

Abstract
The stereoselective synthesis of fused bicyclic ureas 8 is accomplished via enantioselective Pd-catalyzed desymmetrizing carboamination reactions of meso-2,5-diallylpyrroldinyl urea 7c. The reactions generate a C–N bond, a C–C bond, and afford products bearing three stereocenters with good diastereoselectivity (6–12:1 dr) and enantioselectivity (up to 95:5 er). The N-(p-chlorophenyl) group can be cleaved in good yield using a two step sequence. In addition, 8c was transformed to a tricyclic guanidine product using a four-step (two pot) procedure and was converted to 9-epi-batzelladine k in seven steps.
Keywords: Desymmetrization, Heterocycles, Asymmetric Catalysis, Palladium, Stereoselective
Catalytic asymmetric desymmetrization reactions are powerful and efficient tools for the synthesis of chiral molecules.[i] These transformations convert simple achiral substrates into complex enantioenriched products through the differentiation of two enantiotopic groups, and can generate complex structures bearing multiple stereocenters in a highly controlled fashion. As such, the development of asymmetric desymmetrization reactions that allow for the construction of important structural motifs is of considerable utility.
Tricyclic guanidines are an interesting class of compounds that could potentially be accessed via catalytic asymmetric desymmetrization reactions (Figure 1). These scaffolds are displayed in a wide variety of biologically active natural products,[ii] including the batzelladine alkaloids[iii] (e.g. batzelladine K, 1),[iiic] the merobatzelladine alkaloids (e.g., merobatzelladine B, 2),[iv] and the crambescidin alkaloids[v] (e.g., crambescidin 359, 3).[vb] Many synthetic routes to these compounds involve the generation of a fused-bicyclic urea or guanidine derivative (e.g., 4), which is then transformed to the tricyclic guanidine in subsequent steps.[vi,vii,viii] As such, development of a concise asymmetric synthesis of 4 could provide access to a broad array of interesting alkaloids.
Figure 1.

Bioactive guanidine alkaloids prepared from bicyclic urea precursors
We recently reported an asymmetric synthesis of the tricyclic guanidine natural product (+)-merobatzelladine B (2), which featured a new strategy for the construction of bicyclic ureas and polycyclic guanidines via Pd-catalyzed carboamination reactions of enantiomerically enriched 2-allylpyrrolidine-1-carboxamide derivatives 5 (Scheme 1).[viii] These reactions provided bicyclic urea products 6 in good yield with excellent diastereoselectivity, but control of absolute stereochemistry required the chiral-auxiliary mediated introduction of the C2 stereocenter during the fairly lengthy asymmetric synthesis of 5 (7–9 steps).[ix]
Scheme 1.

Synthesis of bicyclic ureas via Pd-catalyzed asymmetric desymmetrization
A potentially more attractive route to enantiomerically enriched bicyclic ureas and related bi- and tricyclic guanidines would involve the asymmetric Pd-catalyzed desymmetrization of meso-2,5-diallylpyrrolidnyl urea 7. This approach would allow for facile introduction of different R1-substituents, and the alkene present in product 8 provides a convenient handle for further elaboration to tricyclic guanidine products or more highly substituted urea derivatives. In addition, the meso substrate 7 can be prepared in only four steps. Our preliminary studies in this area are described in this communication. These transformations represent the first examples of asymmetric desymmetrizations of bis-alkene substrates in intermolecular Pd-catalyzed alkene carboamination reactions, and also the first examples of six-membered ring formation in an asymmetric Pd-catalyzed alkene carboamination.[x,xi,xii]
In initial experiments we elected to employ a catalyst composed of Pd2(dba)3/(S)-Siphos-PE[xiii] for desymmetrization reactions of 7, as we previously illustrated this complex provides good results in related asymmetric carboamination reactions of simple N-allyl urea derivatives.[xiv,xv] We decided to first optimize the structure of the urea N-aryl group, as prior studies in our lab suggested this group may have a significant influence on the level of asymmetric induction.[xiv] Thus, we explored the coupling of Z-1-bromobutene[xvi] with ureas 7 bearing different N-aryl substituents. As shown in Table 1, the use of electron-poor p-cyanophenyl or p-nitrophenyl N-aryl groups resulted in the formation of products 8 with the highest levels of both diastereoselectivity and enantioselectivity. However, these electron-poor substrates were transformed in modest chemical yield due to competing cleavage of the urea moiety (entries 5–6). Use of the electron-rich p-methoxyphenyl group led to improved yields but with lower levels of stereocontrol. After some exploration we found that a substrate bearing a p-chlorophenyl group was transformed to the desired product with both good chemical yield and stereoselectivity (entry 3).[xvii]
Table 1.
