeta cell was checked for presence of air bubbles and if present was removed by tapping. The average zeta potential (mV) was measured after 3 scans.
2.6. Solid-state characterization
2.6.1. X-ray powder diffraction spectroscopy (XRPD)
XRPD diffractograms of pure GLP, PEG 20000 physical mixtures (1:1) and NCs were recorded in X-ray diffractometer (Bruker AXS D8, Germany) with Anton Paar, TTK 450 temperature attachment, using Si (Li) PSD detector. The samples were placed in a glass sample holder and Cu ka radiation was generated at 30 mA and 40 Kv. The samples were scanned from 3° to 80° with a step size of 0.02°.
2.6.2. Differential scanning calorimetry (DSC)
DSC analysis of pure GLP, PEG 20000, physical mixture of drug and polymer (1:1) and NCs were analyzed in a DSC calorimeter (TA Instruments, Q200, USA), equipped with a liquid nitrogen cooling system. About 5 mg of samples were loaded into an aluminum pan, crimped, sealed and further examined at a scanning rate of 10 °C/min from 15 to 200 °C under nitrogen atmosphere (flow rate 100 ml/min) at room temperature. High purity indium was used to calibrate the heat flow and heat capacity of the instruments.
2.6.3. FTIR analysis
Spectra of pure GLP, PEG 20000, physical mixture (1:1) and NCs were recorded in FT-IR spectrophotometer (Thermo Nicolet, Avatar 370, USA). The samples were compressed into a pellet using KBr and scanned for 4 s at a resolution of 4 cm−1 from 4000 to 400 cm−1.
2.6.4. Scanning electron microscopy analysis (SEM)
Morphological evaluation of NCs was performed using a scanning electron microscope (LEO 1530, Gemini, Germany). The samples were mounted to steel stubs (Jeol – 10 mm Dia × 5 mm) using a double sided adhesive tape and sputtered with a thin layer of Au at 20 mA, under 1 × 10−1 bar vacuum for 10 min using a sputter coater (EM S550X – Electron microscopy sciences) and was operated at an acceleration voltage of 3 kV.
2.7. Stability studies
The optimized formulation (Batch F1) was placed in a clean airtight glass vials and stored at different temperature conditions (room temperature and 40 °C/75% RH) over a period of 3 months. During the storage period, the samples were subjected to particle size measurement and observed for any shift in FT-IR spectra.
3. Results
3.1. Photon correlation spectroscopy
3.1.1. Effect of polymer on particle size reduction
The particle size analysis data of precipitated and complexed NCs are shown in Table 1. The particle size was found to vary with polymer concentration in precipitated NCs. The average particle size of pure GLP was found to be 2066 nm, while that of precipitated NCs (F1–F5) were between 30 and 1920 nm. The mean particle size of complexed NCs was in the range of 210–550 nm and was found to decrease after complexation. The PDI of precipitated and complexed NCs were found to be higher in comparison to pure GLP (0.252) and possessed a broader distribution.
Table 1.
Particle characterization data of GLP NCs.
| Batch | Drug:polymer | Precipitated NCs |
Complexed NCs |
||||
|---|---|---|---|---|---|---|---|
| Z.avg (d nm) | PDI (avg.) | Avg. ZP (mV) | Z.avg (d nm) | PDI (avg.) | Avg. ZP (mV) | ||
| Pure GLP | 1:0 | 2066 ± 0.351 | 0.252 ± 0.25 | −40.2 ± 0.01 | – | – | – |
| F1 | 1:1 | 1650 ± 0.017 | 0.367 ± 0.13 | −40.7 ± 0.01 | 240 ± 0.027 | 0.766 ± 0.15 | −59.0 ± 0.05 |
| F2 | 1:2 | 1920 ± 0.042 | 0.460 ± 0.01 | −42.1 ± 0.12 | 550 ± 0.162 | 0.604 ± 0.04 | −54.4 ± 0.03 |
| F3 | 1:4 | 1593 ± 0.138 | 0.673 ± 0.04 | −41.4 ± 0.04 | 281 ± 0.055 | 1.0 ± 0.07 | −51.7 ± 0.16 |
| F4 | 1:8 | 307 ± 0.252 | 0.743 ± 0.06 | −39.1 ± 0.21 | 210 ± 0.039 | 0.847 ± 0.18 | −51.5 ± 0.21 |
| F5 | 2:1 | 30 ± 0.041 | 0.212 ± 0.32 | −31.6 ± 0.47 | 249 ± 0.185 | 1.0±0.06 | −41.7 ± 0.04 |
± Indicates SD (n = 3).
