

Abstract
Background
Crohn disease (CD) and ulcerative colitis (UC) are common forms of inflammatory bowel diseases (IBD). Monozygotic (MZ) twin discordance rates and epidemiologic data implicate that environmental changes and epigenetic factors may play a pathogenic role in IBD. DNA methylation (the methylation of cytosines within CpG dinucleotides) is an epigenetic modification, which can respond to environmental influences. We investigated whether DNA methylation might be connected with IBD in peripheral blood leukocyte (PBL) DNA by utilizing genome-wide microarrays.
Methods
Two different high-throughput microarray based methods for genome wide DNA methylation analysis were employed. First, DNA isolated from MZ twin pairs concordant (CD: 4; UC: 3) and discordant (CD: 4; UC: 7) for IBD was interrogated by a custom made methylation specific amplification microarray (MSAM). Second, the recently developed Illumina Infinium HumanMethylation450 BeadChip arrays were used on 48 samples of PBL DNA from discordant MZ twin pairs (CD:3; UC:3) and treatment naive pediatric cases of IBD (CD:14; UC:8), as well as controls (n=14). The microarrays were validated with bisulfite pyrosequencing.
Results
The Methylation BeadChip approach identified a single DNA methylation association of IBD at TEPP (testis, prostate and placenta-expressed protein) when DNA isolated selectively from peripheral blood mononuclear cells was analyzed (8.6% increase in methylation between CD and control, FDR=0.0065).
Conclusions
Microarray interrogation of IBD dependent DNA methylation from PBLs appears to have limited ability to detect significant disease associations. More detailed and/or selective approaches may be useful for the elucidation of connections between the DNA methylome and IBD in the future.
Keywords: inflammatory bowel disease, DNA methylation, peripheral blood, twin, TEPP
INTRODUCTION
The inflammatory bowel diseases (IBD), including Crohn disease (CD) and ulcerative colitis (UC), are common human disorders with autoimmune characteristics that affect more than four million people worldwide, about a third of whom reside in the United States.1 The disorders are associated with high morbidity leading to a significant healthcare burden.2 The incidence of IBD peaks in young adulthood and its prevalence is on the rise especially in the pediatric population.3–6 Recent observations implicate that the increase in the number of pediatric patients with IBD is a consequence of the overall amplified incidence rather than a shift towards disease onset at younger age.7 Dietary and environmental factors of the industrialized world have been proposed as etiologic culprits for these shifting demographics by imposing critical changes during prenatal and/or pediatric development in the key physiologic components of IBD pathogenesis.8–11
One group of molecular processes that has been shown to dynamically respond to environmental/nutritional influences is designated as epigenetic. Epigenetic mechanisms mediate mitotically heritable changes in gene expression that are not associated with DNA sequence variation.12 The most stable epigenetic alteration is the methylation of cytosines at CpG dinucleotides (DNA methylation). This molecular modification at gene promoters generally correlates with transcriptional down-regulation. DNA methylation is critical for development and differentiation.13 It is catalyzed by DNA methyltransferases and is ultimately dependent upon dietary substrates and co-factors.14 Modification of the prenatal environment can shift levels of DNA methylation at certain genomic loci and can associate with permanent phenotype changes in insects15 and mammals as well.16 Therefore, it has been proposed that epigenetic changes, such as DNA methylation may be an important factor in the developmental origins of common human diseases,17, 18 including IBD.19 The high rate of discordance for IBD within monozygotic (MZ) twin pairs supports the possible role of epigenetic processes in their etiologies.20, 21 Discordant MZ twins have been recognized to provide an opportunity for unraveling the epigenetic aspects of disease etiology in autoimmune diseases, such as systemic lupus erythematosus (SLE).22 Peripheral blood leukocyte (PBL) DNA samples from MZ twins discordant and concordant for SLE have been recently used successfully to identify DNA methylation associations of disease.23 Similar high-throughput observations on PBL DNA methylation are lacking in IBD.
In this study we set out to identify PBL DNA methylation associations of IBD by two independent methods of genome wide microarray interrogation. The study was based on samples collected through the Danish Twin Registry,24, 25 the gene bank of the University of Pecs, Hungary,26 the pediatric gastroenterology tissue bank of Charles University, Prague, Czech Republic, and tissue bank of the Pediatric Inflammatory Bowel Disease Consortium Registry of the Baylor College of Medicine.
MATERIALS AND METHODS
PBL DNA Samples
Peripheral blood leukocyte (PBL) DNA was isolated by different methodologies depending on the tissue bank protocols at each site. The prediction of this work was that uniform DNA methylation changes associate with IBD affecting each human cell lineage equally. Therefore, we did not control for DNA isolation methodology during our initial approach (see Results section on TEPP).