N-Aryl group effects.[a]
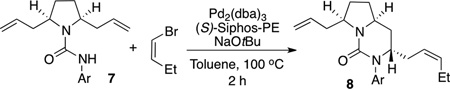 | ||||
|---|---|---|---|---|
| Entry | Ar | Yield(%)[b] | dr[c] | er |
| 1 | 4-MeOC6H4 | 65 | 7:1 | 86:14 |
| 2 | 3,4-MeOC6H4 | 41 | 7:1 | 82:18 |
| 3 | 4-ClC6H4 | 76 | 12:1 (20:1)[d] | 95:5 |
| 4 | 4-BrC6H4 | 12[e] | 18:1 | 94:6 |
| 5 | 4-CNC6H4 | 40[e] | 17:1 | 95:5 |
| 6 | 4-NO2C6H4 | 22[f] | 20:1 | 96:4 |
Conditions: 1.0 equiv substrate, 1.5 equiv (Z)-1-bromobutene, 1.5 equiv NaOtBu, 2 mol % Pd2(dba)3, 8 mol % (S)-Siphos-PE, Toluene (0.2 M), 100 °C, 2h.
Isolated yield (average of two or more runs).
Diastereomeric ratio of the pure isolated material. Diastereomeric ratios of the isolated materials were identical to those of the crude products except for entry 3.
The diastereomeric ratio of the crude material was 12:1. The product was isolated in 76% yield with 20:1 dr.
This material contained a small amount of the corresponding aniline derivative.
The reaction was conducted at 120 °C for 16 h. The isolated material contained ca. 8% of unreacted substrate.
As shown in Table 2, the asymmetric desymmetrization reactions of 7c are effective with a number of different alkenyl and aryl bromide electrophiles. The main side products generated in these reactions were cis-2,5-diallylpyrrolidine (resulting from competing urea cleavage) and an unsaturated bicyclic urea that is generated by competing β-hydride elimination of an intermediate alkylpalladium complex.[xviii] In the reaction of 7c with E-1-bromohexene a regioisomeric side product bearing a 2-hex-1-enyl group was also generated.[xix]
Table 2.
Reaction Scope.[a]
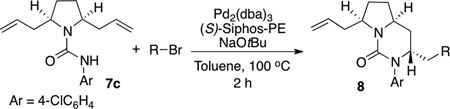 | |||||
|---|---|---|---|---|---|
| Entry | R | Product | Yield(%)[b] | dr[c] | er |
| 1 | Z-butene | 8c | 76 | >20:1[d] | 95:5 |
| 2 | E-hexene | 8g | 50[f] | >20:1[d] | 95:5 |
| 3 | Z-hexene | 8h | 61 | >20:1[d] | 94:6 |
| 4 | 2-methyl propene | 8i | 48 | 10:1 | 94:6 |
| 5 | 2-propene | 8j | 55 | 20:1[d] | 88:12 |
| 6 | 4-MeC6H4 | 8k | 84 | 8:1 | 92:8 |
| 7 | 4-MeOC6H4 | 8l | 70 | 7:1 | 92:8 |
| 8 | Ph | 8m | 83 | 6:1 | 90:10 |
| 9 | 4-F3CC6H4 | 8n | 74 | 5:1 | 85:15 |
| 10 | 4-F3CC6H4 | 8n | 55[g] | 11:1[e] | 90:10 |
| 11 | 4-F3COC6H4 | 8o | 68 | 7:1 | 87:13 |
| 12 | 4-F3COC6H4 | 8o | 51[g] | 18:1[d] | 92:8 |
| 13 | 3-MeOC6H4 | 8p | 72 | 5:1 | 87:13 |
| 14 | 2-naphthyl | 8q | 75 | 7:1 | 88:12 |
| 15 | 2-MeC6H4 | 8r | 81 | 5:1 | 71:29 |
Conditions: 1.0 equiv substrate, 1.5 equiv R–Br, 1.5 equiv NaOtBu, 2 mol % Pd2(dba)3, 8 mol % (S)-Siphos-PE, Toluene (0.2 M), 100 °C, 2h.
