

Abstract
The pharmacological function of heroin requires an activation process which transforms heroin into 6-monoacetylmorphine (6-MAM) which is the most active form. The primary enzyme responsible for this activation process in human plasma is butyrylcholinesterase (BChE). The detailed reaction pathway of the activation process via BChE-catalyzed hydrolysis has been explored computationally, for the first time, in the present study by performing molecular dynamics simulation and first-principles quantum mechanical/molecular mechanical free energy calculations. It has been demonstrated that the whole reaction process includes acylation and deacylation stages. The acylation consists of two reaction steps, i.e. the nucleophilic attack on the carbonyl carbon of 3-acetyl group of heroin by the hydroxyl oxygen of Ser198 side chain and the dissociation of 6-MAM. The deacylation also consists of two reaction steps, i.e. the nucleophilic attack on the carbonyl carbon of the acyl-enzyme intermediate by a water molecule and the dissociation of the acetic acid from Ser198. The calculated free energy profile reveals that the second transition state (TS2) should be rate-determining. The structural analysis reveals that the oxyanion hole of BChE plays an important role in the stabilization of the rate-determining transition state TS2. The free energy barrier (15.9±0.2 or 16.1±0.2 kcal/mol) calculated for the rate-determining step is in good agreement with the experimentally-derived activation free energy (~16.2 kcal/mol), suggesting that the mechanistic insights obtained from the present computational study are reliable. The obtained structural and mechanistic insights could be valuable for use in future rational design of a novel therapeutic treatment of heroin abuse.
Introduction
Heroin (3,6-diacetylmorphine) is an illegal, highly addictive opiate drug.(1) Heroin produces euphoria or pleasurable feelings. A pleasurable euphoria feeling is usually followed by drowsy feeling for several hours due to depression of the central nervous system (CNS). Because of the drowsy feeling and depression of the CNS, the drug may impair judgment and memory, and cloud mental functioning.
Heroin is the most abused one of the opiates.(1) For example, in 2011, 4.2 million Americans reported using heroin at some time in their lives, with 23% of the individuals were classified with dependence on or abuse of heroin.(1) Heroin abuse can result in serious health problems including miscarriages, heart infections, and even death. In addition, heroin abuse also has repercussions that extend far beyond the individual users. The medical and social consequences of heroin abuse include transmission of HIV/AIDS, tuberculosis, crime, violence, and disruptions in family, workplace, and educational environments. These consequences have a devastating impact on our society and cost billions of dollars per year.(1) The disastrous medical and social consequences of heroin addiction have made a high priority the development of an effective pharmacological treatment of heroin abuse.
Heroin is also the most rapidly acting of the opiates.(2) However, heroin actually has a very low μ-opioid receptor binding affinity and, thus, heroin itself has a low potency in activating the G-protein to produce its effects on neurotransmitter systems.(3) Nevertheless, once injected, heroin is very rapidly metabolized via hydrolysis to 6-monoacetylmorphine (6-MAM) (Scheme 1), which can readily cross the blood-brain barrier (BBB) and be rapidly concentrated in the brain.(4–6) More importantly, 6-MAM has a higher μ-opioid receptor affinity than its precursor heroin.(3) It has been recognized that heroin acts principally via 6-MAM.(3, 7–10) These pharmacological characteristics of heroin suggest that heroin actually behaves as a prodrug.(11–12) The prodrug is metabolized into 6-MAM which is actually the most potent form of the drug responsible for the main pharmacological effects of heroin. From this point of view, the hydrolysis of heroin to 6-MAM may be regarded as an activation process, i.e. the activation of heroin itself, which transforms heroin (a relatively inactive form) into 6-MAM (the most active form).
Scheme 1.
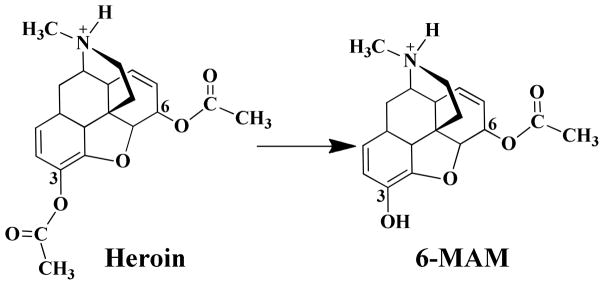
BChE-catalyzed hydrolysis of heroin to 6-MAM
Concerning the endogenous enzymes responsible for the activation process, while carboxylesterases 1 and 2 (hCE-1 and hCE-2) in liver and brain can catalyze the hydrolysis of heroin to 6-MAM,(13) serum butyrylcholinesterase (BChE) and erythrocyte acetylcholinesterase (AChE) can also hydrolyze heroin to 6-MAM in blood.(9, 13–14) Although many tissues have the ability to hydrolyze heroin, blood is generally the first to act on heroin.(9) According to the results from Boix et al.,(15) it is the high blood metabolism rate of heroin and the good BBB permeability of 6-MAM, rather than heroin itself, that could account for the highly efficient delivery of the active metabolites to the brain after heroin is administered. The experimental results reported by Lockridge et al.(9) also indicated that serum should be the major site of 6-MAM production. In comparison, human serum BChE can hydrolyze heroin to 6-MAM with a reaction rate of 4.5/min per μmol/L, which is nine-fold of the heroin hydrolysis rate (0.5/min per μmol/L) by human erythrocyte AChE.(14) Therefore, BChE should be the major enzyme hydrolyzing heroin to 6-MAM in human body. Because BChE can hydrolyze heroin to 6-MAM efficiently with a high catalytic rate constant (kcat = 540 min−1),(14) the half-life of heroin itself in human is only ~2 to 5 minutes.(14, 16–18) In comparison, 6-MAM has a much longer half-life (up to 52 minutes) in human body.(19)
Based on the above background, it is interesting for understanding the molecular mechanism of heroin abuse to uncover the detailed catalytic mechanism concerning how human BChE hydrolyzes heroin to 6-MAM and thus activates heroin. A detailed understanding of the molecular mechanism could provide valuable mechanistic clues for rational design of a novel therapeutic treatment of heroin abuse.
In the present study, the detailed reaction pathway for BChE-catalyzed hydrolysis of heroin to 6-MAM has been explored, for the first time, by performing molecular dynamics (MD) simulation and first-principles quantum mechanical/molecular mechanical (QM/MM)-free energy (QM/MM-FE) calculations.(20–23) In the QM/MM-FE calculations, first-principles QM/MM reaction-coordinate calculations were followed by free energy perturbation (FEP) calculations to account for the dynamic effects of the protein environment on the free energy profile for the catalytic reaction process. Our QM/MM calculations are based on the pseudobond first-principles QM/MM approach,(20–23) which has been demonstrated to be a powerful tool in simulating a variety of enzymes,(24–35) and some theoretical predictions have been confirmed by subsequent experimental studies.(28, 35) The computational results clearly reveal the detailed reaction pathway and the corresponding free energy profile for BChE-catalyzed hydrolysis of heroin. Based on the calculated free energy profile for the favorable reaction pathway of BChE-catalyzed hydrolysis process, we were able to identify the rate-determining step and understand the roles of essential residues in light of the QM/MM-optimized geometries.