3.1.2. Effect of zeta potential and stability of NCs
The average zeta potential of pure GLP, precipitated and complexed NCs are compared in Table 1. The zeta potential of pure GLP was found to be −40.2 mV, while that of precipitated NCs were between −31.6 and −42.1 mV. The zeta potential was found to increase (>−40 mV) in all batches after complexation signifying an increase in stability as compared to pure GLP.
3.2. X-ray powder diffraction spectroscopy
The diffraction spectra of pure GLP, PEG 20000, PM (1:1), precipitated and complexed NCs are illustrated in Fig. 1A–C respectively. Numerous sharp and narrow intense peaks were observed at 17.82°, 20.77°, 21.06°, 22.68°, 24.91°, 26.02° and 53.82° in pure GLP spectra proving its high crystallinity. The XRPD spectra of PM showed less intense peaks with decrease in peak area. Numerous low intense, slightly broadened peaks with reduced sharpness and low peak area were observed in the spectra of all NCs. These spectral changes may be attributed to the changes in crystal size of samples. XRPD peak parameters of GLP, precipitated and complexed NCs are given in Table 2.
Fig. 1.
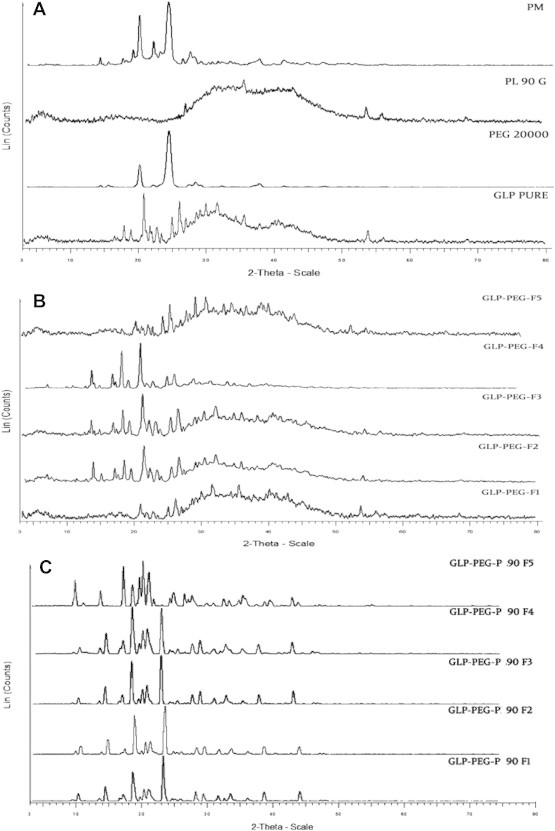
XRPD of pure GLP, PEG 20000, PL90G and PM (1:1) (A), precipitated GLP NCs (F1–F5) (B) and complexed GLP NCs (C) at 2-Theta-scale.
Table 2.