Isolated yield (average of two or more runs).
Diastereomeric ratio of the pure isolated material. Diastereomeric ratios of the isolated materials were identical to those of the crude products unless otherwise noted.
The diastereomeric ratio of the crude material was 10–12:1.
The diastereomeric ratio of the crude material was 6:1
This material contained 15% of the analogous 2-hex-1-enyl regioisomer.
The reaction was conducted using NaOMe as base instead of NaOtBu.
The best enantioselectivities were obtained when either alkenyl bromides, electron-rich aryl bromides, or electron-neutral aryl bromides were employed as substrates. Diastereoselectivities were generally higher with the alkenyl electrophiles than with aryl electrophiles. Use of sterically hindered aryl bromides (entries 14–15) or electron-poor aryl bromides (entries 9, 11, and 13) led to lower diastereo- and enantioselectivities. Selectivities improved when NaOMe was used in place of NaOtBu in reactions of electron-poor aryl bromides (entries 10 and 12), although yields decreased in these cases.
To further demonstrate the utility of the asymmetric desymmetrization reactions, we examined the deprotection of 8c and the conversion of 8c to tricyclic guanidine derivatives. As shown in equation 1, cleavage of the N-p-chlorophenyl group can be accomplished via Pd-catalyzed N-arylation with acetamide[xx, xxi] followed by oxidation of the resulting N-aryl amide with ceric ammonium nitrate. This sequence afforded 9 in 65% yield over two steps.
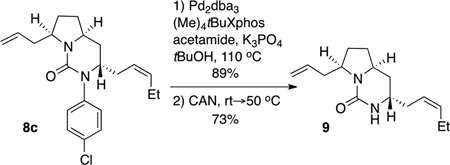 |
(1) |
The conversion of 8c to tricyclic guanidine 12 was carried out as shown in Scheme 2. Treatment of 8c with POCl3 followed by NH3 provided bicyclic guanidine 10 in 78% yield. Wacker oxidation of 10 afforded hemiaminal 11, which was then transformed to tricyclic product 12 in 70% yield with 5:1 dr via reductive amination with NaBH3CN.[xxii] Overall, the synthesis of 12, which is structurally related to the batzelladine and merobatzelladine alkaloids, was accomplished in 5 steps and 41% yield from meso-2,5-diallylpyrrolidinyl urea 7c. In addition, this is the first example of a Wacker oxidation/ring-closure sequence to generate a tricyclic guanidine.[xxiii,xxiv]
Scheme 2.

Conversion of 8c to a tricyclic guanidine derivative
Finally, 8c was converted to tricyclic guanidine 16, which is an unnatural stereoisomer of batzelladine k,[xxv,xxvi] as shown in Scheme 3. To avoid problems with base-mediated epimerization of the C4 stereocenter, the Pd-catalyzed N-arylation with acetamide was carried out prior to Wacker oxidation of the alkene. This two-step sequence provided 13 in 65% yield. Reduction of the alkene followed by CAN deprotection generated urea 14, which was converted to guanidine aminal 15 by O-methylation and treatment with ammonia.[xxvii] The reduction of 15 proceeded with modest diastereoselectivity (3:1 dr), but upon purification 9-epi-batzelladine k was isolated as a single stereoisomer in 48% yield over three steps from 14.
Scheme 3.

Conversion of 8c to 9-epi-batzelladine K.
In conclusion we have developed a concise route to enantiomerically enriched bicyclic ureas via Pd-catalyzed desymmetrizing carboamination reactions of meso-diallylpyrroldinyl ureas. These transformations effect formation of both a C–N and a C–C bond, and provide products bearing three stereocenters with good levels of diasteroselectivity and enantioselectivity. These reactions illustrate the potential utility of asymmetric Pd-catalyzed alkene carboamination for desymmetrization processes and provide synthetically valuable products in a straightforward manner. Further exploration of enantioselective Pd-catalyzed alkene difunctionalization reactions are currently underway.