Computational Methods
Preparation of initial structure of the reaction system
The initial enzyme-substrate (ES) structure was constructed from the X-ray crystal structure of human BChE (PDB ID: 2XQF)(36) and the optimized structure of heroin.(37) Initially, the geometry of heroin was optimized at the HF/6–31G* level using Gaussian03 program.(38) Then, the optimized geometry of heroin was used to calculate the electrostatic potential distribution on the molecular surface at the same HF/6–31G* level. The calculated electrostatic potential distribution was used to determine the partial atomic charges by using the standard restrained electrostatic potential (RESP) fitting procedure(39–40) implemented in the Antechamber module of the AMBER11 program.(41) The determined RESP charges of the heroin atoms were used in the MD simulations described below.
To construct a reasonable binding structure of BChE binding with heroin, molecular docking and MD simulations were performed. Heroin was docked into the active site of BChE by using the AutoDock 4.0 program.(42) The docking process started from a conformational search by using the Solis and Wets local search method and the Lamarkian genetic algorithm (LGA) which was used to deal with the BChE-heroin interactions. The grid size used in the docking was 60 × 60 × 60 with the grid space of 0.375 Å (default). The docked enzyme-ligand complex structure was selected according to the binding energy.
The complex structure of BChE binding with heroin obtained from the molecular docking was used as the starting structures for the energy minimization and MD simulation using the Sander module of AMBER11.(41) The AMBER ff03 force field(43) was used for protein, and the general AMBER force field (gaff)(44) was used for heroin in the energy minimization and MD simulation. The BChE-heroin binding complex was first solvated in an orthorhombic box of TIP3P water molecules(45) with a minimum solute-wall distance of 10 Å and neutralized by adding one chloride ion. The final system size was about 103 Å × 116 Å × 119 Å, composed of 71,173 atoms. The solvated system was subjected to the energy minimization using the AMBER11 program with a non-bonded cutoff of 10 Å and a conjugate gradient energy-minimization method. The energy minimization was performed first for 20,000 steps for the water molecules, and then 6,000 steps for the backbone atoms, followed by additional 20,000 steps for the side chain atoms of the enzyme together with water molecules. Finally, the whole system was energy-minimized until a convergence criterion of 0.001 kcal ·mol−1·Å−1 was achieved.
Starting from the energy-minimized complex structure, MD simulation was performed by using the Sander module of AMBER11.(41) The energy-minimized complex structure was gradually heated to 300 K by using the weak-coupling method with a constrained force constant of 50 kcal·mol−1·Å−2 on the backbone of BChE. The constrained force constant was gradually decreased during a period of 600 ps equilibration and finally removed for the production MD simulation. Throughout the MD simulation, a 10 Å non-bonded interaction cutoff was used and the non-bonded list was updated once every 1,000 steps. The particle mesh Ewald (PME) method(46) was applied to the calculation of the long-range electrostatic interactions. During the MD simulation, the SHAKE algorithm(47) was used to fix the lengths of covalent bonds involving a hydrogen atom, enabling the use of a 2-fs time step to numerically integrate the equations of motion. The production MD run was kept for 2 ns with a periodic boundary condition in the NTP ensemble at T = 300 K using Berendsen temperature coupling and at P = 1 atm using anisotropic molecule-based scaling.
As seen in Scheme 2, there are two reaction stages (acylation and deacylation) in the BChE-catalyzed hydrolysis of heroin. The reaction product 6-MAM leaves the protein after the acylation stage. Consequently, the initial structure of INT2′ was constructed by removing the 6-MAM from the QM/MM-optimized INT2 structure. The constructed INT2′ structure was then relaxed by performing a ~2 ns MD simulation. The simulated INT2′ system was solvated in an orthorhombic box of TIP3P water molecules,(45) with a minimum solute-wall distance of 10 Å.
Scheme 2.

Fundamental reaction pathway for BChE-catalyzed hydrolysis of heroin to 6-MAM. The atoms in blue belong to the QM subsystem, the atoms in red are treated with the improved pseudobond parameters, and the other atoms belong to the MM subsystem in the pseudobond first-principles QM/MM calculations.
For each reaction stage (acylation or deacylation), the last snapshot of the MD simulation was used to prepare the pseudobond first-principles QM/MM calculation as we noted that the structure of the last snapshot was close to the average structure simulated. As we are mainly interested in the reaction center, the water molecules beyond 50 Å of the hydroxy oxygen (Oγ) of Ser198 were removed. Thus, the QM/MM system consisted of 3,760 water molecules and a total of 19,696 atoms for the acylation, and 4,232 water molecules and a total of 21,065 atoms for the deacylation. The QM/MM partition strategy was based on the treatment in previous QM/MM calculations on the hydrolysis reactions of other substrates catalyzed by BChE or another serine esterase.(23–24, 26, 28, 48) The QM subsystem in the acylation stage consisted of heroin and the catalytic triad (side chains of Ser198, His438, and Glu325), whereas the QM subsystem in the deacylation stage included the reactive water molecule and side chains of acetyl-Ser198, His438, and Glu325. A pseudobond approach(20–23) was used to deal with the QM-MM interface; see below for the used boundary of the QM-MM system. For QM/MM geometry optimization on each state of the reaction system, the initial geometry was energy-minimized first with the MM method by using the AMBER11 program(41) until the convergence criterion for energy gradient of 0.1 kcal·mol−1·Å−1 was achieved.
Minimum-energy path of the reaction
With a reaction-coordinate driving method and an iterative energy minimization procedure,(21) the enzymatic reaction path was determined by performing the pseudobond QM/MM calculations at the B3LYP/6–31G*:AMBER level. Specifically, the QM calculations were performed with the B3LYP functional(49–51) and 6–31G* basis set by using a modified version of Gaussian03 program(38) and the MM calculations were carried out by using a modified version of the AMBER8 program.(52) Normal mode analysis was performed to characterize the geometries optimized for the reactant, intermediates, transition sates, and the final product of the reaction process. The QM/MM-optimized structures were used to perform single-point QM/MM energy calculations with the QM part performed at the B3LYP/6–31++G** level. The B3LYP functional has been used extensively in previous QM/MM calculations on enzymatic reaction systems by many researchers including our own group,(25, 27, 31, 33–34, 53–54) and the calculated results are generally in good agreement with the experimental data. For comparison, the QM part of the single-point QM/MM energy calculations were also performed at the B3LYP/6–31+G* and B3LYP/6–311++G** levels for the rate-determining step. In the QM/MM calculations, the boundary carbon atoms were treated with previously improved pseudobond parameters,(20–23) and no cutoff was applied to the non-bonded interactions. The convergence criterion used in the geometry optimization on the QM subsystem was the same as the original Gaussian03 default, and the convergence criterion used in the geometry optimization on the MM subsystem was that the root-mean-square deviation (RMSD) of energy gradient is ≤ 0.1 kcal·mol−1·Å−1. In all QM/MM geometry optimizations, the atoms within 20 Å of Oγ atom of Ser198 side chain (Scheme 2) were allowed to move while all the other atoms outside this range were frozen, and the QM and MM subsystems were energy-minimized iteratively. For each iteration step of the QM/MM geometry optimization, the MM subsystem was energy-minimized while the QM subsystem was kept frozen, whereas the QM subsystem was energy-minimized while the MM subsystem was kept frozen.
