

Abstract
Background
Prostasin, a serine protease, is suggested to be a novel mechanism regulating the epithelial sodium channel expressed in the distal nephron. This study aimed to evaluate whether the human prostasin gene is a novel candidate gene underlying blood pressure (BP) elevation.
Methods
In a sample of healthy African American (AA) and European American (EA) twin subjects aged 17.6±3.3 years (n=920, 45% AAs), race-specific tagging single nucleotide polymorphisms (tSNPs) were identified to tag all the available SNPs ± 2Kb up- and downstream of the prostasin gene from HapMap at r2 of 0.8 – 1.0. Selection yielded four tSNPs in AAs and one in EAs, with one tSNP (rs12597511: C to T) present in both AAs and EAs.
Results
For rs12597511, CT and TT genotypes exhibited higher systolic BP than CC genotype (115.9±1.1 mmHg vs. 113.7±0.6 mmHg, p=0.025 [AAs]; and 110.7±0.5 mmHg vs. 109.6±0.6 mmHg, p=0.115 [EAs]). CT and TT genotypes compared to CC genotype showed a significant increase in diastolic BP in both racial groups (62.5±0.7 mmHg vs. 60.4±0.4 mmHg, p=0.003 [AAs]; and 58.2±0.3 mmHg vs. 56.7±0.4 mmHg, p=0.007 [EAs]). Furthermore, there was an increase in radial pulse wave velocity (PWV) in subjects with CT and TT genotype as compared to those with CC genotype (6.5±0.1 vs. 6.1±0.1 m/s, p<0.0001) [EAs]; and 6.7±0.1 vs. 6.6±0.1 m/s, p=0.354 [AAs]). Analyses combining AAs and EAs consistently demonstrated a statistical significance of rs1259751 on all the phenotypes including systolic/diastolic BP, and PWV.
Conclusion
Genetic variation of the prostasin gene may be implicated in the development of hypertension in youths.
Keywords: Prostasin, ENaC, Polymorphisms, Blood Pressure, Arterial stiffness
INTRODUCTION
The epithelial sodium channel (ENaC) consisting of α-, β-, and γ- subunits is expressed in the renal distal tubules, and constitutes the rate-limiting step for sodium reabsorption.1 Two monogenic disorders, Liddle’s syndrome (salt retaining hypertension) and pseudohypoaldosteronism type 1 (salt wasting hypotension) highlight the significance of genetic defects of ENaC in sodium homeostasis and blood pressure (BP) regulation.1 Furthermore, genetic variants of αENaC, βENaC, and γENaC were found to be associated with population BP variation or the presence of essential hypertension in some populations, although not in others.2–12
ENaC activity is tightly regulated by a network of accessory regulatory proteins. In particular, two regulatory proteins have gained great interest: neural precursor cell-expressed developmentally down-regulated 4 (Nedd4L) and serum glucocorticoid-inducible kinase (SGK-1).1 Nedd4L, an ubiquitin ligase, ubiquitinates ENaC to tag the channel for degradation, leading to its removal from the cell membrane. SGK1, an aldosterone-dependent positive regulator, inhibits the capacity of Nedd4L to down-regulate ENaC through phosphorylation. Previously, chromosomal regions harbouring Nedd4L or SGK1 genes were linked to BP phenotypes, and genetic variants of Nedd4L and SGK1 genes have been recently associated with BP and hypertension.13–17 For example, a significant association between several Nedd4L polymorphisms and hypertension was observed in three populations with essential hypertension including AAs and EAs.16 SGK1 polymorphisms, alone or synergistically, were associated with cross-sectional and longitudinal BP in over 4,000 EA subjects.14
Studies in the late 1990s suggested a novel ENaC regulatory mechanism by a glycosylphosphatidylinositol (GPI)-anchored serine protease expressed in the distal nephron, called prostasin.18, 19 The expression of prostasin was enhanced by aldosterone both in vitro and in vivo, which increases sodium reabsorption in the distal nephron through ENaC.20 Further, prostasin was believed to induct cleavage of γ-ENaC to fully activate the channel.21 Adenovirus-mediated human prostasin gene delivery caused a marked increase in BP and electrolyte imbalance, which was accompanied by increased plasma aldosterone level, and reduced plasma renin activity in rats.22 These data suggest that prostasin participates in BP and electrolyte homeostasis by interacting with the renin-angiotensin-aldosterone system. Data on humans, however, are scant.20, 23 In three patients with primary aldosteronism, urinary excretion of prostasin was abnormally elevated and normalized after adrenalectomy. After adrenalectomy, reduction in the urinary excretion of prostasin in these patients was correlated with the increase in the urinary Na+/K+ ratio.20 By conducting spironolactone or saline/Florinef suppression tests in eight patients with raised aldosterone to renin ratio in comparison with ten healthy volunteers, Olivieri et al 23 suggested that urinary prostasin may be a candidate marker of ENaC activation in humans. These findings encouraged us to hypothesize that the human prostasin gene denoted as PRSS8 (chromosome 16p11.2)24 is a novel candidate gene of the ENaC complex underlying the variation of BP in the general population, and thus, the development of essential hypertension.