Supplementary Material
Acknowledgments
The authors acknowledge the NIH-NIGMS (GM 098314) and the University of Michigan Associate Professor Support Fund for financial support of this work. Additional funding was provided by GlaxoSmithKline and Amgen.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Nicholas R. Babij, Department of Chemistry, University of Michigan, 930 N. University Ave, Ann Arbor, MI 48109-1055, USA
John P. Wolfe, Department of Chemistry, University of Michigan, 930 N. University Ave, Ann Arbor, MI 48109-1055, USA, jpwolfe@umich.edu
References
- i.For reviews, see: Enriquez-Garcia A, Kündig EP. Chem. Soc. Rev. 2012;41:7803–7831. doi: 10.1039/c2cs35049a. Diaz de Villegas MD, Galvez JA, Etayo P, Badorrey R, Lopez Ram de Viu P. Chem. Soc. Rev. 2011;40:5564–5587. doi: 10.1039/c1cs15120g. Rovis T. In: New Frontiers in Asymmetric Catalysis. Mikami K, Lautens M, editors. Hoboken, NJ: Wiley; 2007. pp. 275–309. Mikami K, Yoshida A. J. Synth. Org. Chem., Jpn. 2002;60:732–739. Trost BM. Isr. J. Chem. 1997;37:109–118.
- ii.a) Berlinck RGS, Trindade-Silva AE, Santos MFC. Nat. Prod. Rep. 2012;29:1382–1406. doi: 10.1039/c2np20071f. [DOI] [PubMed] [Google Scholar]; b) Berlinck RGS, Burtoloso ACB, Trindade-Silva AE, Romminger S, Morais RP, Bandeira K, Mizuno CM. Nat. Prod. Rep. 2010;27:1871–1907. doi: 10.1039/c0np00016g. [DOI] [PubMed] [Google Scholar]; c) Berlinck RGS, Burtoloso ACB, Kossuga MH. Nat. Prod. Rep. 2008;25:919–954. doi: 10.1039/b507874c. [DOI] [PubMed] [Google Scholar]; d) Berlinck RGS, Kossuga MH. Nat. Prod. Rep. 2005;22:516–550. doi: 10.1039/b209227c. [DOI] [PubMed] [Google Scholar]; e) Bewley CA, Ray S, Cohen F, Collins SK, Overman LE. J. Nat. Prod. 2004;67:1319–1324. doi: 10.1021/np049958o. [DOI] [PubMed] [Google Scholar]
- iii.a) Laville R, Thomas OP, Berrue F, Marquez D, Vacelet J, Amade P. J. Nat. Prod. 2009;72:1589–1594. doi: 10.1021/np900244g. [DOI] [PubMed] [Google Scholar]; b) Gallimore WA, Kelly M, Scheuer PJ. J. Nat. Prod. 2005;68:1420–1423. doi: 10.1021/np050149u. [DOI] [PubMed] [Google Scholar]; c) Hua H–M, Peng J, Dunbar DC, Schinazi RF, de Castro Andrews AG, Cuevas C, Garcia-Fernandez LF, Kelly M, Hamann MT. Tetrahedron. 2007;63:11179–11188. [Google Scholar]; d) Patil AD, Kumar NV, Kokke WC, Bean MF, Freyer AJ, De Brosse C, Mai S, Truneh A, Faulkner DJ, Carte B, Breen AL, Hertzberg RP, Johnson RK, Westley JW, Potts BCM. J. Org. Chem. 1995;60:1182–1188. [Google Scholar]
- iv.a) Takishima S, Ishiyama A, Iwatsuki M, Otoguro K, Yamada H, Omura S, Kobayashi H, van Soest RWM, Matsunaga S. Org. Lett. 2009;11:2655–2658. doi: 10.1021/ol9006794. [DOI] [PubMed] [Google Scholar]; b) Takishima S, Ishiyama A, Iwatsuki M, Otoguro K, Yamada H, Omura S, Kobayashi H, van Soest RWM, Matsunaga S. Org. Lett. 2010;12:896. doi: 10.1021/ol9006794. [DOI] [PubMed] [Google Scholar]