Free energy perturbation
Free energy perturbation (FEP) is known as a reliable computational method in combination with the MD or Monte Carlo (MC) simulation to evaluate free energy change caused by a small structural change.(55–58) This method has been used extensively in computational studies on organic reactions,(55, 58–60) protein-ligand interaction,(61–66) and protein stability.(67–68) In the present study, after the minimum-energy path was determined by the QM/MM calculations, the free energy changes associated with the QM-MM interactions were evaluated by using the FEP method.(21) The sampling of the MM subsystem was performed with the QM subsystem frozen at each state along the reaction path in the FEP calculations. During the FEP calculations, the used atomic point charges on the frozen QM atoms were determined by fitting the electrostatic potential (ESP) in the QM part of the QM/MM single-point calculations. The total free energy change from the reactant to the transition state was evaluated by using the same procedure as used in our previous computational studies on other enzymatic reaction systems.(24–34) Using our own version of the FEP implementation(24), the FEP calculations enabled us to more reasonably evaluate the relative free energy changes due to the QM-MM interactions. The final relative free energy predicted by the QM/MM-FE calculations was the QM part of the QM/MM energy (excluding the Coulumbic interaction energy between the point charges of the MM atoms and the ESP charges of the QM atoms) plus the relative free energy change obtained from the FEP calculations at 298.15 K. The time step used in the FEP calculations was 2 fs, and the lengths of all covalent bonds involving a hydrogen atom were constrained. Each MD-based FEP calculation consisted of 50 ps of equilibration and 300 ps of sampling.
All of the MD simulations and QM/MM-FE calculations were performed on a Dell supercomputer (X-series Cluster with 384 nodes or 4,768 processors) at University of Kentucky’s Computer Center. The other modeling and computations were performed on SGI Fuel workstations in our own laboratory at University of Kentucky.
Results and Discussion
BChE-heroin binding structure from MD simulation
The docked BChE-heroin complex structure revealed that the 3-acetyl group of heroin was positioned in the oxyanion hole consisting of Gly116, Gly117, and Ala199, and the positively charged amino-group was positioned in the choline binding site nearby Trp82. Further energy minimizations and a ~2 ns MD simulation were performed on the docked enzyme-substrate (ES) complex to refine the BChE-heroin binding structure. Depicted in Figure 1 are plots of the key internuclear distances and the root-mean-square deviation (RMSD) of the positions of the all backbone atoms vs the simulation time in the MD-simulated ES complex. Traces D1, D2, and D3 represent the internuclear distances between the carbonyl oxygen O1 of the 3-acetyl group of heroin and the backbone NH hydrogen atoms of residues Gly116, Gly117, and Ala199, respectively. Trace D4 refers to the internuclear distance between the hydroxyl oxygen Oγ of Ser198 side chain and carbonyl carbon C1 of the 3-acetyl group of heroin, and trace D5 represents the internuclear distance between the hydroxyl hydrogen Hγ of Ser198 side chain and Nε atom of His438 side chain.
Figure 1.

Plots of key internuclear distances and RMSD vs the simulation time in the MD-simulated ES structure.
As seen in Figure 1B, in the MD-simulated ES complex structure, the average values of D1, D2, and D3 are 2.49, 1.97, and 3.10 Å, respectively, indicating that only two hydrogen bonds are formed between the carbonyl oxygen of 3-acetyl group of heroin and the oxyanion hole (consisting of the backbone NH groups of Gly116, Gly117, and Ala199). As shown in Figure 1C, the average value (3.35 Å) of D4 indicates that the hydroxyl oxygen Oγ of Ser198 side chain is in an appropriate position to initiate the nucleophilic attack on the C1 of heroin. The average value of D5 is 1.87 Å, showing that a strong hydrogen bond is formed between the hydroxyl group of Ser198 side chain and the Nε atom of His438 side chain. RMSD of the positions of all backbone atoms from those in the initial structure was also monitored to represent the overall conformational change of the protein during the MD simulation. As shown in Figure 1C, the MD trajectory was stabilized at ~100 ps, suggesting that 2 ns is long enough for the MD simulation on the ES complex.
Reaction pathway for BChE-catalyzed hydrolysis of heroin to 6-MAM
As shown in Figure 1B and 1C, the MD simulation led to a dynamically stable ES complex structure. The QM/MM reaction-coordinate calculations at the B3LYP/6–31G*:AMBER level starting from the MD-simulated ES complex structure revealed that the BChE-catalyzed heroin hydrolysis reaction pathway should consist of four reaction steps. The whole reaction is initiated by the nucleophilic attack of Oγ of Ser198 side chain on the carbonyl carbon C1 of heroin, followed by the dissociation between acetyl-enzyme intermediate and 6-MAM. Then, a water molecule initiates the nucleophilic attack on the carbonyl carbon (C1) of the acylated Ser198. Finally, the dissociation of the acetic acid regenerates Ser198 of BChE. The QM/MM-optimized structures of the reactant, intermediates, transition states, and final product are depicted in Figure 2. Below we discuss each of the reaction steps in detail.
Figure 2.

(A) Division of the QM/MM system. Atoms in blue color belong to the QM subsystem, the boundary carbon atoms in red were treated with the pseudobond parameters, and all of the other atoms were included in the MM subsystem. (B–F) Geometries optimized at the QM/MM(B3LYP/6–31G*:AMBER) level for the key states during the acylation stage of BChE-catalyzed hydrolysis of heroin. The key distances indicated in the figures are given in Å. The carbon, oxygen, nitrogen, and hydrogen atoms are in green, red, blue, and white, respectively. Represented as balls and sticks are the QM atoms, with the surrounding residues rendered as sticks and the protein rendered as cartoon.
Step 1. The initial nucleophilic attack on the carbonyl carbon of heroin
The initial structure of the ES complex was first optimized at the QM/MM(B3LYP/6–31G*:AMBER) level prior to the reaction-coordinate calculations. As shown in Figure 2B for the QM/MM-optimized ES structure, D1, D2, and D3 are 1.88, 1.87, and 2.54 Å, respectively, suggesting that two strong hydrogen bonds are formed between the carbonyl oxygen O1 of heroin and the oxyanion hole. One is between the backbone NH group of Gly116 and O1 of heroin, and the other is between the NH group of Gly117 and O1 of heroin. In comparison, the D3 = 2.54 Å, indicating that the interaction between the backbone NH of Ala199 and the carbonyl oxygen O1 of heroin is much weaker.