This study attempted to evaluate the association between genetic variants in the prostasin gene and BP phenotypes in a large sample of normotensive youth including AAs and EAs from the Georgia Cardiovascular Twin Study. Means and ranges of quantitative phenotypes in twins, including cardiovascular traits, have shown to be similar to age-matched individuals from the general population.25, 26 As illustrated in numerous gene association studies, twins can be efficiently used to study specific candidate genes underlying complex traits such as BP and related phenotypes.
METHODS
Study Population
A total of 920 AA and EA twins (mean age: 17.6 ± 3.3 years; 45% AAs) from the Georgia Cardiovascular Twin Study participated in this study. These included monozygotic (MZ, 50%), and dizygotic (DZ, 50%) pairs of same- as well as opposite-gender. Recruitment and ethnic classification have been described previously.27 All participants were apparently healthy based upon (parental) report of the child’s medical history. All subjects in the Georgia Cardiovascular Twin Study were recruited from within a 120 mile radius of the study location, Augusta, Georgia. African and European ancestry was strictly defined for each group. Subjects were classified as AA if 1) both parents reported being of African heritage; 2) they and the child were born and raised in the United States; and 3) parents considered themselves and their child to be AA. Subjects were classified as EA if: 1) both parents reported that they were of European ancestry; 2) they and the child were born and raised in the United States; and 3) they considered themselves and their child to be EA and not of Hispanic, Native American, or Asian descent. The study was approved by the institutional review board at the Medical College of Georgia and all subjects (and parents if subjects were <18 years) provided written informed consent. The subject characteristics by race and gender are shown in Table 1.
Table 1.
Description of the study population
| Characteristics | AAs | EAs | Race & Gender Effects | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Overall | Males | Females | Overall | Males | Females | Race | Gender | ||
| N | 416 | 187 | 229 | 504 | 252 | 252 | ― | ― | |
| Age, years | 17.2(3.2) | 17.3(3.0) | 17.1(3.4) | 17.8(3.3) | 17.7(3.4) | 17.9(3.2) | 0.012 | 0.924 | |
| Height, cm | 167.3(9.7) | 173.2(9.9) | 162.3(6.6) | 167.9(10.2) | 173.3(9.4) | 162.6(7.3) | 0.539 | <0.001 | |
| BMI, kg/m2 | 24.7(6.3) | 23.7(5.1) | 25.5(7.0) | 23.2(5.4) | 23.5(5.5) | 22.9(5.3) | <0.001 | 0.007 | |
| SBP, mmHg | 114.1(11.0) | 117.6(10.4) | 111.3(10.6) | 110.3(10.1) | 113.8(12.7) | 106.8(8.5) | <0.001 | <0.001 | |
| DBP, mmHg | 60.9(7.2) | 59.3(6.7) | 62.1(7.4) | 57.8(6.3) | 56.7(6.3) | 58.9(6.2) | <0.001 | <0.001 | |
| PP, mm Hg | 63.7(11.0) | 59.6(10.8) | 66.9(10.0) | 64.0(10.9) | 61.5(10.4) | 66.6(10.8) | 0.173 | <0.001 | |
| Radial PWV, m/s | 6.7(1.0) | 6.7(1.0) | 6.7(1.0) | 6.3(1.1) | 6.4(1.1) | 6.3(1.0) | <0.001 | 0.835 | |
BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; PWV, pulse wave velocity.
Radial PWV values were available for 198 and 182 EA males and females, and 123 and 151 for AA males and females.
Age was adjusted for comparisons of height, BMI, SBP, DBP, PP and radial PWV.
Measures
After arrival in the laboratory, participants were engaged in a standard battery of anthropometric evaluations using established protocols.28 SBP and DBP measurements (Dinamap 1846 SX; Criticon Inc., Tampa, FL) were taken at 11, 13 and 15 minutes, during a 15-minute relaxation period in which subjects were instructed to relax as completely as possible while laying (supine) on a hospital bed. The average of the last two measurements was used to represent SBP and DBP at rest. Pulse pressure (PP) was calculated as the difference between SBP and DBP. Aorto-radial PWV was measured noninvasively with applanation tonometry (Millar Instruments)29 and analysis software (SphygmoCor, AtCor Medical, Sydney, Australia). Pressure waves were recorded at the common carotid and radial arteries. PWV was then automatically calculated from measurements of pulse transit time and the distance traveled by the pulse between the two recording sites: PWV=Distance (meters)/Transit Time (seconds).