- v.a) Martin V, Vale C, Bondu S, Thomas OP, Vieytes MR, Botana LM. Chem. Res. Toxicol. 2013;26:169–178. doi: 10.1021/tx3004483. [DOI] [PubMed] [Google Scholar]; b) Braekman JC, Daloze D, Tavares R, Hajdu E, Van Soest RWM. J. Nat. Prod. 2000;63:193–196. doi: 10.1021/np990403g. [DOI] [PubMed] [Google Scholar]; c) Jares-Erijman EA, Sakai R, Rinehart KL. J. Org. Chem. 1991;56:5712–5715. [Google Scholar]
- vi.For examples of bicyclic urea as intermediates in the synthesis of tricyclic guanidines, see: Evans PA, Qin J, Robinson JE, Bazin B. Angew. Chem., Int. Ed. 2007;46:7417–7419. doi: 10.1002/anie.200700840. Aron ZD, Overman LE. J. Am. Chem. Soc. 2005;127:3380–3390. doi: 10.1021/ja042875+. Rama Rao AV, Gurjar MK, Vasudevan J. J. Chem. Soc., Chem. Commun. 1995:1369–1370. Overman LE, Rabinowitz MH, Renhowe PA. J. Am. Chem. Soc. 1995;117:2657–2658.
- vii.For examples of bicyclic guanidines as intermediates in the synthesis of tricyclic guanidines, see: Arnold MA, Day KA, Duron SG, Gin DY. J. Am. Chem. Soc. 2006;128:13255–13260. doi: 10.1021/ja063860+. Cohen F, Overman LE. J. Am. Chem. Soc. 2006;128:2604–2608. doi: 10.1021/ja057433s. Collins SK, McDonald AI, Overman LE, Rhee YH. Org. Lett. 2004;6:1253–1255. doi: 10.1021/ol0498141. Ishiwata T, Hino T, Koshino H, Hashimoto Y, Nakata T, Nagasawa K. Org. Lett. 2002;4:2921–2924. doi: 10.1021/ol026303a.
- viii.Babij NR, Wolfe JP. Angew. Chem. 2012;124:4204–4206. Angew. Chem. Int. Ed.2012, 51, 4128–4130. [Google Scholar]
- ix.Synthesis of the substrate needed for the preparation of (+)-merobatzelladine B, which contained a functionalized side chain bearing a stereocenter) required 9 steps. Synthesis of an enantiopure substrate 5 where R = CH2CH=C(H)TMS required 7 steps. See the Supporting information for details on the preparation of this latter compound, which was used to assign absolute stereochemistry of products 8 generated in catalytic reactions.
- x.One single report has described asymmetric desymmetrization reactions via Cu-catalyzed intramolecular carboaminations, which afford benzo-fused indolizidine products. See: Miao L, Haque I, Manzoni MR, Tham WS, Chemler SR. Org. Lett. 2010;12:4739–4741. doi: 10.1021/ol102233g.
- xi.Although there is an intramolecular component to the reactions described herein, the aryl/alkenyl halide and urea substrate are separate components coupled in an intermolecular process. This contrasts with the prior work cited in reference 10, in which all components involved in C–C or C–N bond formation are tethered together.
- xii.For examples of asymmetric Pd-catalyzed desymmetrization reactions that afford enantiomerically enriched nitrogen-heterocycles, see: Snell RH, Durbin MJ, Woodward RL, Willis MC. Chem. Eur. J. 2012;18:16754–16764. doi: 10.1002/chem.201203150. Snell RH, Woodward RL, Willis MC. Angew. Chem. 2011;123:9282–9285. doi: 10.1002/anie.201103864. Angew. Chem. Int. Ed.2011, 50, 9116–9119. Chapsal BD, Ojima I. Org. Lett. 2006;8:1395–1398. doi: 10.1021/ol060181v. Trost BM, Patterson DE. Chem. Eur. J. 1999;5:3279–3284. Trost BM, Patterson DE. J. Org. Chem. 1998;63:1339–1341.