The nucleophilic attack process proceeds as the hydroxyl oxygen Oγ of Ser198 side chain gradually approaches to the carbonyl carbon C1 of heroin, while the hydroxyl hydrogen Hγ of Ser198 gradually transfers to the Nε atom of His438 side chain. This reaction step involves the breaking of the Hγ–Oγ bond and the formation of both the Hγ– Nε and C1–Oγ bonds, as shown in Scheme 2. So, the changes of the distances Hγ–Oγ (RHγ-Oγ), Hγ–Nε(RHγ-Nε), and C1–Oγ (RC1-Oγ) can reflect the nature of reaction step 1. Therefore, RHγ-Oγ–RHγ-Nε–RC1-Oγ was used as the reaction coordinate in the QM/MM reaction-coordinate calculations for this reaction step. As shown in Figure 2, RC1-Oγ shortens from 2.74 Å in ES to 1.81 Å in TS1 and then to 1.62 Å in INT1. Simultaneously, RHγ-Oγ elongates from 1.00 Å in ES to 1.61 Å in TS1 and then to 1.70 Å in INT1, while RHγ-Nε shortens from 1.79 Å in ES to 1.07 Å in TS1 and then to 1.06 Å in INT1. These structural variations indicate that the C1–Oγ bond has formed and the hydroxyl hydrogen Hγ has transferred to Nε atom of His438 in intermediate INT1. Noteworthy, it is interesting to know the catalytic role of the oxyanion hole in this reaction step, although the distance D1 (between the hydrogen of NH group in Gly117 and O1 of heroin) is elongated from 1.88 Å in ES to 1.99 Å in TS1 and then to 2.05 Å in INT1, the distances D2 (between the hydrogen of NH group in Gly116 and O1 of heroin) and D3 (between the hydrogen of NH group in Ala199 and O1 of heroin) are shortened. In particular, D3 = 2.54 Å in ES, 2.02 Å in TS1, and 1.94 Å in INT1. In other words, the interaction between the oxyanion hole and the negatively charged carbonyl oxygen (O1) of heroin is strengthened, which helps to stabilize the transition state TS1 and intermediate INT1. Moreover, accompanied by the proton (Hγ) transfer from the hydroxyl oxygen (Oγ) of Ser198 to the Nε atom of His438, the imidazole ring of His438 side chain becomes more and more positively charged. However, the developed positive charge on the imidazole ring of His438 can be stabilized by the negative charge of Glu325 side chain, which is demonstrated by the shortened hydrogen-bond distance between the carboxylate anion of Glu325 and the proton (Hδ) on Nδ of His438 (the Hδ···O distance in the Nδ–Hδ ···O hydrogen bond: 1.66 Å in ES, 1.57 Å in TS1, and 1.56 Å in INT1).
Step 2. Dissociation of 6-MAM from the enzyme
In this reaction step, the C1–O2 bond is gradually broken while the proton (Hγ) transfers from Nε of His438 to O2, resulting in the dissociation of 6-MAM from the enzyme. This reaction step involves the breaking of the Nε–Hγ and C1–O2 bonds and the formation of the O2–Hγ bond. The nature of this reaction step can be represented by the changes of the Nε–Hγ distance (RNε-Hγ), the C1–O2 distance (RC1–O2), and the O2–Hγ distance (RO2-Hγ). Thus, the reaction coordinate in the QM/MM calculations for this step was chosen as RNε-Hγ+RC1–O2–RO2-Hγ. In the INT1 structure, the proton (Hγ) of Ser198 side chain has been transferred to the Nε of His438 in reaction step 1 while the distance ROγ-Hγ between the Oγ atom of Ser198 and the Hγ atom of His438 is 1.70 Å, indicating a very strong hydrogen bond (Nε–Hγ ···Oγ) between the oxygen of Ser198 side chain and the NH group of His438 side chain. The distance (RO2-Hγ) between the Hγ atom and the leaving ester oxygen O2 atom to which Hγ is about to be transferred is 2.73 Å. However, accompanied by the elongation of RC1–O2 (1.57 Å in INT1 and 1.86 Å in TS2), RO2-Hγ is gradually shortened (2.73 Å in INT1 and 2.48 Å in TS2). Our reaction-coordinate calculations reveal that it is a one-step reaction process consisting of two substeps. In the first substep, the C1–O2 bond is broken. In the second substep, the proton (Hγ) gradually transfers to O2. It should be noted that the interaction between the oxyanion hole and the negatively charged carbonyl oxygen (O1) of heroin is enhanced from INT1 to TS2, indicating that the oxyanion hole plays an important role in stabilization of the transition state TS2. In addition, associated with these structural variations, the positive charge on the imidazole ring of His438 is decreased and, therefore, the hydrogen bond between the carboxylate anion of Glu325 and the proton (Hδ) of His438 is weakened during this reaction step (Nδ–Hδ ···O distance: 1.56 Å in INT1, 1.54 Å in TS2, and 1.74 Å in INT2).
Step 3. Nucleophilic attack on the carbonyl carbon of heroin by a water molecule
The generated reaction product 6-MAM was removed from the above-discussed QM/MM-optimized geometry of INT2 to construct the initial structure of INT2′ which was then relaxed by performing the MD simulation. A water molecule which is close to the carbonyl carbon (C1) of heroin was selected as the nucleophile and was included in the QM part of the QM/MM calculation. As shown in Figure 3, two strong hydrogen bonds are formed between the carbonyl oxygen (O1) and the oxyanion hole in the QM/MM-optimized geometry of INT2′.
Figure 3.

Geometries optimized at the QM/MM(B3LYP/6–31G*:AMBER) level for the key states of the reaction system in the deacylation stage of BChE-catalyzed hydrolysis of heroin. The color code used here is the same as that of Figure 2.
The current nucleophilic attack process involves the breaking of the Oω–Hω bond and the formation of the C1–Oω and Ne–Hω bonds (Scheme 2). Thus, the distances ROω–Hω, RC1–Oγ, and RNε–Hω were chosen to represent the reaction coordinate as ROω–Hω–RC1–Oω–RNε–Hω in the QM/MM calculations for the current reaction step. During this reaction process, the distance ROω–Hω elongates from 0.98 Å in INT2′ to 1.43 Å in TS3 and then to 2.28 Å in INT3, while the distances RC1–Oω and RNε–Hω respectively shorten from 2.62 and 1.89 Å in INT2′ to 1.76 and 1.13 Å in TS3 and then to 1.49 and 1.05 Å in INT3. Noteworthy, similar to reaction step 1 in the acylation stage, the interaction between the carbonyl oxygen (O1) and the oxyanion hole is strengthened during this reaction step, which helps to stabilize the transition state TS3 and intermediate INT3. The developed positive charge on the imidazole ring of His438 is also stabilized by the strengthened hydrogen bond between the carboxylate anion of Glu325 and the proton (Hδ) of His438 (Nδ–Hδ ···O distance: 1.79 Å in INT2′, 1.69 Å in TS3, and 1.61 Å in INT3).