Tagging Single Nucleotide Polymorphism (tSNP) Selection and Genotyping
A 7-kb PRSS fragment was previously reported including a 1.4-kb 5’-flanking region, the 4.4-kb PRSS gene containing 6 exons and five introns, and a 1.2-kb 3’-flanking region (Figure 1).24 As such, genotypes of 90 CEU (white) and YRI (black) parent-offspring trio subjects from the PRSS8 locus, including 2.0 kb upstream and 2.0 kb downstream of the gene were downloaded from Hapmap phase II database into Haploview. The Tagger algorithm (http://www.broad.mit.edu/hapview/) was used to select tSNPs. In January 2008, the database only contained 3 SNPs in the CEU population (rs1549293, rs12597511, rs2855475, minor allele frequency between 0.386–0.392). One tSNP (rs12597511) was selected which captures all 3 common SNPs at r2 of 0.8. In the YRI population, 5 SNPs are listed with minor allele frequency between 0.03–0.27. Four tSNPs (rs1259711, rs1549294, rs1549295, and rs2855475) were selected which capture those 5 SNPs at r2 ≥ 0.8, using the aggressive multi-marker tagging mode. Four tSNPs (rs1259711, rs1549294, rs1549295, and rs2855475) were selected which capture those 5 SNPs at r2 ≥ 0.8, using the aggressive multi-marker tagging mode. In this approach, a multimarker haplotype is reconstructed from the selected tSNPs, which can act as effective predictors for some un-genotyped SNPs that are not explained by a single tSNP. Including multimarker tests for the SNP selection can reduce the genotyping efforts further than the single marker test only. The tSNPs were genotyped by allelic discrimination Taqman assays (Applied Biosystems, Foster City, CA). PCR reactions were performed in a 96-well format in a total of 5 ul reaction volume using 10 ng of genomic DNA and FAM/VIC dye labeled allelic probes with the Taqman Universal Fast Master mix (Applied Biosystems). The Taqman assay plates were transferred to an ABI 7500 Fast Real Time PCR system in which the fluorescence intensity in each well of the plate was recorded and genotypes were analyzed using Sequence Detection Software 1.3. Genotyping quality control procedures included genotyping 10% duplicates for accuracy checking; inclusion of both positive and non-template controls in each 96-well plate.
Figure 1. Structure of the Prostasin gene. Exons are shown as boxes, introns and intergenic regions as lines.
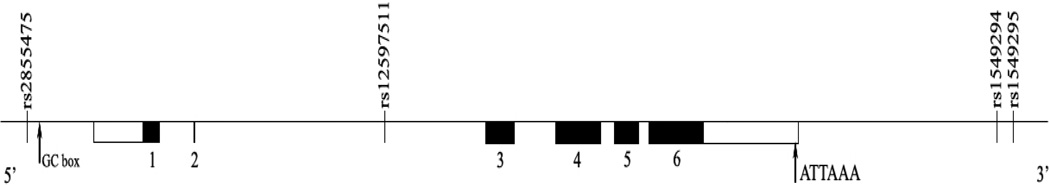
rs2855475 is in the 5’ region close to a GC box, rs12597511 is in the intron 2 region, and rs1549294 and rs1549295 are in the 3’ region.
Analytical Approach
Generalized Estimating Equations (GEE)30 were applied to test the association between each individual tSNPs and phenotype in AAs and EAs separately, after the effects of age, gender and body mass index (BMI) were adjusted. For related individuals, conventional statistical analyses lead to inflated significance. GEE accounts for dependency of the observations within pairs and yields unbiased SEs and P values. We first modeled the effects of age, gender and BMI on SBP and DBP, PP, and PWV. After arriving at the most parsimonious full ‘environmental’ model including only significant terms, polymorphisms or haplotypes were then added to test for main effects and interactions. To reduce the numbers of tests, we first performed a 2-degree of freedom overall test (codominant models) of genotypic association. Only in the presence of a significant association, 1-degree of freedom models including the additive, recessive and dominant effects were further tested to find the best mode of inheritance. Simultaneously modeling gender as a factor in our model is more powerful than subgroup analyses, and it also allows us to statistically test for gender-specific effects of the polymorphisms and haplotypes. However, AAs and EAs were analyzed separately, because selected tSNPs were race-specific. The haplotype trend regression (HTR) approach was used to test for associations of statistically inferred haplotypes with the above-mentioned phenotypes.30 Hardy-Weinberg equilibrium and racial differences in allele frequencies were tested by χ2 tests in subjects including only one of each twin pair chosen randomly to prevent inflated significance. Single locus association analyses and HTR were performed with STATA 8 (StataCorp, College Station, Texas).
RESULTS
Study Population
Table 1 describes the study population. Boys were taller than girls in each racial group. EA participants were slightly older than AA participants. AA girls had greater BMI than AA boys, yet EA boys had greater BMI than EA girls. AAs had higher BMI, SBP, DBP, and radial PWV than EAs. In both AAs and EAs, males had higher SBP, yet lower DBP as compared to females.
Allele Frequencies
The minor allele frequency of rs12597511 was more common in EAs than AAs, 35% vs. 11% (p<0.0001). The minor allele frequencies of rs1549294 (C/T) and rs2855475 (C/T) were 10% in AAs and 35% in EAs, respectively. All the tSNPs except rs1549295 (C/T) were in Hardy Weinberg equilibrium. The rs1549295 SNP in AAs deviated significantly from Hardy-Weinberg Equilibrium, possibly due to its low allele frequency (4%).