- xiii.(S)-Siphos-PE = (11aS)-(+)-10,11,12,13-Tetrahydrodiindeno[7,1-de:1’,7’fg]-[1,3,2]dioxaphosphocin-5-bis[(R)-1-phenylethyl]amine
- xiv.Hopkins BA, Wolfe JP. Angew. Chem. 2012;124:10024–10028. Angew. Chem. Int. Ed.2012, 51, 9886–9890. [Google Scholar]
- xv.For asymmetric Pd-catalyzed alkene carboamination reactions that afford pyrrolidines, see: Mai DN, Wolfe JP. J. Am. Chem. Soc. 2010;132:12157–12159. doi: 10.1021/ja106989h. Mai DN, Rosen BR, Wolfe JP. Org. Lett. 2011;13:2932–2935. doi: 10.1021/ol2009895. For asymmetric Pd-catalyzed cascade oxidative cyclizations that proceed via syn-aminopalladation, see: He W, Yip K–T, Zhu N–Y, Yang D. Org. Lett. 2009;11:5626–5628. doi: 10.1021/ol902348t. For asymmetric Cu-catalyzed alkene carboamination reactions, see: Zeng W, Chemler SR. J. Am. Chem. Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. Liwosz TW, Chemler SR. J. Am. Chem. Soc. 2012;134:2020–2023. doi: 10.1021/ja211272v. For a discussion of the influence of alkene aminopalladation mechanism on asymmetric induction, see: Weinstein AB, Stahl SS. Angew. Chem. 2012;124:11673–11677. Angew. Chem. Int. Ed.2012, 51, 11505–11509.
- xvi.This electrophile was chosen for optimization studies as it had proven to be a satisfactory coupling partner in our synthesis of merobatzelladine B; see reference 8.
- xvii.The reaction of the analogous p-bromophenyl derivative proceeded in low yield due to competing oligomerization of the substrate (Table 1, entry 4).
- xviii.In some instances (Table 2, entries 2, 3, and 5) modest yields were due to product losses during the chromatographic separation of diastereomers.
- xix.This product results from competing β-hydride elimination/isomerization of the alkenylpalladium intermediate generated from oxidative addition of E-1-bromohexene. For further discussion of the origin of this side product, see: Ney JE, Hay MB, Yang Q, Wolfe JP. Adv. Synth. Catal. 2005;347:1614–1620. doi: 10.1002/adsc.200505172.
- xx.Ikawa T, Barder TE, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2007;129:13001–13007. doi: 10.1021/ja0717414. [DOI] [PubMed] [Google Scholar]
- xxi.(Me)4tBuXPhos = 2-di-t-butylphosphino-3,4,5,6-tetramethyl-2’,4’,6’-tri-i-propylbiphenyl.
- xxii.Snider BB, Chen J. Tetrahedron Lett. 1998;39:5697–5700. [Google Scholar]
- xxiii.For synthesis of bicyclic guanidines via Pd-catalyzed alkene diamination, see: Hövelmann CH, Streuff J, Brelot L, Muñiz K. Chem. Commun. 2008:2334–2336. doi: 10.1039/b719479j.
- xxiv.Efforts to cleave the N-aryl group from 12 have thus far been unsuccessful.
- xxv.Batzelladine k isolation: Hua H–M, Peng J, Dunbar DC, Schinazi RF, de Castro Andrews AG, Cuevas C, Garcia-Fernandez LF, Kelly M, Hamann MT. Tetrahedron. 2007;63:11179–11188.
- xxvi.For syntheses of batzelladine k, see: Sekine M, Iijima Y, Iwamoto O, Nagasawa K. Heterocycles. 2010;80:395–408. Ahmed N, Brahmbhatt KG, Singh IP, Bhutani KK. Synthesis. 2010;15:2567–2570. Ahmed N, Brahmbhatt KG, Khan SI, Jacob M, Tekwani BL, Sabde S, Mitra D, Singh IP, Kahn IA, Bhutani KK. Chem. Biol. Drug. Des. 2013;81:491–498. doi: 10.1111/cbdd.1427.
- xxvii.Coffey DS, McDonald AI, Overman LE, Rabinowitz MH, Renhowe PA. J. Am. Chem. Soc. 2000;122:4893–4903. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.