De-identified PBL DNA samples were obtained through the Danish Twin Registry,24, 25 the gene bank of the University of Pecs, Hungary,26 the pediatric gastroenterology tissue bank of Charles University, Prague, Czech Republic, and the Pediatric Inflammatory Bowel Disease Consortium Registry of the Baylor College of Medicine, which were established in agreement with local and federal regulations. DNA isolation from the twin samples was performed with Maxwell® 16 system from Promega (Madison, WI) with the Blood DNA purification kit. In order to increase the concentration of the extracted DNA, Millipore microcon filters were used (http://www.millipore.com/userguides/tech1/99394). PBL DNA at the University of Pecs was extracted with standard salting out methods. For the PBL DNA of the pediatric cohorts (used on the Illumina arrays, other than the twins) either Gentra Puregene (Qiagen, Valencia, CA) for whole blood DNA isolation was used according to manufacturers recommendation on the samples from the Charles University, or the QuiAmp DNA mini kit (Qiagen, Valencia, CA) was utilized on isolated peripheral blood mononuclear cells (PBMCs) form the cohort of the Baylor College of Medicine. BD Vacutainer® CPT™ Cell Preparation Tube with Sodium Citrate was used in the later case to isolate peipheral blood mononuclear cells (PBMCs) from PBL. Only age, gender, and disease status were collected with the samples (except for the pediatric cohort where pharmacologic therapy was also recorded), the details of which are shown in Table 1. Adult controls were healthy, free from any chronic diseases. Pediatric controls included children who underwent colonoscopic evaluation for diagnoses of hematochezia, diarrhea, or abdominal pain, but whose endoscopy was grossly and histologically normal.
Table 1.
Summary of PBL DNA samples.
| Cohorts | Diagnosis | Age (years) | Gender | ||
|---|---|---|---|---|---|
| Mean | Median | Min to Max | (M/F) | ||
| Danish Twin Registry (n=number of twin pairs) |
CD discordant (n=4) UC discordant (n=7) CD concordant (n=4) UC concordant (n=3) |
44.75 45.71 46.5 41 |
43.5 49 46 43 |
43–49 33–56 44–50 36–44 |
2/2 2/5 2/2 0/3 |
| Hungarian cohort (n=80) |
C-CD C-UC CD UC |
36.85 37.6 36.85 36.85 |
37 37.5 37 37 |
24–52 22–56 24–52 24–52 |
10/10 10/10 10/10 10/10 |
| Illumina cohort (n=36) |
H CD UC |
12.14 14.43 12.88 |
13.25 15.5 13.5 |
3.5–17.5 9–17 9–17 |
6/8 8/6 5/3 |
| BCM - Crohn's disease cohort (PBMC DNA) (n=55) |
H CD new CD Rx |
11.94 14.50 14.55 |
13 15.05 14.9 |
0.1–17.5 9–18 9–18 |
6/15 13/11 4/6 |
BCM: Baylor College of Medicine; PBMC: peripheral blood mononuclear cell
Methylation Specific Amplification Microarray (MSAM)
MSAM was carried out as previously described.18, 27 MSAM is based upon serial digestion of the DNA with methylation sensitive and insensitive restriction endonuclease isochizomers: SmaI and XmaI, respectively. The endonuclease digestion is followed by PCR mediated amplification and processing towards differentially labelled microarray hybridization (co-hybridization) to compare the relative difference in DNA methylation between two samples at a large number of SmaI/XmaI genomic intervals. 18 independent MZ twin-twin co-hybridizations (see Table 1) were performed. We designed a custom microarray (Agilent Technologies, Santa Clara, CA). SmaI/XmaI intervals between 100-2,000bp, along with negative control intervals in the range 10-15kbp, were identified based on the human genome assembly (NCBI36/UCSC hg18) excluding chromosome X and Y. Intervals containing single nucleotide polymorphisms, based on dbSNP 129 (http://www.ncbi.nlm.nih.gov/projects/SNP/), which disrupted or introduced SmaI/XmaI cut sites were removed from consideration. The intervals were uploaded to Agilent eArray (https://earray.chem.agilent.com/earray/) in order to identify high density probes contained within the intervals. eArray filtering for probes in known copy number variation regions was also applied. The final array design consisted of 99,071 probes interrogating 41,079 (100-2,000bp length) intervals and 100 probes interrogating 10-15kbp negative controls in addition to the standard Agilent array controls. Therefore, the custom array covered 45.31% (41,079 out of 90,657 in the human genome) of the potentially informative 100-2,000bp SmaI/XmaI intervals. To exclude falsely significant findings arising from the massive numbers of loci interrogated, multiple testing correction of the p-values was performed using Benjamini and Hochberg’s method28 (false discovery rate: FDR). Based on our previous experience,18 we first examined inter-group differences exceeding 1.8 fold with p<0.0002 for significant association. A secondary criteria of significance was determined at FDR<0.5 at intervals where inter-group differences at select SmaI/XmaI intervals were >1.2 fold.
Infinium Methylation Assay Microarrays
48 PBL DNA (3 MZ twin discordant for CD, 3 MZ twin discordant for UC; and pediatric samples: 14 control, 14 CD and 8 UC collected at the time of diagnostic endoscopy in untreated patients) samples following quality control with PicoGreen (http://probes.invitrogen.com/media/pis/mp07581.pdf) were processed by Infinium HumanMethylation450 BeadChip Kits (Illumina San Diego, CA, USA; http://www.illumina.com/products/methylation_450_beadchip_kits.ilmn) according to the manufacturer’s recommendations through automated processes in the Core Laboratory for Translational Genomics of the Baylor College of Medicine. Arrays were imaged with BeadArray Reader using standard recommended Illumina scanner settings. GenomeStudio software version 2010.3.0.30128 was used to generate beta values normalized to internal control probes. Internal controls determined the array processing to be of good quality. Only the 482,421 CpG probes on the array were used for subsequent analysis. The R LIMMA package29 was employed to compare beta values between normal controls and CD or UC affected individuals. Limma fitted a linear model to beta values for each probe in the compared samples and then calculated t-statistic using an empirical Bayesian model that moderates the standard errors across probes. P-values were calculated from the moderated t-statistics and FDR was determined to identify differentially methylated probes. The criterion for significant association was set at FDR<0.2. The raw data of the microarrays was uploaded to Gene Expression Omnibus (GEO; Series GSE32148) and is accessible at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=pjszvekkmmaeyzu&acc=GSE32148.