In addition, we also tested the QM/MM reaction-coordinate calculations using alternative reaction coordinates in which two internuclear distances involving Oγ of Ser198 were added into the reaction coordinate. The first alternative reaction coordinate tested was RC1–Oγ–RC1–Oω+ROω–Hω–ROγ–Hω–RNε–Hω. The QM/MM reaction-coordinate calculations using such a reaction coordinate may lead to a reaction pathway with the proton (Hγ) of the water molecule transferred to either Nε of His438 (the main reaction pathway discussed above) or Oγ of Ser198 (an alternative reaction pathway). It turned out that the QM/MM calculations using this reaction coordinate led to exactly the same results as shown in Scheme 2 and Figure 3, i.e. the proton (Hω) was still transferred to Nω of His438 rather than Oγ of Ser198 during the reaction process.
Further, we performed the QM/MM reaction-coordinate calculations using RC1–Oγ–RC1–Oω+ROω–Hω–ROγ–Hω as the reaction coordinate. This reaction coordinate involves the breaking of the C1–Oγ and Oω–Hω bonds and formation of the C1–Oω and Oγ–Hω bonds. The QM/MM reaction-coordinate calculations using such a reaction coordinate may only lead to a reaction pathway (as shown in Scheme 3) with the proton (Hω) transferred to Oγ of Ser198 during the nucleophilic attack of Oω on C1 of Ser198. In fact, the QM/MM reaction-coordinate calculations using this reaction coordinate led to an unfavorable, high-energy transition state structure, denoted as TS3a in Figure 4, with a four-membered ring (see below for the energetic results); we stopped the unnecessary further reaction-coordinate calculations after the transition state TS3a was identified.
Scheme 3.

Possible reaction pathway in the deacylation stage via the hypothetical transition state TS3a with a four-membered ring.
Figure 4.
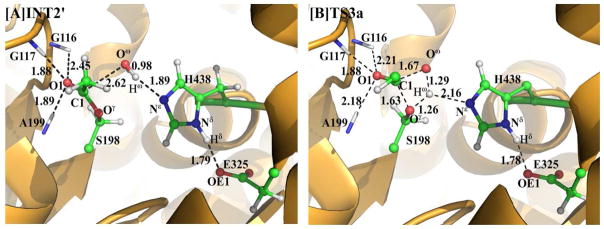
Geometries of INT2′ and TS3a optimized at the QM/MM(B3LYP/6–31G*:AMBER) level. The color code used here is the same as that of Figure 2.
Step 4. Dissociation between the acetic acid and Ser198 of BChE
As seen from Scheme 2, this reaction step involves the breaking of the C1–Oγ and Nε–Hω bonds and the formation of the Oγ–Hω bond. The changes in the distances RC1–Oγ, RNε–Hω, and ROγ–Hω reflect the nature of this reaction step. Thus, the reaction coordinate RC1–Oγ+RNε–Hω–ROγ–Hω was set in the QM/MM calculations for this reaction step. In the formed intermediate INT3, the distance ROγ–Hω is 1.79 Å, indicating that a strong hydrogen bond has formed between the oxygen of Ser198 side chain and the H atom. As shown in Figure 3, the distances RC1–Oγ and RNε–Hω respectively elongate from 1.60 and 1.05 Å in INT3 to 1.85 and 1.14 Å in TS4 and then to 2.40 and 1.80 Å in PD, while the distance ROγ–Hω shortens from 1.79 Å in INT3 to 1.45 Å in TS4 and then to 1.00 Å in PD.
Roles of the catalytic triad and oxyanion hole
According to the above reaction pathways examined by our pseudobond first-principles QM/MM-FE calculations, the catalytic triad and oxyanion hole of the enzyme are the most important factors in BChE-catalyzed hydrolysis of heroin to 6-MAM. The catalytic triad consists of Ser198, His438, and Glu325, in which Ser198 acts as a nucleophile in reaction step 1 to initiate the hydrolysis reaction, His438 serves as a general base which accepts the proton from the nucleophile to facilitate the two nucleophilic attack processes, i.e. reaction steps 1 and 3, and Glu325 helps to stabilize the transition states through increasing the hydrogen-bonding interaction with His438 throughout the hydrolysis reaction. The oxyanion hole consists of the backbone NH groups of Gly116, Gly117, and Ala199, and helps to stabilize the transition states throughout the hydrolysis reaction. Based on the QM/MM reaction-coordinate calculations, in the initial ES complex, the carbonyl oxygen (O1) of heroin only forms two hydrogen bonds with the oxyanion hole: the of N–H···O1 hydrogen bond with the backbone NH group of Gly116; the N–H···O1 hydrogen bond with the backbone NH group of Gly117. As shown in Figure 2B, the two H···O1 distances are all ~1.9 Å. In comparison, the H···O1 distance between the backbone NH group of Ala199 and the carbonyl oxygen (O1) of heroin (~2.5 Å) is relatively longer. However, in the rate-determining transition state TS2 (see below for the free energy profile) shown in Figure 2E, the hydrogen bond between the backbone NH group of Ala199 and the carbonyl oxygen (O1) of heroin is strengthened significantly (with the H···O1 distance changing from ~2.5 Å to ~1.9 Å). For the other two hydrogen bonds with the backbone NH groups of Gly116 and Gly117, the changes in the H···O1 distances (~1.8 Å and ~2.0 Å, respectively) are relatively small during the hydrolysis reaction. All of the three hydrogen bonds stabilize the negative charge on the carbonyl oxygen (O1) of heroin that develops during the hydrolysis reaction. Thus, it can be concluded that, for BChE-catalyzed hydrolysis of heroin, the catalytic triad initiates the reaction and the oxyanion hole helps to accelerate the reaction. The roles of the catalytic triad and oxyanion hole noted in the BChE-catalyzed hydrolysis of heroin are essentially similar to those observed in previous QM/MM reaction-coordinate calculations on acetylcholine (ACh) hydrolysis or other ester hydrolysis reactions catalyzed by either AChE(69–70) or BChE.(26, 29–30) In fact, AChE and BChE have the same catalytic triad and oxyanion hole. The main difference between AChE and BChE exists in the size of the active-site gorge, with BChE having a relatively larger active-site gorge, resulting in different substrate selectivity.(71–78) With a larger active-site gorge, BChE can accommodate relatively larger substrates compared to AChE. For a common substrate such as ACh, the two enzymes are expected to follow the same fundamental reaction pathway,(26, 29, 48, 70) but with different energy barriers.
Free energy profiles
The aforementioned QM/MM reaction-coordinate calculations at the B3LYP/6–31G*:AMBER level have revealed that the main reaction pathway consists of four reaction steps (steps 1 and 2 belong to the acylation stage, and steps 3 and 4 belong to the deacylation stage) in the BChE-catalyzed hydrolysis of heroin to 6-MAM. For an alternative reaction pathway, the deacylation stage becomes a single reaction step (the third reaction step). Further, to determine the free energy profiles for the reaction pathways, we carried out the single-point QM/MM energy calculations with the QM part treated at the B3LYP/6–31++G** level for each QM/MM optimized geometry along the minimum-energy path. Concerning the results of the FEP calculations, the relative free energy of the reaction system was taken as the average of the “forward” and “backward” perturbation results in which the difference between the “forward” and “backward” perturbation results may represent the error bar of the FEP calculations.