XRPD peak parameters of GLP and formulations.
| Batch | Precipitated NCs |
Complexed NCs |
||||
|---|---|---|---|---|---|---|
| 2θ position | Peak intensity (d) | FWHM (deg) | 2θ position | Peak intensity (d) | FWHM (deg) | |
| Pure GLP | 17.82 | 4.97 | – | – | – | – |
| 20.77 | 4.27 | – | – | – | – | |
| 22.68 | 3.91 | – | – | – | – | |
| 24.91 | 3.57 | – | – | – | – | |
| 26.02 | 3.42 | – | – | – | – | |
| 53.82 | 1.70 | – | – | – | – | |
| F1 | 20.77 | 4.27 | – | 18.92 | 4.66 | 0.42 |
| 22.64 | 3.92 | – | – | – | – | |
| 24.91 | 3.57 | – | 23.55 | 3.77 | 3.31 | |
| 26.00 | 3.42 | – | 28.43 | 3.13 | – | |
| 53.73 | 1.70 | – | – | – | – | |
| F2 | 18.02 | 4.91 | – | 18.90 | 4.66 | 0.36 |
| 20.92 | 4.23 | – | – | – | – | |
| 25.11 | 3.54 | – | 23.66 | 3.75 | 0.43 | |
| 26.23 | 3.39 | – | – | – | – | |
| F3 | 17.92 | 4.94 | – | – | – | – |
| 18.92 | 4.63 | – | 19.01 | 4.66 | 0.34 | |
| 20.80 | 4.25 | – | 20.70 | 4.41 | 0.32 | |
| 22.78 | 3.90 | – | 23.63 | 3.76 | 0.34 | |
| 25.0 | 3.5 | – | – | – | – | |
| 26.10 | 3.41 | – | – | – | – | |
| F4 | 18.04 | 4.91 | 0.28 | 19.06 | 4.65 | 0.43 |
| 20.97 | 4.23 | 0.33 | – | – | – | |
| 22.89 | 3.88 | – | 23.67 | 3.75 | 0.38 | |
| 26.24 | 3.39 | 0.30 | – | – | – | |
| 22.51 | 3.93 | – | – | – | – | |
| F5 | 24.84 | 3.58 | – | 21.60 | 11.24 | 0.41 |
| 25.94 | 3.43 | – | 24.41 | 2.67 | 0.38 | |
FWHM – full width half maximum.
3.3. Differential scanning calorimetry
DSC thermograms of GLP, PEG 20000, PM (1:1), precipitated and complexed NCs are compared in Fig. 2A–C respectively. A sharp endothermic peak at 213.8 °C (ΔH = 43.1 J/g) in the pure GLP thermogram indicated its high crystallinity. An endothermic peak at 65.24 °C in PEG 20000 thermogram revealed it is crystalline in nature (ΔH = 187.6 J/g). A single endothermic peak at 69.9 °C (ΔH = 71.0 J/g) were observed in the thermogram of physical mixture (1:1), due to the fusion of the components and indicated some modifications due to the presence of PEG 20000.
Fig. 2.
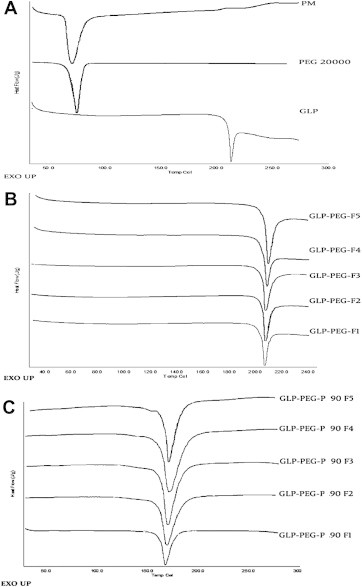
DSC thermograms of pure GLP, PEG 20000 and PM (1:1) (A), precipitated GLP NCs (B) and complexed GLP NCs (C).
The endothermic peak values of all precipitated NCs were between 212.6 and 213.3 °C, and were comparable to the peak values of pure GLP. The ΔH (J/g) of NCs was found to slightly vary within the batches. Incase of complexed NCs, the endothermic peak values were found to be lower and were between 166.7 and 167.7 °C.