Bisulfite-pyrosequencing
Analytical validation using the same cohort as the array studies was performed by bisulfite-pyrosequencing at select loci. This analysis was followed by validation of differential methylation status using expanded or independent cohorts (see PBL DNA Samples paragraph above). PBL DNA was bisulfite converted with EZ DNA Methylation-Gold Kit (D5006, Zymo Research, Orange, CA, USA). DNA was amplified with traditional PCR methodology following bisulfite conversion and including a biotinylated primer in the sets (Supplementary Table 1). A quantitative bisulfite pyrosequencing protocol was used for all methylation analyses with the utilization of the Pyro Q CpG program (QIAGEN GmbH, QIAGEN Strasse 1. 40724 Hilden, Germany). Methylation measurements at (ANO1 associated SmaI/XmaI: chr11:69597807-69598085, EHD1 associated SmaI/XmaI: chr11:64377161-64377539) or in the vicinity (MGRN1 associated SmaI/XmaI: chr16:4654296-4655675) of both of the SmaI/XmaI sites of the gene associated intervals were performed in the case of the validation measurements of the MSAMs. An algorithm was used to calculate and compare methylation ratios at these intervals.27 Illumina array validation was performed at three candidate loci: FAM53B associated CpG site (chr10:126360669-126360669); SLC6A9 associated CpG site (chr1:44466601-44466601); TEPP associated CpG site (chr16: 58019367-58020365). Only the results of assays that passed quality control were used. The criterion for significant association for bisulfite pyrosequencing results was p<0.05 with non-parametric U test. The primer sequences for the bisulfite pyrosequencing measurements are described in Supplementary Table 1.
Statistical analyses
Statistical analysis of the Illumina arrays was described above. Pearson’s correlation analysis was used in the correlation studies. The non-parametric Mann-Whitney U test was employed in the intergroup comparisons. Significance was set at p<0.05.
RESULTS
MSAM in Discordant MZ Twins Found No Significant IBD Association
None of the discordant MZ twin comparisons yielded average disease to healthy ratio comparisons that met our criteria of significance (see Materials and Methods). Nevertheless, we wished to test whether relaxing the selection criteria may still provide significant findings. The selection criteria were relaxed by different approaches and top candidates were selected for bisulfite pyrosequencing. MRGN1 showed over 1.4 average fold difference between disease and healthy (p<0.05); ANO1 showed 2 out of 4 comparisons exceeding 1.95 fold difference between disease and healthy; and EHD1 showed 3 out of 4 comparisons exceeding 1.5 fold difference between disease and healthy. Pyrosequencing failed to confirm the significant DNA methylation differences between MZ twins discordant for IBD indicated by the arrays (correlation at MGRN: r=0.28, p=0.72, at ANO1: r= −0.68, p=0.32; and at EHD1: r=0.3, p=0.7, respectively). Therefore, MSAM did not yield significant results.
With the assumption that disease specific epigenetic differences between MZ twins may be suppressed,30 and the MSAM might have been more sensitive than our validation method, we measured the methylation of candidate loci in an independent cohort of adults with IBD and healthy controls. None of our measurements showed disease specific DNA methylation in the PBL DNA samples from this independent cohort (see Supplementary Figure 1 as an example).
Infinium Methylation Assay Microarrays Did Not Show Significant IBD Association
We first examined the genetically unrelated pediatric samples on the Illumina arrays. After correcting for multiple testing, none of the CD to control comparisons was significant (FDR>0.2 for all). Similar relaxation of the false discovery rate was done with success when assessing PBL DNA methylation associations of SLE.23
We found a total of 6 CpG sites in the human genome where UC to control comparisons resulted in over 4% mean methylation difference (this value approaches the reliability of the bisulfite pyrosequencing validation method19) with FDR<0.2 (Table 2) in the pediatric groups of the Illumina arrays. Bisulfite pyrosequencing validation at select loci from this UC specific group gave excellent correlation with the microarrays (Figure 1). However, group comparisons between UC and control were not significant (Supplementary Figure 2). This later finding is likely the result of the low (less than 15% difference in 5 out of the 6 candidates) inter-group methylation differences detected by the microarrays in addition to relatively large intra-group variation that was predicted by the overall high FDRs.
Table 2.
Genomic loci (coordinates) of UC associated CpG candidates where disease specific DNA methylation occurred according to the Illumina arrays from the pediatric cohort (loci meeting criteria for >4% mean methylation difference between UC and control: C; and Benjamini-Hochberg FDR<0.2). Numbers represent percent methylation. Gene associations are based on the Infinium HumanMethylation450 v1.1 manifest supplied by Illumina.
| Coordinates | Associated gene | C | UC | UC-C | FDR |
|---|---|---|---|---|---|
| chr13:20771319-20771319 | 56.19 | 27.33 | −28.86 | 0.07 | |
| chr20:60915009-60915009 | LAMA5 | 65.17 | 51.74 | −13.43 | 0.16 |
| chr6:31803832-31803832 | C6orf48,SNORD52 | 41.81 | 46.46 | 4.65 | 0.08 |
| chr2:34719565-34719565 | 68.06 | 73.05 | 4.99 | 0.19 | |
| chr1:44466601-44466601 | SLC6A9 | 75.83 | 81.34 | 5.51 | 0.16 |
| chr10:126360669-126360669 | FAM53B | 42.40 | 55.00 | 12.60 | 0.07 |
Figure 1.