Depicted in Figure 5 is the free energy profile calculated for the main reaction pathway by the QM/MM-FE calculations at the B3LYP/6–31++G**:AMBER level without the zero-point and thermal corrections for the QM subsystem, and depicted in Figure 6 is the free energy profile determined for the alternative third reaction step by using the same computational approach. The values in parentheses are the corresponding relative free energies including the zero-point and thermal corrections for the QM subsystem. The error bars given to the relative free energies in the figures are associated with the free energy differences between the “forward” and “backward” perturbation calculations. The largest error bar was ±0.2 kcal/mol, suggesting that the FEP calculations converged well in terms of the number of the FEP windows used.
Figure 5.
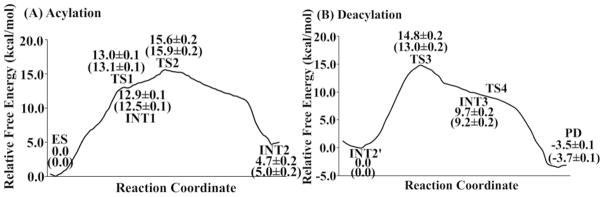
Free energy profile determined by the QM/MM-FE calculations for the acylation (A) and deacylation (B) stages of the reaction pathway. Given in the parentheses are the relative free energies with the zero-point and thermal corrections for the QM subsystem. The QM/MM-FE calculations on for all QM/MM optimized geometries along the reaction path were performed at the B3LYP/6–31++G**:AMBER level.
Figure 6.
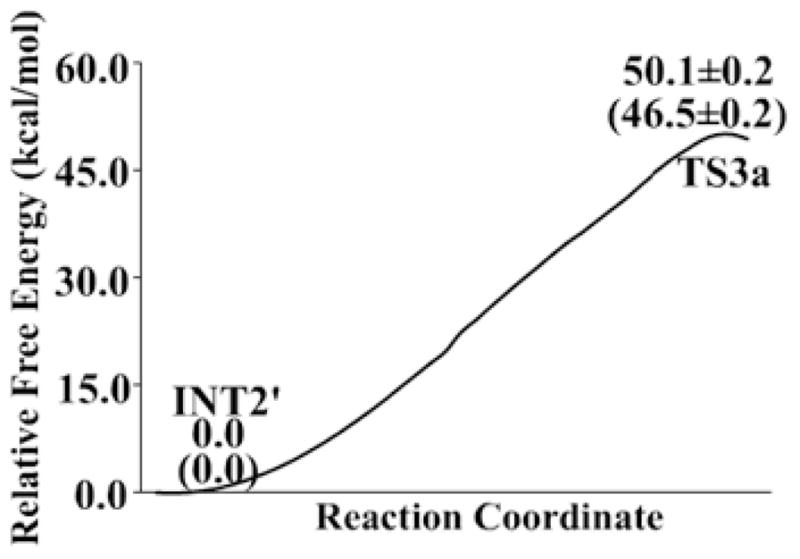
Free energy profile for the alternative pathway of the third reaction step according to the QM/MM-FE calculations. Given in the parentheses are the relative free energies with the zero-point and thermal corrections for the QM subsystem. The QM/MM-FE calculations on for all QM/MM optimized geometries along the reaction path were performed at the B3LYP/6–31++G**:AMBER level.
As seen in Figure 6, the free energy barrier calculated for the alternative pathway of the third reaction step (deacylation) is as high as 46.5±0.2 kcal/mol and, thus, this alternative reaction pathway can be ignored compared to the main reaction pathway. So, in the discussion below, we focus on the free energy profile (Figure 5) calculated for the main reaction pathway.
As can be seen from Figure 5, the calculated results reveal that the relative energy of TS2 is higher than TS1, indicating that the calculated overall free energy barrier for the entire acylation stage is the free energy change from ES to TS2. Similarly, the higher energy of TS3 relative to TS4 indicates that the overall free energy barrier calculated for the deacylation is the free energy change from INT2′ to TS3. According to the results, the free energy barrier for the acylation is higher than that for the deacylation, demonstrating that the free energy change associated with the structural transformation from ES to TS2 is rate-determining for the BChE-catalyzed hydrolysis of heroin to 6-MAM. Noteworthy, the energy barrier of the fourth step, i.e. the dissociation of the acetic acid and Ser198, is only ~0.2 kcal/mol according to the potential energy surface calculated at the QM/MM(B3LYP/6–31G*:AMBER) level. The negligible energy barrier disappears according to the final QM/MM-FE calculations performed at the B3LYP/6–31++G**:AMBER level, suggesting that the final step of the enzymatic reaction is actually barrier-less after the FEP calculations.
Finally, we examined the basis set dependence of the QM/MM-FE calculations results for the rate-determining step associated with TS2 (i.e. the free energy change from ES to TS2). When the basis set used in the QM part of the QM/MM changed from 6–31+G* to 6–31++G**, the overall free energy barrier calculated for the rate-determining step (i.e. the free energy change from ES to TS2) changed significantly from 13.8±0.2 kcal/mol to 15.9±0.2 kcal/mol, with a net change of 2.1 kcal/mol. However, when the basis set further changed from 6–31++G** to 6–311++G**, the calculated overall free energy barrier for the rate-determining step changed from 15.9±0.2 kcal/mol to 16.1±0.2 kcal/mol, with a net change of only 0.2 kcal/mol, suggesting that the 6–31++G** or 6–311++G** basis set is large enough. So, the overall free energy barrier calculated for the rate-determining step associated with transition state TS2 is 15.9±0.2 kcal/mol (using the 6–31++G** basis set) or 16.1±0.2 kcal/mol (using the 6–311++G** basis set). According to the reported experimental data,(14) kcat = 540 min−1 which is associated with an activation free energy of ~16.2 kcal/mol at room temperature (25°C) according to the conventional transition state theory.(79) The calculated free energy barrier of 15.9±0.2 or 16.1±0.2 kcal/mol is very close to the experimentally derived activation free energy of ~16.2 kcal/mol, which suggests that the computational results are reasonable.
Insights from the mechanistic understanding for rational design of novel therapeutic agents
Currently used therapeutic treatments of heroin abuse all target opiate receptors. As noted above, the pharmacological function of heroin requires an activation process which transforms heroin into 6-MAM (the most active form), and BChE is the major enzyme responsible for this activation process. It might be interesting to develop a novel therapeutic treatment targeting the activation process, particularly BChE-catalyzed hydrolysis of heroin to 6-MAM. One possibility is to design a small-molecule allosteric inhibitor which can significantly increase the free energy barrier for BChE-catalyzed hydrolysis of heroin to 6-MAM without significantly affecting the other functions of the enzyme. The desirable allosteric inhibitor should bind to a binding site (other than the active site), such as the well-known peripheral anionic site of BChE (around Asp70),{Masson, 1996 #80} so that it does not block the active site. The allosteric inhibitor should be designed to increase the overall free energy barrier, i.e. the free energy change from ES to TS2 depicted in Figure 2. For the purpose of increasing the free energy change from ES to TS2, one should aim to design an allosteric inhibitor which can more favorably stabilize the ES structure and less favorably stabilize (or destabilize) the TS2 structure.