3.4. FT-IR analysis
FT-IR spectra of pure GLP, PEG 20000, PM (1:1) and precipitated NCs are compared in Fig. 3A and B respectively. Pure GLP exhibited characteristic bands at 3366 cm−1 and 2946 cm−1 (NH and C–H aromatic stretching), 1683 cm−1 (methylene cyclohexane), 1532 cm−1 and 1443 cm−1 (N O stretching), 1146 cm−1 and 1081 cm−1 (C–O stretching and sulfoxide group respectively).18 The presence of characteristic peaks of GLP in all NC formulations proved the absence of interaction between drug and the polymer.
Fig. 3.
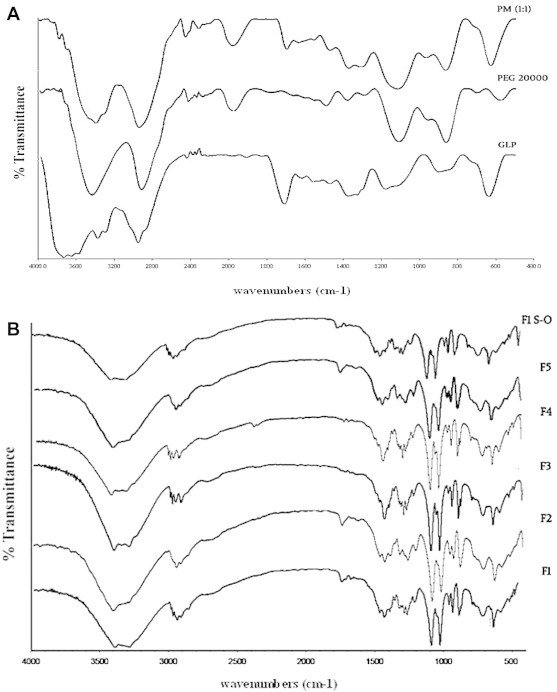
FTIR spectra of pure GLP, PEG 20000 and PM (1:1) (A), precipitated GLP NCs (F1–F5) and optimized formulation (F1–S–O) after 3 months of storage (B).
3.5. Surface characteristic analysis
The SEM images of pure GLP (Fig. 4A) showed numerous irregularly shaped particles of large size (>5 μm). The precipitated NCs were polyhedron in shape and were aggregated before complexation (Fig. 4B). SEM images of complexed NCs show numerous uniform spherically shaped particles below 1000 nm with a lipid coat onto the surface (Fig. 4C). Fig. 4D shows images of complexed NCs (Batch F1) of reduced size after being dispersed in water and then air dried. Fig. 4E shows aggregated NCs before complexation and after microscopical examination. These observations reveal a distinct difference in surface morphology of pure GLP and NCs.
Fig. 4.

SEM images of pure GLP (A), precipitated F1 NCs (B), complexed FI NCs (C), complexed F1 NCs dispersed in water and air dried (D) and aggregated NCs before complexation and after microscopical examination (E).
3.6. Stability testing
The stability data of optimized batch (F1) is given in Table 3. No significant change in particle size was observed during the storage period. FTIR spectra of optimized formulation (batch F1) were found to possess the characteristic peaks of pure GLP at specific positions and are given in Fig. 3B (GLP F1 SO).
Table 3.
Stability data of optimized NCs (Batch F1).
| Stability conditions | Observation (months) |
|||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Room temperature | 240.4 ± 0.027 | 240.1 ± 0.73 | 241.5 ± 0.085 | 241.7 ± 0.161 |
| 40 °C (RH = 75%) | 240.4 ± 0.019 | 240.8 ± 0.17 | 241.5 ± 0.179 | 242.7 ± 0.158 |
Values represent Z.avg (d nm), ± indicates SD (n = 3).
4. Discussion
GLP NCs were formulated by precipitation technique and complexed using P90G (lipid). Particle and solid-state characterization studies were performed on NCs at various stages of its development and the factors were optimized. The effect of PEG 20000 and P90G on particle size reduction, crystallinity and stability of NCs were also assessed.