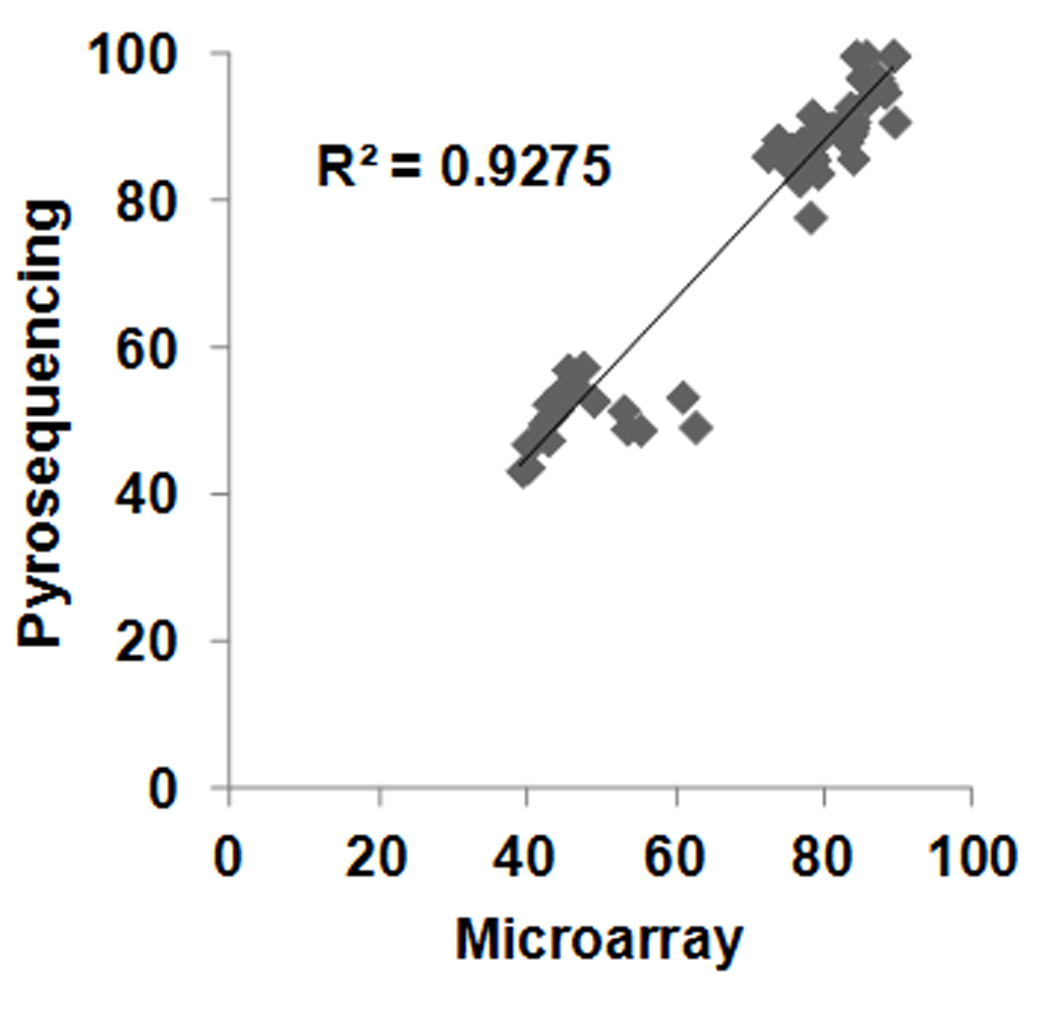
Scatter plot of methylation levels obtained by Infinium HumanMethylation450 Microarrays (Microarray) versus bisulfite pyrosequencing at the SCL6A9, FAM53B, and TEPP associated CpG sites. Significant correlation was observed. r=0.963; p<<0.00001
Selective PBMC DNA Analysis Identifies TEPP CpG Methylation Association with IBD
The DNA samples of this study originated from different tissue banks utilizing various extraction methods. To address the possibility that the sample variation originating from technical and geographic disparity masked disease specific DNA methylation, we performed a selective bioinformatic analysis of the samples from the Baylor College of Medicine cohort. In this case, the DNA was extracted from PBMCs excluding neutrophil granulocytes with uniform methodology. This analysis showed one single CD specific DNA methylation change associated with TEPP with FDR<0.2 (chr16:58019866-58019866, CD-C average methylation difference: 8.6%; FDR=0.0065). Bisulfite pyrosequencing validated the results (included in Figure 1) and showed a significant (p=0.011) CD associated increase in methylation at this locus. However, this increase was not specific to CD, but was present in untreated UC patients as well (p=0.013; Figure 2A). When the control and treatment naïve CD groups were expanded, CpG methylation at this locus showed an even more significant association with CD (p<0.0001; Figure 2B). In the meantime, the amplified DNA methylation in treatment naïve CD was absent in treated children (Figure 2C).
Figure 2.