For practical design of an allosteric inhibitor, one may computationally test variety of possible small molecules. For each of the possible small molecules, one may first computationally evaluate its binding affinity with the ES structure of BChE. If the small molecule is evaluated to have a high affinity with the ES structure, then one may further carry out QM/MM-FE calculations on the BChE-catalyzed hydrolysis of heroin in the presence of the small molecule to predict the free energy barrier (i.e. the free energy change from ES to TS2). The small molecule predicted to significantly increase the free energy barrier may be recommended for wet experiments including chemical synthesis and in vitro activity assays.
Conclusion
The first-principles QM/MM-FE calculations performed in this study have demonstrated the detailed reaction pathway for the BChE-catalyzed hydrolysis of heroin to 6-MAM. According to the QM/MM calculations, the whole reaction pathway for the BChE-catalyzed hydrolysis of heroin consists of four reaction steps. The reaction is initiated by the nucleophilic attack of hydroxyl oxygen (Oγ) atom of Ser198 side chain on the carbonyl carbon (C1) of 3-acetyl group of heroin, accompanied by the proton (Hγ) transfer from the Ser198-Oγ atom to the His438-Nε atom. The tetrahedral intermediate INT1 is thus formed, in which the negatively charged carbonyl oxygen (O1) is stabilized by the backbone NH groups of Gly116, Gly117, and Ala199 in the oxyanion hole. Subsequently, the C1–O2 bond is gradually broken while the proton (Hγ) transfers from His438-Nε to the O2 atom, leading to the formation of 6-MAM and the acyl-enzyme intermediate. The third reaction step is initiated by the nucleophilic attack of a water molecule on the carbonyl carbon (C1) atom of the acyl-enzyme intermediate. This process is coupled with a proton (Hω) transfer from the water oxygen (Oω) to His438-Nε. Finally, the acetyl group is released with the regeneration of Ser198.
The calculated free energy profile reveals that the second transition state (TS2) should be the rate-determining transition state, and that the overall free energy barrier for the reaction should be the free energy change from ES and TS2. The structural analysis reveals that the oxyanion hole plays an important role in stabilization of the rate-determining transition state TS2. The calculated overall free energy barrier of 15.9±0.2 or 16.1±0.2 kcal/mol is in good agreement with the experimentally derived activation free energy of ~16.2 kcal/mol, suggesting that the mechanistic insights obtained from the QM/MM-FE calculations are reliable. The obtained structural and mechanistic insights could be valuable for use in future rational design of a novel therapeutic strategy which aims to control heroin mechanism and, thus, treat heroin abuse.
Acknowledgments
Funding. This work was supported in part by the NIH (grants R01 DA035552, R01 DA032910, R01 DA013930, and R01 DA025100) and the NSF (grant CHE-1111761).
Qiao worked in Zhan’s laboratory for this project at University of Kentucky as an exchange graduate student (from Dalian Institute of Chemical Physics, Chinese Academy of Science) supported by the China Scholarship Council. The authors also acknowledge the Computer Center at University of Kentucky for supercomputing time on a Dell X-series Cluster with 384 nodes or 4,768 processors.
References
- 1.National Insititute on Drug Abuse. The Science of Drug Abuse & Addiction. 2013 http://www.drugabuse.gov/publications/drugfacts/heroin.
- 2.Oldendorf WH, Hyman S, Braun L, Oldendorf SZ. Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science. 1972;178:984–986. doi: 10.1126/science.178.4064.984. [DOI] [PubMed] [Google Scholar]
- 3.Selley DE, Cao CC, Sexton T, Schwegel JA, Martin TJ, Childers SR. mu Opioid receptor-mediated G-protein activation by heroin metabolites: evidence for greater efficacy of 6-monoacetylmorphine compared with morphine, Biochem. Pharmacol. 2001;62:447–455. doi: 10.1016/s0006-2952(01)00689-x. [DOI] [PubMed] [Google Scholar]
- 4.Way EL, Kemp JW, Young JM, Grassetti DR. The pharmacologic effects of heroin in relationship to its rate of biotransformation. J Pharmacol Exp Ther. 1960;129:144–154. [PubMed] [Google Scholar]
- 5.Way EL, Young JM, Kemp JW. Metabolism of Heroin and Its Pharmacologic Implications. B Narcotics. 1965;17:25–33. [Google Scholar]
- 6.Goldberger BA, Cone EJ, Grant TM, Caplan YH, Levine BS, Smialek JE. Disposition of heroin and Its metabolites in heroin-related deaths. J Anal Toxicol. 1994;18:22–28. doi: 10.1093/jat/18.1.22. [DOI] [PubMed] [Google Scholar]
- 7.Eddy NB, Howes HA. Studies of morphine, codeine and their derivatives VIII Monoacetyl- and diacetylmorphine their hydrogenated derivatives. J Pharmacol Exp Ther. 1935;53:430–439. [Google Scholar]
- 8.Wright CI, Barbour FA. The respiratory effects of morphine, codeine and related substances IV. The effect of alpha-monoacetylmorphine, monoacetyldihydromorphine, diacetylmorphine (heroin) and diacetyldihydromorphine on the respiratory activity of the rabbit. J Pharmacol Exp Ther. 1935;54:25–33. [Google Scholar]
- 9.Lockridge O, Mottershaw-Jackson N, Eckerson HW, La Du BN. Hydrolysis of diacetylmorphine (heroin) by human serum cholinesterase. J Pharmacol Exp Ther. 1980;215:1–8. [PubMed] [Google Scholar]
- 10.Andersen JM, Ripel A, Boix F, Normann PT, Morland J. Increased locomotor activity induced by heroin in mice: pharmacokinetic demonstration of heroin acting as a prodrug for the mediator 6-monoacetylmorphine in vivo. J Pharmacol Exp Ther. 2009;331:153–161. doi: 10.1124/jpet.109.152462. [DOI] [PubMed] [Google Scholar]
- 11.Inturrisi CE, Schultz M, Shin S, Umans JG, Angel L, Simon EJ. Evidence from Opiate Binding-Studies That Heroin Acts through Its Metabolites. Life Sci. 1983;33:773–776. doi: 10.1016/0024-3205(83)90616-1. [DOI] [PubMed] [Google Scholar]
- 12.Strandberg JJ, Kugelberg FC, Alkass K, Gustavsson A, Zahlsen K, Spigset O, Druid H. Toxicological analysis in rats subjected to heroin and morphine overdose. Toxicol Lett. 2006;166:11–18. doi: 10.1016/j.toxlet.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J Pharmacol Exp Ther. 1996;279:713–717. [PubMed] [Google Scholar]
- 14.Salmon AY, Goren Z, Avissar Y, Soreq H. Human erythrocyte but not brain acetylcholinesterase hydrolyses heroin to morphine. Clin Exp Pharmacol Physiol. 1999;26:596–600. doi: 10.1046/j.1440-1681.1999.03090.x. [DOI] [PubMed] [Google Scholar]