The average particle size was found to vary (30–1950 nm) with polymer content in precipitated NCs. This could be due to instabilization of particles as a result of crystal growth. The average particle size was found to be lower in complexed NCs, possibly due to less aggregation of particles.19 The increase in PDI before and after complexation could be as a result of application of less energy during the process of milling. All the batches were polydisperse in nature after complexation. It can be inferred that batch F1 was found to be the optimum batch in terms of lowest particle size and stability, and a drug–polymer ratio of 1:1 was most suitable to develop stable NCs.
The average zeta values were found to increase after complexation. This is because of formation of a lipid coat on the surface of NCs. Lipid (P90G) imparts a negative charge, makes the drug particles to get dissociated and provides maximum stability to NCs. These findings also suggest that a repulsive force is associated with the molecules which reduce the particle agglomeration and it confirms the stability of the formulation.20
XRPD studies show no change in crystallinity in precipitated and complexed NCs. This was evident from relative intensity values (d-value), as it decreased initially and later became constant. A shift in diffraction pattern was noticed at a longer end in 2θ scale, indicating the change in crystal size due to PEG 20000.21 It was observed that all NCs exhibited a similar characteristic diffraction pattern as that of pure GLP at 2θ positions revealing the absence of interaction between drug and polymer.
DSC studies confirmed that crystallinity was maintained in all complexed NCs but with a slight degree of disorientation in their crystal structure. This could be due to the change in ΔH (J/g) values. The variation in peak and ΔH value of NCs supports the above suggestions.
The surface morphology of precipitated and complexed NCs was easily distinguishable by SEM analysis. The complexed NCs were spherical in nature and the particle size was below 1 μm. Dispersion of these NCs in water exhibited less particle aggregation due to the presence of lipids on the surface of NCs. Stability assessment on optimized formulation (Batch F1) by FTIR analysis and particle size measurement showed no change in characteristic peak and particle size. Thus the chemical identity of GLP was preserved in the samples and the formulation was stable during the period.
The particle analysis report and solid-state characterization studies reveals that in vitro stability of NCs were improved and the chemical identity of the drug was not altered during the process of complexation. This confirms that the formulation is stable and complexed NCs can be effectively used in design of various formulations. However, for assessing the complete safety and efficacy in drug delivery in vivo studies ought to be performed, as there is a possibility for the NCs to experience a change in surface properties in presence of body fluids. This could be assessed by performing permeability studies and by analyzing the biodistribution properties of NCs.
5. Conclusion
Particle analysis and solid-state characterization studies portray the drug properties, its stability, and compatibility of drug and excipients. These studies are essential at various stages of development of lipid nanocrystals, as it would assist in design of an uncomplicated formulation. These complexed NCs are promising carriers for development of various formulations in treatment of diabetes. To conclude, PEG 20000 is an excellent polymer for nanonization process and P90G could be effectively used for complexation as it can alter the surface properties, maintain crystallinity and improve in vitro stability.
Conflicts of interest
All authors have none to declare.
Acknowledgments
The authors would like to thank Faculty of Pharmacy and Faculty of Applied Sciences, AIMST University, Malaysia and SAIF-STIC, Cochin, India for their laboratory and instrumentation analysis support.