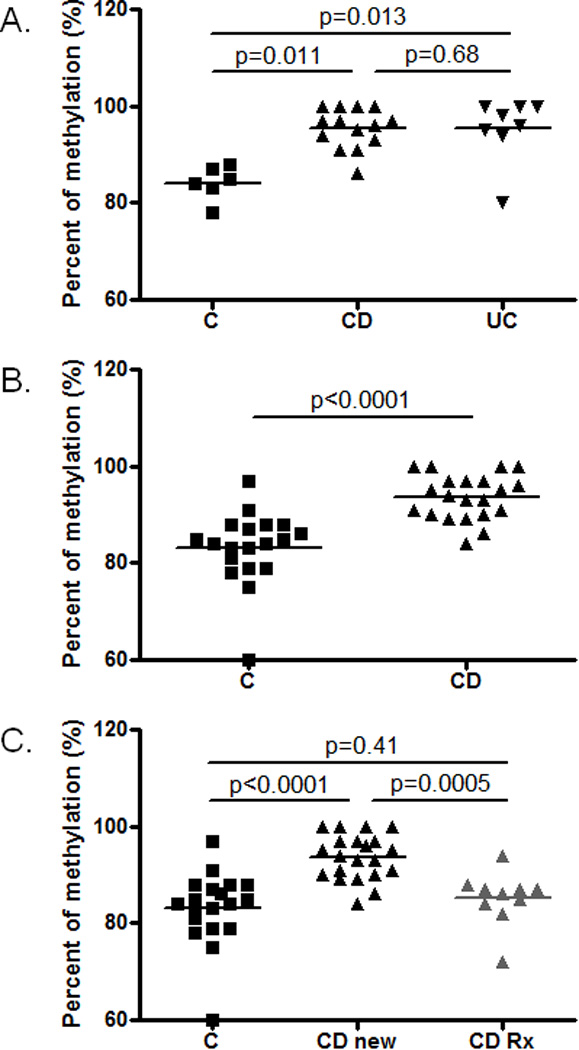
Methylation levels at the TEPP associated CpG site in the select pediatric PBMC cohort of the study (see Table 2). A. Bisulfite pyrosequencing results from the PBMC samples of the Infinium HumanMethylation450 Microarrays. The significant increase in methylation was confirmed by bisulfite pyrosequencing in the CD and UC cohorts compared to controls (n=6–14). B. Extension of the measurements to larger groups of PBMC DNA from controls and treatment naïve CD patients further supported the validity of our findings (n=19–22). C. The increased level of methylation in treatment naïve (CD) patients was lost when compared to CD patients receiving therapy (CD-rx; 3 biologic monotherapy; 2 biologic+immunomodulator, 2 mesalamine alone, 1 steroid alone, 1 immunomodulator alone, 1 steroid+immunomodulator) (n=10–22).
DISCUSSION
Identification of disease specific epigenetic (including DNA methylation) changes is a very extensive area of current biomedical research. Epigenetic associations could support the unraveling of disease etiology, provide novel biomarkers, and aid the development of potential preventative and/or therapeutic measures for disorders, such as IBD.31, 32 Based on MZ twin discordance rates, the relative genetic contribution to CD and UC is low33 supporting the importance of non-genetic (including epigenetic and/or other molecular processes within the large biological components of gastrointestinal disease10) processes in their etiology as discussed in the introduction.
Peripheral blood (PB) is the most commonly obtained and stored biological specimen and PBL DNA constitutes the vast majority of gene-bank samples. Therefore, identification of disease specific molecular characteristics from PB or PBL is highly feasible. Recent findings indicate that PB micro-RNAs (small RNA molecules modulating gene expression through epigenetic mechanisms) may serve as useful bio-markers of CD.36
In case a DNA methylation modification at a disease susceptibility locus occurs before gastrulation, it can be detected in all tissues of the body as has been observed at metastable epialleles in mice.16 Therefore, if such loci exist in humans for certain diseases, then the disease specific DNA methylation change could be detected from any type of cell, including leukocytes obtained from peripheral blood specimens. Our approach to DNA methylation associations of IBD in PBL DNA was based on this prediction.
We first used the MSAM technique on MZ twin samples, but could not identify any significant IBD association. These results indicated that either the MZ twin based approach, or our MSAM technique is insufficient to identify IBD specific DNA methylation from PBL DNA, or that uniform epigenetic changes may not exist in association with the disease group in peripheral white blood cells. One limitation of the MSAM technique is that it does not provide information on the actual levels of DNA methylation, just the relative difference in methylation between two different (disease-healthy, for example) samples at the same genomic region (SmaI/XmaI interval in this case). Therefore, at genomic loci where DNA methylation is low, even subtle changes may show up as highly significant (for example 1% methylation in healthy individuals and 3% methylation in disease would result in a 3 fold difference on the array). Subtle methylation differences are of questionable physiologic relevance and are very difficult to validate with sequencing methodologies. Therefore, we turned to the Infinum methylation assay based Infinium HumanMethylation450 BeadChip Kits.
Secondary to our negative findings with the MSAM approach in the twin cohort, only a subset of MZ twins discordant for CD and UC were examined by the Illumina arrays. One reason for this approach was the prediction based on healthy twin observations37 that a twin with a disease in discordant MZ pairs may be epigenetically less different from the healthy twin sibling at critical pathogenic loci, as compared to unrelated healthy individuals. However, at least according to one publication, SLE associated DNA methylation was not influenced by relatedness (i.e. disease specific DNA methylation difference was as high between unrelated patients and controls, as between affected twins and healthy counterparts in MZ pairs). Nevertheless, we decided to first study unrelated individuals with IBD where epigenetic differences could be higher than in MZ twins. Therefore, PBL DNA samples collected at diagnosis from untreated pediatric cases of IBD were tested and compared to controls. The untreated pediatric cases eliminated the possible confounding factor of therapeutic interventions inducing non-specific DNA methylation changes.38 However, because the patients were unrelated their comparisons carried the potential for genetic variation induced DNA methylation differences being identified. Therefore, our plan was to overlap candidate loci determined in the pediatric cohort with loci identified through the discordant MZ twin comparisons.