Genotype and Phenotype Association Analyses
None of the interactions between gender and individual tSNPs were significant for all the phenotypes including SBP, DBP, PP and PWV. A main effect of the T allele of rs12597511 in a dominant mode on the phenotypes was observed in both AAs and EAs (Table 2 and Figure 2). CT and TT genotypes exhibited higher SBP than CC genotype, although it was only statistically significant in AAs (115.9 vs. 113.7 mmHg, p=0.025 for AAs; 110.7 vs. 109.6 mmHg, p=0.115 for EAs). In both racial groups, CT and TT genotypes compared to CC genotype showed a significantly increased DBP (62.5 vs. 60.4 mmHg, p=0.003 for AAs; and 58.2 vs. 56.7 mmHg, p=0.007 for EAs), even after the Bonferroni correction. In addition, there was a non-significant increasing trend in PP with CT and TT genotypes when compared to CC genotype in EAs (p=0.06) and AAs (p=0.218). Finally, in EAs, there was an increase in PWV in youths with CT or TT genotype as compared to those with CC genotype (6.6 vs. 6.4m/s, p<0.0001), which remained statistically significant even after adjustment of SBP (p<0.0001), DBP (p=0.001) and mean arterial pressure (MAP) (p=0.001). This remained significant after Bonferroni correction for multiple comparisons. In AAs, there was a non-significant increase of PWV in CT and TT genotypes when compared to CC genotype (6.7±0.1 vs. 6.6±0.1 m/s, p=0.354). The haplotypic analysis findings in AAs with respect to the rs12597511 tSNP were supportive, yet not more informative. The haplotype data thus were not presented. We did not find any associations between rs1549294, rs1549295, rs2855475 with any of the phenotypes.
Table 2.
The associations between rs12597511 with SBP, DBP, PP and radial PWV
| Phenotypes | AAs | p | EAs | p | ||
|---|---|---|---|---|---|---|
| CC (n=224) |
CT+TT (n=50) |
CC (n=163) |
CT+TT (n=217) |
|||
| SBP, mm Hg | 113.7(0.6) | 115.9(1.1) | 0.025 | 109.6(0.6) | 110.7(0.5) | 0.115 |
| DBP, mm Hg | 60.4(0.4) | 62.5(0.7) | 0.003 | 56.7(0.4) | 58.2(0.3) | 0.007 |
| PP, mm Hg | 63.0(0.6) | 66.0(1.1) | 0.218 | 63.3(0.7) | 65.0(0.6) | 0.06 |
| Radial PWV, m/s | 6.6(0.1) | 6.7(0.1) | 0.354 | 6.1(0.1) | 6.5(0.1) | <0.0001* |
SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; PWV, pulse wave velocity
Values are mean (SE) after adjustment of age, gender and BMI.
Difference of means between genotypes remains significant after adjustment of SBP (p<0.0001) or DBP (p=0.001) or MAP (p=0.001).
Figure 2. SBP and DBP in AAs and EAs according to rs12597511.

CT +TT genotype carriers compared with CC carriers had higher SBP and DBP in AAs and higher DBP in EAs, after adjustment of age, gender and BMI
For rs12597511that AAs and EAs had in common, we decided to conduct an analysis combining both races and test for potential interaction between race and rs12597511. Thus, all individuals including AAs and EAs were pooled for additional analyses, as we did previously in association studies of bi-racial cohorts. 30 The main effect of rs12597511 in the dominant mode was consistently observed on all phenotypes, after adjustment of age, gender, race, and BMI. Individuals with CT and TT genotypes (n=376) compared to those with CC genotype (n=542) had increased SBP (112.9±0.5 mmHg vs. 111.4±0.4 mmHg, p=0.009), and DBP (60.2±0.3 mmHg vs. 58.5±0.3 mmHg, p=0.001). Similarly, youths with CT and TT genotypes exhibited elevated PP than those with CC genotype (65.2±0.5 vs. 62.9±0.4 mmHg, p=0.007). Moreover, there was an increase in radial PWV in youths with CT and TT genotypes as compared to those with CC genotype (6.6±0.1 vs. 6.4±0.1 m/s, p=0.008). Thus, effects of rs12597511 became clearly significant in this combined analysis for all phenotypes. However, none of the interactions between race and rs12597511 were significant for SBP, DBP, PP or PWV.
DISCUSSION
The human prostasin gene (PRSS8) is located on chromosome 16,24 where several loci have shown strong or suggestive linkage to BP and related phenotypes by genome-wide linkage scans.31–34 It is accepted that a gene-wide examination by a minimal subset of tSNPs can effectively capture information of all common variants and haplotypes by taking into account patterns of linkage disequilibrium across the gene.35, 36 Our gene-wide analyses using tSNPs disclose the contribution of the genetic variance of human prostasin gene to BP elevation and arterial stiffness in normotensives, and therefore, the potential impact on the future development of hypertension.
The present study demonstrates consistencies for our gene association findings. First, because of the race-specific tSNP selection, genotype and phenotype association analyses were done separately in AAs and EAs. One of the tSNPs, rs12597511, was associated with BP and related phenotypes in both racial groups. Although some of the associations only reached a statistical significance in one of the two racial groups, similar trends were yet seen in both racial groups. The low minor allele frequency in AAs compared to EAs may be one explanation why effects on PP and PWV were not significant in AAs. Of note, when AAs and EAs were combined, the main effect of rs1259751 in the dominant mode was consistently observed and significant for all phenotypes including SBP, DBP and PWV. Second, a considerable number of SNPs in previously studied candidate genes in the literature are either associated with SBP, or DBP, whereas rs12597511 was found to be associated with both SBP and DBP. The SNP located in intron 2 might not change the expression or activity of prostasin, unless there are some unknown regulatory elements in intron 2. However, it can be in tight linkage disequilibrium with one or more causal variants. The increase in the levels or function of prostasin caused by functional genetic variants might amplify or result in the prostasin-dependent release of an inhibitory peptide from γ-ENaC, subsequently cleaving the channel and causing an increased channel gating or open probability.21 ENaC activation in the distal nephron augments tubular sodium reabsorption, and extracellular volume expansion, which leads to BP elevation.