References
- 1.Jens U.A., Rainer H.M. Nanocrystal technology drug delivery and clinical applications. Int J Nanomed. 2008;3:295–299. doi: 10.2147/ijn.s595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huabing C., Chalermchai K., Xiangliang Y., Xueling C., Jinming G. Nanonization strategies for poorly water soluble drugs. Drug Discov Today. 2011;16:354–360. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Marcato P.D., Duran N. New aspects of nanopharmaceutical delivery systems. J Nanosci Nanotech. 2008;8:2216–2229. doi: 10.1166/jnn.2008.274. [DOI] [PubMed] [Google Scholar]
- 4.Rainer H.M., Sven G., Cornelia M.K. State of art nanocrystals – special features, production, nanotoxicity aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78:1–9. doi: 10.1016/j.ejpb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Banu S.Z., Nakissa S. Regulatory perspective on the importance of ADME assessment of nanoscale material containing drugs. Adv Drug Deliver Rev. 2009;6:422–427. doi: 10.1016/j.addr.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Mohanraj V.J., Chen Y. Nanoparticles – a review. Trop J Pharm Res. 2006;5:561–573. [Google Scholar]
- 7.Tom A., Max Z. Formulation technologies to overcome poor drug like properties. Drug Discov Today. 2012;9:e71–e72. doi: 10.1016/j.ddtec.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Lai F., Sinico C., Ennas G., Marogiu F., Marogiu G., Fadda A.M. Diclofenac nanosuspensions: influence of preparation procedure and crystal form on drug dissolution behaviour. Int J Pharm. 2009;373:124–132. doi: 10.1016/j.ijpharm.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 9.Faris N.B., Müller R.H. Nanocrystals of poorly soluble drugs for oral administration. Topics of PhD thesis and details on technologies, products, IP. New Drugs. 2002;2:20–21. [Google Scholar]
- 10.Jaime S., Antoine G., Rainer H.M., Jan P.M. Nanocrystals: comparison of the size reduction effectiveness of a novel combinative method with conventional top-down approaches. Eur J Pharm Biopharm. 2012;81:82–90. doi: 10.1016/j.ejpb.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 11.Sandrine D., Lucie S., Jean L.C. Physico-chemical parameters that govern nanoparticles fate also dictate rules for their molecular evolution. Adv Drug Deliver Rev. 2012;64:179–189. doi: 10.1016/j.addr.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 12.Zeng N., Gao X., Hu Q., et al. Lipid-based liquid crystalline nanoparticles as oral drug delivery vehicles for poorly water-soluble drugs: cellular interaction and in vivo absorption. Int J Nanomed. 2012;7:3703–3718. doi: 10.2147/IJN.S32599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müller R.H., Jacobs C., Kayser O. Nanosuspensions as particulate drug formulations in therapy, rationale for development and what we can expect for the future. Adv Drug Deliver Rev. 2001;47:3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 14.Shelesh J., Swarnalata S. Type 2 diabetes mellitus – Its global prevalence and therapeutic strategies. Diabetes Metab Syndr Clini Res Rev. 2010;4:48–56. [Google Scholar]
- 15.Vyas M., Galani V.J. In vivo and in vitro drug interactions study of glimepiride and atorvastatin and rosuvastatin. J Young Pharmacist. 2010;2:196–200. doi: 10.4103/0975-1483.63169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sajeev K.B., Saraswathi R., Dilip C., Venkates K., Jha S.K. Formulation and evaluation of controlled release glimepiride osmotic systems. Int J Pharm Res. 2011;3:79–84. [Google Scholar]
- 17.Rhoban T. The efficacy and safety of glimepiride in the management of Type II diabetes in Muslim patients during Ramadan. Diabetes Care. 2005;28:421–422. doi: 10.2337/diacare.28.2.421. [DOI] [PubMed] [Google Scholar]
- 18.Baliar S., Biswal S., Sahoo J., Murthy P.N. Physiochemical properties of glimepiride in solid dispersions with polyethylene glycol 20000. Int J Pharm Sci Nano. 2009;2:537–543. [Google Scholar]
- 19.Marie G., Angelica V., Robert G., Florence D. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69:1–9. doi: 10.1016/j.ejpb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Jane W., Sabine G., Heather O., Lakshmy N., Thomas G., Philip W.G. Physicochemical stability of phospholipid-dispersed suspensions of crystalline itraconazole. Eur J Pharm Biopharm. 2008;69:1104–1113. doi: 10.1016/j.ejpb.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 21.Anjan K., Mahapatra Dissolution enhancement and physiochemical characterization of fenofibrate in solid dispersion with polyethylene glycol 40000 and 20000. Int J Pharm Sci Tech. 2010;4:21–31. [Google Scholar]