We could not identify IBD specific PBL DNA methylation association in unrelated children where even IBD linked genetic variation induced DNA methylation changes could have been present. Consequently, we did not proceed with overlapping the pediatric cohort results with the independent adult MZ twin samples on the Illumina arrays.
Studies with high-throughput methodologies applied on clinical samples frequently bare limitations. Some of the confounding factors of our analyses may have been: the low power secondary to small sample sizes, the limited coverage and/or resolution of the microarrays employed, and the clinical heterogeneity of both the control and IBD groups (non-healthy control children, healthy adults, twins, adult treated IBD patients likely with different disease sub-types) studied. However, as stated earlier, the original hypothesis of our work was that uniform DNA methylation changes may exist in humans, which predispose to the development of IBD independently from disease sub-types within CD and UC.
Our largely negative findings with MSAMs and Infinum Methylation Assays implicate that microarray profiling of DNA methylation from PBL is less likely to yield positive results in IBD. Sub-fractionation of PBL into lymphocyte subsets and performing microarray comparisons with such DNA samples may be valuable in the future since DNA methylation is cell type specific. The one positive IBD association at TEPP when PBMCs were selectively analyzed would support this conclusion. Indeed, interferon-gamma (IFNG) methylation in isolated peripheral T cells correlated with IBD, for example.39 However, IFNG methylation difference between IBD and controls was less than 4% in this case, which would have been likely missed by the resolution of the currently available high-throughput interrogation methods.
TEPP encodes for testis, prostate and placenta-expressed protein with uncertain function.40 It is poorly expressed in bone marrow, whole blood, thymus, monocytes, and T cell subsets, as well as in the appendix (expression atlas 2 data from U133A and GNF1H chips) implicating that the physiologic relevance of the small average DNA methylation increase (from 83% in controls to 94% in CD, Figure 2A) detected from PBMCs is questionable. One explanation for this finding is that PBMC subset modifications (altered ratios of macrophages, T and B cell subsets, etc. with differing TEPP methylation levels) characteristic for IBD at onset are lost upon treatment. However, there is no flow cytometric data available for our patients, which could specifically address this prediction. Nevertheless, we can conclude that IBD associated DNA methylation increase in PBMC DNA at TEPP, a gene poorly expressed in peripheral blood leukocytes, is small and is not disease specific. Additionally, only treatment naïve patients showed this difference. Therefore, the functional relevance and the potential diagnostic value of this finding appear limited.
Numerous observations on links between gene and cell type specific DNA methylation and IBD have been made, where either the low fraction of the specific cell type (regulatory T cells, for example41) or the unique location of the DNA methylation correlate (such as in the interleukin 17A: IL17A42) provide limitations to detectability from PBL DNA by microarray methodology. Increasing the depth of interrogation with methods such as genome wide direct sequencing43 may also increase the chances of determining DNA methylation associations of IBD in PBL, and PBL subtypes. Unfortunately, the financial aspects of such interrogation in reliable sample sizes provide significant constraint to such studies currently.
Intestinal biopsies may offer more readily identifiable DNA methylation correlates of IBD through genome wide microarray analysis. Both targeted gene methylation44 and DNA methylation microarray45 assessments have detected numerous colonic mucosal associates of inflammation in IBD. Techniques with higher resolution may offer even more insight into the intestinal mucosal DNA methylation aspects of the disease group.
In summary, our largely negative study can significantly guide epigenetic investigations in IBD regarding technical planning and tissue selection. Our findings on TEPP associated DNA methylation may also prove of significance in the future.
Supplementary Material
Acknowledgements
The authors would like to thank the patients, the clinical colleagues contributing to the collection of tissue, and specifically Stefi Lee, Alfred Balasa, Harry Siegele, Sahna Reddy and Sabina Mir for their contribution to this work.