Third, rs12597511 is associated with increased PWV. Arterial stiffness can be evaluated by measuring PWV between two sites in the arterial tree, with a higher PWV indicating stiffer arteries.37 There is a clear relationship between PWV and BP.38–40 BP elevation is one of the major determinants of arterial stiffness, which is related to a number of molecular changes of the load-bearing media of elastic arteries.38–40 Elevated BP in youths carrying the T allele of rs12597511 could result in arterial stiffness and related preclinical cardiovascular disease. On the other hand, after adjustment of SBP, DBP and MAP, the association of rs12597511 with PWV remained significant. It is known that increased PWV is a significant independent predictor of adverse cardiovascular events in hypertensive adults.39, 40 We thus speculate that genetic variance of the human prostasin gene might be directly involved in arterial stiffness.
The contribution of increased PWV to the pathogenesis of hypertension, cardiovascular damage, and renal dysfunction has stimulated interest in defining the determinants of arterial stiffness.39, 40 In addition to transmural pressure, structural components within the arterial wall such as collagen, elastin and cell-matrix interactions are important determinants of arterial stiffness. Furthermore, a degree of functional regulation of smooth muscle tone by circulating and locally generated vasoactive substances may also influence elastic and muscular artery stiffness.41 ENaC is expressed in endothelial cells and smooth muscle cells.42, 43 In vascular endothelium, aldosterone induces ENaC expression and insertion into the plasma membrane. Upon functional blockage with amiloride, the ENaC channels disappear from the cell surface and from intracellular pooles , indicating either channel degradation and/or membrane pinch-off.42 Recently, expression of ENaC proteins was found to be involved in normal smooth muscle cell migration, suggesting a potential new role for ENaC proteins in vascular tissue repair.43 In fact, ENaC has been recognized as a mechanosensor in sensory neurons and vascular smooth muscles, and required for vessel responses to pressure in isolated rat arteries.44, 45 Prostasin is widely distributed in various tissues including the heart and vessel wall.22, 46–48 Expression and activity of prostasin determined by gene and environment interaction may affect ENaC activity, and subsequently cause pressure-independent and/or pressure-dependent arterial alteration and stiffness. Alternatively, because of the interactions between prostasin and the renin-angiotensin-aldosterone system, prostasin increase could lead to the elevation of levels of aldosterone,22 which is a major stimulus for arterial fibrosis and collagen accumulation, thus favoring arterial stiffness.49, 50
There are concerns and limitations in the present study. First, replication in an independent population is requisite for gene association studies for complex diseases such as hypertension. Specially, this is the first gene association study for the prostasin gene. However, as discussed above, the tSNP selection was race-specific, such that race-specific genotype-phenotype association analyses were conducted. The positive findings from one racial group were able to be replicated in the other racial group, albeit some of the associations only showed non-significance statistically. We call on additional studies to potentially replicate these findings either through cross-sectional or longitudinal study design. In particular, studies investigating the association of variation in the human prostasin gene with the presence of essential hypertension in adults are warranted. Second, the functionality of the genetic variants of the human prostasin gene needs to be further elucidated using in vivo and in vitro approaches. For example, the relationships between levels of urinary prostasin and rs12597511 can be examined in vivo. Lastly, although we undertook a gene-wide study design (tSNPs) based on the human HapMap database, the gene coverage is incomplete. Re-sequencing of the prostasin gene would be more informative.
In summary, this is the first study to investigate the effect of variation in the human prostasin gene on BP and PWV. BP elevation and arterial damage tracks from late childhood into adulthood with a relatively consistent progression. Although the rs12597511 SNP seems to account for only an over 2 mmHg increase in BP and a lesser difference in PWV, affected young individuals may be prone to the future development of arterial hypertension. Finding causative genes of BP elevation and arterial stiffness in youths is of great significance and will help to identify individuals at risk for cardiovascular disease and events. This study introduces a novel candidate gene for hypertension and arterial stiffness, which would endorse further investigations including longitudinal study design, population with established hypertension, biomarker discovery, functional studies in vivo and in vitro, and potential therapeutic approaches.
Acknowledgments
This study was supported by grants from the National Heart, Lung, and Blood Institute (HL77230, HL076723, HL85817 and HL69999) and the American Heart Association (0430078N and 0435146N).
Footnotes
DISCLOSURES
The authors declare that they have no conflicts of interest.