R.K. was supported in part by the Broad Medical Research Program, the Broad Foundation (IBD-0252), the Crohn’s and Colitis Foundation of America-Children’s Digestive Health and Nutrition Foundation/North American Society of Pediatric Gastroenterology Hepatology and Nutrition (CCFA Ref #2426); the Child Health Research Career Development Agency of the Baylor College of Medicine (NIH # 5K12 HD041648); and a Public Health Service grant DK56338, funding the Texas Medical Center Digestive Diseases Center
REFERENCES
- 1.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 2.Park KT, Bass D. Inflammatory bowel disease-attributable costs and cost-effective strategies in the United States: A review. Inflamm Bowel Dis. 2011;17:1603–1609. doi: 10.1002/ibd.21488. [DOI] [PubMed] [Google Scholar]
- 3.Benchimol EI, Guttmann A, Griffiths AM, Rabeneck L, Mack DR, Brill H, Howard J, Guan J, To T. Increasing incidence of paediatric inflammatory bowel disease in Ontario, Canada: evidence from health administrative data. Gut. 2009;58:1490–1497. doi: 10.1136/gut.2009.188383. [DOI] [PubMed] [Google Scholar]
- 4.Abramson O, Durant M, Mow W, Finley A, Kodali P, Wong A, Tavares V, McCroskey E, Liu L, Lewis JD, Allison JE, Flowers N, Hutfless S, Velayos FS, Perry GS, Cannon R, Herrinton LJ. Incidence, Prevalence, and Time Trends of Pediatric Inflammatory Bowel Disease in Northern California, 1996 to 2006. J Pediatr. 2010;157:233.e1–239.e1. doi: 10.1016/j.jpeds.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 5.Gupta N, Bostrom AG, Kirschner BS, Ferry GD, Gold BD, Cohen SA, Winter HS, Baldassano RN, Abramson O, Smith T, Heyman MB. Incidence of stricturing and penetrating complications of Crohn's disease diagnosed in pediatric patients. Inflamm Bowel Dis. 2010;16:638–644. doi: 10.1002/ibd.21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunde S, Prasad M, Kugathasan S. Rising incidence of inflammatory bowel disease in young children: what does the future hold? J Pediatr Gastroenterol Nutr. 2011;53:128. doi: 10.1097/MPG.0b013e318225cde5. [DOI] [PubMed] [Google Scholar]
- 7.Braegger CP, Ballabeni P, Rogler D, Vavricka SR, Friedt M, Pittet V. Epidemiology of Inflammatory Bowel Disease: Is There a Shift Towards Onset at a Younger Age? J Pediatr Gastroenterol Nutr. 2011;53:141–144. doi: 10.1097/MPG.0b013e318218be35. [DOI] [PubMed] [Google Scholar]
- 8.Barnett M, Bermingham E, McNabb W, Bassett S, Armstrong K, Rounce J, Roy N. Investigating micronutrients and epigenetic mechanisms in relation to inflammatory bowel disease. Mutat Res. 2010;690:71–80. doi: 10.1016/j.mrfmmm.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol. 2010;106:563–573. doi: 10.1038/ajg.2011.44. [DOI] [PubMed] [Google Scholar]
- 10.Kellermayer R. Genetic drift. "Omics" as the filtering gateway between environment and phenotype: The inflammatory bowel diseases example. Am J Med Genet A. 2010;152A:3022–3025. doi: 10.1002/ajmg.a.33726. [DOI] [PubMed] [Google Scholar]
- 11.Kugathasan S, Amre D. Inflammatory bowel disease--environmental modification and genetic determinants. Pediatr Clin North Am. 2006;53:727–749. doi: 10.1016/j.pcl.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 13.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 14.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 15.Lyko F, Foret S, Kucharski R, Wolf S, Falckenhayn C, Maleszka R. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000506. e1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolinoy DC, Das R, Weidman JR, Jirtle RL. Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatr Res. 2007;61:30R–37R. doi: 10.1203/pdr.0b013e31804575f7. [DOI] [PubMed] [Google Scholar]
- 17.Hochberg Z, Feil R, Constancia M, Fraga M, Junien C, Carel JC, Boileau P, Le Bouc Y, Deal CL, Lillycrop K, Scharfmann R, Sheppard A, Skinner M, Szyf M, Waterland RA, Waxman DJ, Whitelaw E, Ong K, Albertsson-Wikland K. Child health, developmental plasticity, and epigenetic programming. Endocr Rev. 2011;32:159–224. doi: 10.1210/er.2009-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, Zhang W, Torskaya MS, Zhang J, Shen L, Manary MJ, Prentice AM. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001252. e1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balasa A, Gathungu G, Kisfali P, Smith EO, Cho JH, Melegh B, Kellermayer R. Assessment of DNA methylation at the interferon regulatory factor 5 (IRF5) promoter region in inflammatory bowel diseases. Int J Colorectal Dis. 2010;25:553–556. doi: 10.1007/s00384-010-0874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petronis A, Petroniene R. Epigenetics of inflammatory bowel disease. Gut. 2000;47:302–306. doi: 10.1136/gut.47.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petronis A. Epigenetics and twins: three variations on the theme. Trends Genet. 2006;22:347–350. doi: 10.1016/j.tig.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat Rev Rheumatol. 2011;7:263–271. doi: 10.1038/nrrheum.2011.16. [DOI] [PubMed] [Google Scholar]
- 23.Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M, Fraga MF, O'Hanlon TP, Rider LG, Jacinto FV, Lopez-Longo FJ, Dopazo J, Forn M, Peinado MA, Carreno L, Sawalha AH, Harley JB, Siebert R, Esteller M, Miller FW, Ballestar E. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jess T, Riis L, Jespersgaard C, Hougs L, Andersen PS, Orholm MK, Binder V, Munkholm P. Disease concordance, zygosity, and NOD2/CARD15 status: follow-up of a population-based cohort of Danish twins with inflammatory bowel disease. Am J Gastroenterol. 2005;100:2486–2492. doi: 10.1111/j.1572-0241.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 25.Halfvarson J, Jess T, Bodin L, Jarnerot G, Munkholm P, Binder V, Tysk C. Longitudinal concordance for clinical characteristics in a Swedish-Danish twin population with inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1536–1544. doi: 10.1002/ibd.20242. [DOI] [PubMed] [Google Scholar]
- 26.Csongei V, Jaromi L, Safrany E, Sipeky C, Magyari L, Farago B, Bene J, Polgar N, Lakner L, Sarlos P, Varga M, Melegh B. Interaction of the major inflammatory bowel disease susceptibility alleles in Crohn's disease patients. World J Gastroenterol. 2010;16:176–183. doi: 10.3748/wjg.v16.i2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waterland RA, Kellermayer R, Rached MT, Tatevian N, Gomes MV, Zhang J, Zhang L, Chakravarty A, Zhu W, Laritsky E, Zhang W, Wang X, Shen L. Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development. Hum Mol Genet. 2009;18:3026–3038. doi: 10.1093/hmg/ddp241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjamini YaH Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995;57:289–300. [Google Scholar]
- 29.Smyth GK. Limma: linear models for microarray data. In: Gentleman VC R, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. 2005 ed: [Google Scholar]
- 30.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 31.de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology. 2010;138:583–594. doi: 10.1053/j.gastro.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaible TD, Harris RA, Dowd SE, Smith CW, Kellermayer R. Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Hum Mol Genet. 2011;20:1687–1696. doi: 10.1093/hmg/ddr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halfvarson J. Genetics in twins with Crohn's disease: less pronounced than previously believed? Inflamm Bowel Dis. 2011;17:6–12. doi: 10.1002/ibd.21295. [DOI] [PubMed] [Google Scholar]
- 34.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D, Wijmenga C, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Cohen A, Colombel JF, Cottone M, Stronati L, Denson T, De Vos M, D'Inca R, Dubinsky M, Edwards C, Florin T, Franchimont D, Gearry R, Glas J, Van Gossum A, Guthery SL, Halfvarson J, Verspaget HW, Hugot JP, Karban A, Laukens D, Lawrance I, Lemann M, Levine A, Libioulle C, Louis E, Mowat C, Newman W, Panes J, Phillips A, Proctor DD, Regueiro M, Russell R, Rutgeerts P, Sanderson J, Sans M, Seibold F, Steinhart AH, Stokkers PC, Torkvist L, Kullak-Ublick G, Wilson D, Walters T, Targan SR, Brant SR, Rioux JD, D'Amato M, Weersma RK, Kugathasan S, Griffiths AM, Mansfield JC, Vermeire S, Duerr RH, Silverberg MS, Satsangi J, Schreiber S, Cho JH, Annese V, Hakonarson H, Daly MJ, Parkes M. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, Lagace C, Scott R, Amininejad L, Bumpstead S, Baidoo L, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Colombel JF, Denson LA, De Vos M, Dubinsky M, Edwards C, Ellinghaus D, Fehrmann RS, Floyd JA, Florin T, Franchimont D, Franke L, Georges M, Glas J, Glazer NL, Guthery SL, Haritunians T, Hayward NK, Hugot JP, Jobin G, Laukens D, Lawrance I, Lemann M, Levine A, Libioulle C, Louis E, McGovern DP, Milla M, Montgomery GW, Morley KI, Mowat C, Ng A, Newman W, Ophoff RA, Papi L, Palmieri O, Peyrin-Biroulet L, Panes J, Phillips A, Prescott NJ, Proctor DD, Roberts R, Russell R, Rutgeerts P, Sanderson J, Sans M, Schumm P, Seibold F, Sharma Y, Simms LA, Seielstad M, Steinhart AH, Targan SR, van den Berg LH, Vatn M, Verspaget H, Walters T, Wijmenga C, Wilson DC, Westra HJ, Xavier RJ, Zhao ZZ, Ponsioen CY, Andersen V, Torkvist L, Gazouli M, Anagnou NP, Karlsen TH, Kupcinskas L, Sventoraityte J, Mansfield JC, Kugathasan S, Silverberg MS, Halfvarson J, Rotter JI, Mathew CG, Griffiths AM, Gearry R, Ahmad T, Brant SR, Chamaillard M, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zahm AM, Thayu M, Hand NJ, Horner A, Leonard MB, Friedman JR. Circulating MicroRNA is a biomarker of pediatric Crohn disease. J Pediatr Gastroenterol Nutr. 2011;53:26–33. doi: 10.1097/MPG.0b013e31822200cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, Feldcamp LA, Virtanen C, Halfvarson J, Tysk C, McRae AF, Visscher PM, Montgomery GW, Gottesman II, Martin NG, Petronis A. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 38.Yuan B, O'Connor TR, Wang Y. 6-Thioguanine and S-methylthioguanine are mutagenic in human cells. ACS Chem Biol. 2010;5:1021–1027. doi: 10.1021/cb100214b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonsky R, Deem RL, Landers CJ, Derkowski CA, Berel D, McGovern DP, Targan SR. Distinct IFNG methylation in a subset of ulcerative colitis patients based on reactivity to microbial antigens. Inflamm Bowel Dis. 2011;17:171–178. doi: 10.1002/ibd.21352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bera TK, Hahn Y, Lee B, Pastan IH. TEPP, a new gene specifically expressed in testis, prostate, and placenta and well conserved in chordates. Biochem Biophys Res Commun. 2003;312:1209–1215. doi: 10.1016/j.bbrc.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 41.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, Hancock WW. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 42.Kim SW, Kim ES, Moon CM, Park JJ, Kim TI, Kim WH, Cheon JH. Genetic polymorphisms of IL-23R and IL-17A and novel insights into their associations with inflammatory bowel disease. Gut. 2011;60(11):1527–1536. doi: 10.1136/gut.2011.238477. [DOI] [PubMed] [Google Scholar]
- 43.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saito S, Kato J, Hiraoka S, Horii J, Suzuki H, Higashi R, Kaji E, Kondo Y, Yamamoto K. DNA methylation of colon mucosa in ulcerative colitis patients: Correlation with inflammatory status. Inflamm Bowel Dis. 2011;17:1955–1965. doi: 10.1002/ibd.21573. [DOI] [PubMed] [Google Scholar]
- 45.Lin Z, Hegarty JP, Cappel JA, Yu W, Chen X, Faber P, Wang Y, Kelly AA, Poritz LS, Peterson BZ, Schreiber S, Fan JB, Koltun WA. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet. 2011;80:59–67. doi: 10.1111/j.1399-0004.2010.01546.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.