Reference
- 1.Gormley K, Dong Y, Sagnella GA. Regulation of the epithelial sodium channel by accessory proteins. Biochem J. 2003;371:1–14. doi: 10.1042/BJ20021375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Persu A, Barbry P, Bassilana F, Houot AM, Mengual R, Lazdunski M, Corvol P, Jeunemaitre X. Genetic analysis of the beta subunit of the epithelial Na+ channel in essential hypertension. Hypertension. 1998;32:129–137. doi: 10.1161/01.hyp.32.1.129. [DOI] [PubMed] [Google Scholar]
- 3.Baker EH, Dong YB, Sagnella GA, Rothwell M, Onipinla AK, Markandu ND, Cappuccio FP, Cook DG, Persu A, Corvol P, Jeunemaitre X, Carter ND, MacGregor GA. Association of hypertension with T594M mutation in beta subunit of epithelial sodium channels in black people resident in London. Lancet. 1998;351:1388–1392. doi: 10.1016/s0140-6736(97)07306-6. [DOI] [PubMed] [Google Scholar]
- 4.Persu A, Coscoy S, Houot AM, Corvol P, Barbry P, Jeunemaitre X. Polymorphisms of the gamma subunit of the epithelial Na+ channel in essential hypertension. J Hypertens. 1999;17:639–645. doi: 10.1097/00004872-199917050-00007. [DOI] [PubMed] [Google Scholar]
- 5.Ambrosius WT, Bloem LJ, Zhou L, Rebhun JF, Snyder PM, Wagner MA, Guo C, Pratt JH. Genetic variants in the epithelial sodium channel in relation to aldosterone and potassium excretion and risk for hypertension. Hypertension. 1999;34:631–637. doi: 10.1161/01.hyp.34.4.631. [DOI] [PubMed] [Google Scholar]
- 6.Dong YB, Zhu HD, Baker EH, Sagnella GA, MacGregor GA, Carter ND, Wicks PD, Cook DG, Cappuccio FP. T594M and G442V polymorphisms of the sodium channel beta subunit and hypertension in a black population. J Hum Hypertens. 2001;15:425–430. doi: 10.1038/sj.jhh.1001182. [DOI] [PubMed] [Google Scholar]
- 7.Matsubara M, Metoki H, Suzuki M, Fujiwara T, Kikuya M, Michimata M, Ohkubo T, Hozawa A, Tsuji I, Hisamichi S, Araki T, Imai Y. Genotypes of the betaENaC gene have little influence on blood pressure level in the Japanese population. Am J Hypertens. 2002;15:189–192. doi: 10.1016/s0895-7061(01)02266-x. [DOI] [PubMed] [Google Scholar]
- 8.Nkeh B, Samani NJ, Badenhorst D, Libhaber E, Sareli P, Norton GR, Woodiwiss AJ. T594M variant of the epithelial sodium channel beta-subunit gene and hypertension in individuals of African ancestry in South Africa. Am J Hypertens. 2003;16:847–852. doi: 10.1016/s0895-7061(03)01016-1. [DOI] [PubMed] [Google Scholar]
- 9.Hollier JM, Martin DF, Bell DM, Li JL, Chirachanchai MG, Menon DV, Leonard D, Wu X, Cooper RS, McKenzie C, Victor RG, Auchus RJ. Epithelial sodium channel allele T594M is not associated with blood pressure or blood pressure response to amiloride. Hypertension. 2006;47:428–433. doi: 10.1161/01.HYP.0000200704.45994.ff. [DOI] [PubMed] [Google Scholar]
- 10.Busst CJ, Scurrah KJ, Ellis JA, Harrap SB. Selective genotyping reveals association between the epithelial sodium channel gamma-subunit and systolic blood pressure. Hypertension. 2007;50:672–678. doi: 10.1161/HYPERTENSIONAHA.107.089128. [DOI] [PubMed] [Google Scholar]
- 11.Wong ZY, Stebbing M, Ellis JA, Lamantia A, Harrap SB. Genetic linkage of beta and gamma subunits of epithelial sodium channel to systolic blood pressure. Lancet. 1999;353:1222–1225. doi: 10.1016/S0140-6736(98)10118-6. [DOI] [PubMed] [Google Scholar]
- 12.Iwai N, Baba S, Mannami T, Ogihara T, Ogata J. Association of a sodium channel alpha subunit promoter variant with blood pressure. J Am Soc Nephrol. 2002;13:80–85. doi: 10.1681/ASN.V13180. [DOI] [PubMed] [Google Scholar]
- 13.Fava C, von Wowern F, Berglund G, Carlson J, Hedblad B, Rosberg L, Burri P, Almgren P, Melander O. 24-h ambulatory blood pressure is linked to chromosome 18q21-22 and genetic variation of NEDD4L associates with cross-sectional and longitudinal blood pressure in Swedes. Kidney Int. 2006;70:562–569. doi: 10.1038/sj.ki.5001590. [DOI] [PubMed] [Google Scholar]
- 14.von Wowern F, Berglund G, Carlson J, Mansson H, Hedblad B, Melander O. Genetic variance of SGK-1 is associated with blood pressure, blood pressure change over time and strength of the insulin-diastolic blood pressure relationship. Kidney Int. 2005;68:2164–2172. doi: 10.1111/j.1523-1755.2005.00672.x. [DOI] [PubMed] [Google Scholar]
- 15.Busjahn A, Aydin A, Uhlmann R, Krasko C, Bahring S, Szelestei T, Feng Y, Dahm S, Sharma AM, Luft FC, Lang F. Serum- and glucocorticoid-regulated kinase (SGK1) gene and blood pressure. Hypertension. 2002;40:256–260. doi: 10.1161/01.hyp.0000030153.19366.26. [DOI] [PubMed] [Google Scholar]
- 16.Russo CJ, Melista E, Cui J, DeStefano AL, Bakris GL, Manolis AJ, Gavras H, Baldwin CT. Association of NEDD4L ubiquitin ligase with essential hypertension. Hypertension. 2005;46:488–491. doi: 10.1161/01.HYP.0000178594.63193.c0. [DOI] [PubMed] [Google Scholar]
- 17.Trochen N, Ganapathipillai S, Ferrari P, Frey BM, Frey FJ. Low prevalence of nonconservative mutations of serum and glucocorticoid-regulated kinase (SGK1) gene in hypertensive and renal patients. Nephrol Dial Transplant. 2004;19:2499–2504. doi: 10.1093/ndt/gfh417. [DOI] [PubMed] [Google Scholar]
- 18.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–610. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 19.Vallet V, Horisberger JD, Rossier BC. Epithelial sodium channel regulatory proteins identified by functional expression cloning. Kidney Int Suppl. 1998;67:S109–S114. doi: 10.1046/j.1523-1755.1998.06721.x. [DOI] [PubMed] [Google Scholar]
- 20.Narikiyo T, Kitamura K, Adachi M, Miyoshi T, Iwashita K, Shiraishi N, Nonoguchi H, Chen LM, Chai KX, Chao J, Tomita K. Regulation of prostasin by aldosterone in the kidney. J Clin Invest. 2002;109:401–408. doi: 10.1172/JCI13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma-subunit. J Biol Chem. 2007;282:6153–6160. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Chao J, Chao L. Adenovirus-mediated human prostasin gene delivery is linked to increased aldosterone production and hypertension in rats. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1031–R1036. doi: 10.1152/ajpregu.00660.2002. [DOI] [PubMed] [Google Scholar]
- 23.Olivieri O, Castagna A, Guarini P, Chiecchi L, Sabaini G, Pizzolo F, Corrocher R, Righetti PG. Urinary prostasin: a candidate marker of epithelial sodium channel activation in humans. Hypertension. 2005;46:683–688. doi: 10.1161/01.HYP.0000184108.12155.6b. [DOI] [PubMed] [Google Scholar]
- 24.Yu JX, Chao L, Ward DC, Chao J. Structure and chromosomal localization of the human prostasin (PRSS8) gene. Genomics. 1996;32:334–340. doi: 10.1006/geno.1996.0127. [DOI] [PubMed] [Google Scholar]
- 25.Andrew T, Hart DJ, Snieder H, de Lange M, Spector TD, MacGregor AJ. Are twins and singletons comparable? A study of disease-related and lifestyle characteristics in adult women. Twin Res. 2001;4:464–477. doi: 10.1375/1369052012803. [DOI] [PubMed] [Google Scholar]
- 26.Kupper N, Willemsen G, Riese H, Posthuma D, Boomsma DI, de Geus EJ. Heritability of daytime ambulatory blood pressure in an extended twin design. Hypertension. 2005;45:80–85. doi: 10.1161/01.HYP.0000149952.84391.54. [DOI] [PubMed] [Google Scholar]
- 27.Snieder H, Treiber FA. The Georgia Cardiovascular Twin Study. Twin Res. 2002;5:497–498. doi: 10.1375/136905202320906354. [DOI] [PubMed] [Google Scholar]
- 28.Kapuku GK, Treiber FA, Davis HC, Harshfield GA, Cook BB, Mensah GA. Hemodynamic function at rest, during acute stress, and in the field: predictors of cardiac structure and function 2 years later in youth. Hypertension. 1999;34:1026–1031. doi: 10.1161/01.hyp.34.5.1026. [DOI] [PubMed] [Google Scholar]
- 29.Nichols W, O'Rourke MF. McDonalds's Blood Flow in Arteries: Theoretical, Experimental and Clinical Principles. 4th ed. London, UK: Arnold; 1998. [Google Scholar]
- 30.Ge D, Zhu H, Huang Y, Treiber FA, Harshfield GA, Snieder H, Dong Y. Multilocus analyses of Renin-Angiotensin-aldosterone system gene variants on blood pressure at rest and during behavioral stress in young normotensive subjects. Hypertension. 2007;49:107–112. doi: 10.1161/01.HYP.0000251524.00326.e7. [DOI] [PubMed] [Google Scholar]
- 31.de Lange M, Spector TD, Andrew T. Genome-wide scan for blood pressure suggests linkage to chromosome 11, and replication of loci on 16, 17, and 22. Hypertension. 2004;44:872–877. doi: 10.1161/01.HYP.0000148994.89903.fa. [DOI] [PubMed] [Google Scholar]
- 32.Bell JT, Wallace C, Dobson R, Wiltshire S, Mein C, Pembroke J, Brown M, Clayton D, Samani N, Dominiczak A, Webster J, Lathrop GM, Connell J, Munroe P, Caulfield M, Farrall M. Two-dimensional genome-scan identifies novel epistatic loci for essential hypertension. Hum Mol Genet. 2006;15:1365–1374. doi: 10.1093/hmg/ddl058. [DOI] [PubMed] [Google Scholar]
- 33.Bella JN, Tang W, Kraja A, Rao DC, Hunt SC, Miller MB, Palmieri V, Roman MJ, Kitzman DW, Oberman A, Devereux RB, Arnett DK. Genome-wide linkage mapping for valve calcification susceptibility loci in hypertensive sibships: the Hypertension Genetic Epidemiology Network Study. Hypertension. 2007;49:453–460. doi: 10.1161/01.HYP.0000256957.10242.75. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmann K, Mattheisen M, Dahm S, Nurnberg P, Roe C, Johnson J, Cox NJ, Wichmann HE, Wienker TF, Schulze J, Schwarz PE, Lindner TH. A German genome-wide linkage scan for type 2 diabetes supports the existence of a metabolic syndrome locus on chromosome 1p36.13 and a type 2 diabetes locus on chromosome 16p12.2. Diabetologia. 2007;50:1418–1422. doi: 10.1007/s00125-007-0658-4. [DOI] [PubMed] [Google Scholar]
- 35.Neale BM, Sham PC. The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004;75:353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson GC, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, Twells RC, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SC, Clayton DG, Todd JA. Haplotype tagging for the identification of common disease genes. Nat Genet. 2001;29:233–237. doi: 10.1038/ng1001-233. [DOI] [PubMed] [Google Scholar]
- 37.Ter Avest E, Stalenhoef AF, de Graaf J. What is the role of non-invasive measurements of atherosclerosis in individual cardiovascular risk prediction? Clin Sci (Lond) 2007;112:507–516. [PubMed] [Google Scholar]
- 38.Willum-Hansen T, Staessen J, C T-P, Rasmussen S, Thijs L, Ibsen H, Jeppesen J. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation. 2006;113:664–670. doi: 10.1161/CIRCULATIONAHA.105.579342. [DOI] [PubMed] [Google Scholar]
- 39.Laurent S, Boutouyrie P. Recent advances in arterial stiffness and wave reflection in human hypertension. Hypertension. 2007;49:1202–1206. doi: 10.1161/HYPERTENSIONAHA.106.076166. [DOI] [PubMed] [Google Scholar]
- 40.Safar ME, Levy BI, Struijker-Boudier H. Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation. 2003;107:2864–2869. doi: 10.1161/01.CIR.0000069826.36125.B4. [DOI] [PubMed] [Google Scholar]
- 41.Avolio A, Jones D, Tafazzoli-Shadpour M. Quantification of alterations in structure and function of elastin in the arterial media. Hypertension. 1998;32:170–175. doi: 10.1161/01.hyp.32.1.170. [DOI] [PubMed] [Google Scholar]
- 42.Kusche-Vihrog K, Sobczak K, Bangel N, Wilhelmi M, Nechyporuk-Zloy V, Schwab A, Schillers H, Oberleithner H. Aldosterone and amiloride alter ENaC abundance in vascular endothelium. Pflugers Arch. 2007 doi: 10.1007/s00424-007-0341-0. [DOI] [PubMed] [Google Scholar]
- 43.Grifoni SC, Gannon KP, Stec DE, Drummond HA. ENaC proteins contribute to VSMC migration. Am J Physiol Heart Circ Physiol. 2006;291:H3076–H3086. doi: 10.1152/ajpheart.00333.2006. [DOI] [PubMed] [Google Scholar]
- 44.Drummond HA, Gebremedhin D, Harder DR. Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension. 2004;44:643–648. doi: 10.1161/01.HYP.0000144465.56360.ad. [DOI] [PubMed] [Google Scholar]
- 45.Benos DJ. Sensing tension: recognizing ENaC as a stretch sensor. Hypertension. 2004;44:616–617. doi: 10.1161/01.HYP.0000144467.43205.ed. [DOI] [PubMed] [Google Scholar]
- 46.Yu JX, Chao L, Chao J. Prostasin is a novel human serine proteinase from seminal fluid. Purification, tissue distribution, and localization in prostate gland. J Biol Chem. 1994;269:18843–18848. [PubMed] [Google Scholar]
- 47.Yu JX, Chao L, Chao J. Molecular cloning, tissue-specific expression, and cellular localization of human prostasin mRNA. J Biol Chem. 1995;270:13483–13489. doi: 10.1074/jbc.270.22.13483. [DOI] [PubMed] [Google Scholar]
- 48.Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, Patapoutian A, Hampton GM, Schultz PG, Hogenesch JB. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lacolley P, Safar ME, Lucet B, Ledudal K, Labat C, Benetos A. Prevention of aortic and cardiac fibrosis by spironolactone in old normotensive rats. J Am Coll Cardiol. 2001;37:662–667. doi: 10.1016/s0735-1097(00)01129-3. [DOI] [PubMed] [Google Scholar]
- 50.Safar ME, Cattan V, Lacolley P, Nzietchueng R, Labat C, Lajemi M, de Luca N, Benetos A. Aldosterone synthase gene polymorphism, stroke volume and age-related changes in aortic pulse wave velocity in subjects with hypertension. J Hypertens. 2005;23:1159–1166. doi: 10.1097/01.hjh.0000170378.08214.13. [DOI] [PubMed] [Google Scholar]