- 15.Boix F, Andersen JM, Morland J. Pharmacokinetic modeling of subcutaneous heroin and its metabolites in blood and brain of mice. Addict Biol. 2013;18:1–7. doi: 10.1111/j.1369-1600.2010.00298.x. [DOI] [PubMed] [Google Scholar]
- 16.Martin WR, Fraser HF. A comparative study of physiological and subjective effects of heroin and morphine administered intravenously in postaddicts. J Pharmacol Exp Ther. 1961;133:388–399. [PubMed] [Google Scholar]
- 17.Nakamura GR, Thornton JI, Noguchi TT. Kinetics of heroin deacetylation in aqueous alkaline solution and in human serum and whole blood. J Chromatogr. 1975;110:81–89. doi: 10.1016/s0021-9673(00)91213-5. [DOI] [PubMed] [Google Scholar]
- 18.Owen JA, Nakatsu K. Diacetylmorphine (Heroin) Hydrolases in Human-Blood. Can J Physiol Pharm. 1983;61:870–875. doi: 10.1139/y83-132. [DOI] [PubMed] [Google Scholar]
- 19.Rook EJ, Huitema AD, van den Brink W, van Ree JM, Beijnen JH. Pharmacokinetics and pharmacokinetic variability of heroin and its metabolites: review of the literature. Curr Clin Pharmacol. 2006;1:109–118. doi: 10.2174/157488406775268219. [DOI] [PubMed] [Google Scholar]
- 20.Zhang YK, Lee TS, Yang WT. A pseudodond approach to combining quantum mechanical and molecular mechanical methods. J Chem Phys. 1999;110:46–54. [Google Scholar]
- 21.Zhang YK, Liu HY, Yang WT. Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio QM/MM potential energy surface. J Chem Phys. 2000;112:3483–3492. [Google Scholar]
- 22.Zhang YK. Improved pseudobonds for combined ab initio quantum mechanical/molecular mechanical methods. J Chem Phys. 2005;122:024114. doi: 10.1063/1.1834899. [DOI] [PubMed] [Google Scholar]
- 23.Zhang YK. Pseudobond ab initio QM/MM approach and its applications to enzyme reactions. Theor Chem Acc. 2006;116:43–50. [Google Scholar]
- 24.Liu JJ, Hamza A, Zhan CG. Fundamental Reaction Mechanism and Free Energy Profile for (−)-Cocaine Hydrolysis Catalyzed by Cocaine Esterase. J Am Chem Soc. 2009;131:11964–11975. doi: 10.1021/ja903990p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu JJ, Zhang YK, Zhan CG. Reaction Pathway and Free-Energy Barrier for Reactivation of Dimethylphosphoryl-Inhibited Human Acetylcholinesterase. J Phys Chem B. 2009;113:16226–16236. doi: 10.1021/jp9055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Fang L, Liu J, Zhan CG. Reaction Pathway and Free Energy Profile for Butyrylcholinesterase-Catalyzed Hydrolysis of Acetylcholine. J Phys Chem B. 2010;115:1315–1322. doi: 10.1021/jp110709a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Zhao X, Xiong Y, Liu J, Zhan CG. Fundamental reaction pathway and free energy profile for hydrolysis of intracellular second messenger adenosine 3′,5′-cyclic monophosphate (cAMP) catalyzed by phosphodiesterase-4. J Phys Chem B. 2011;115:12208–12219. doi: 10.1021/jp205509w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JJ, Zhao XY, Yang WC, Zhan CG. Reaction Mechanism for Cocaine Esterase-Catalyzed Hydrolyses of (+)- and (−)-Cocaine: Unexpected Common Rate-Determining Step. J Phys Chem B. 2011;115:5017–5025. doi: 10.1021/jp200975v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Fang L, Liu J, Zhan C-G. Reaction Pathway and Free Energy Profiles for Butyrylcholinesterase-Catalyzed Hydrolysis of Acetylthiocholine. Biochemistry. 2012;51:1297–1305. doi: 10.1021/bi201786s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Zhan CG. Reaction pathway and free energy profile for cocaine hydrolase-catalyzed hydrolysis of (−)-cocaine. J Chem Theory Comput. 2012;8:1426–1435. doi: 10.1021/ct200810d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei DH, Lei BL, Tang MS, Zhan CG. Fundamental Reaction Pathway and Free Energy Profile for Inhibition of Proteasome by Epoxomicin. J Am Chem Soc. 2012;134:10436–10450. doi: 10.1021/ja3006463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao Y, Liu J, Zhan CG. Why does the G117H mutation considerably improve the activity of human butyrylcholinesterase against sarin? Insights from quantum mechanical/molecular mechanical free energy calculations. Biochemstry. 2012;51:8980–8992. doi: 10.1021/bi3009246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li D, Huang X, Lin J, Zhan C-G. Catalytic mechanism of cytochrome P450 for N-methylhydroxylation of nicotine: Reaction pathways and regioselectivity of the enzymatic nicotine oxidation. Dalton Trans. 2013;42:3812–3820. doi: 10.1039/c2dt32106h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei D, Huang X, Liu J, Tang M, Zhan C-G. Reaction Pathway and Free Energy Profile for Papain-Catalyzed Hydrolysis of N-Acetyl-Phe-Gly 4-Nitroanilide. Biochemistry. 2013;52:5145–5154. doi: 10.1021/bi400629r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng F, Yang WC, Ko MC, Liu JJ, Cho H, Gao DQ, Tong M, Tai HH, Woods JH, Zhan CG. Most efficient cocaine hydrolase designed by virtual screening of transition states. J Am Chem Soc. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wandhammer M, Carletti E, Van der Schans M, Gillon E, Nicolet Y, Masson P, Goeldner M, Noort D, Nachon F. Structural study of the complex stereoselectivity of human butyrylcholinesterase for the neurotoxic V-agents. J Biol Chem. 2011;286:16783–16789. doi: 10.1074/jbc.M110.209569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balchin E, Malcolme-Lawes DJ, Rowe MDS, Smith JA, Bearpark MJ, Steed JW, Wu W, Horsewill AJ, Stephenson D. The unusual solid state structure of heroin hydrochloride monohydrate and its selective detection using NQR spectroscopy. New J Chem. 2004;28:1309–1314. [Google Scholar]
- 38.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, udin KN, Burant JC, Millam JM, yengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, shida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Version C.02. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 39.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges - the resp model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 40.Cieplak P, Cornell WD, Bayly C, Kollman PA. Application of the multimolecule and multiconformational resp methodology to biopolymers - charge derivation for DNA, RNA, and proteins. J Comput Chem. 1995;16:1357–1377. [Google Scholar]
- 41.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER11. University of California; San Francisco: 2010. [Google Scholar]
- 42.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 43.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 44.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 45.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 46.Toukmaji A, Sagui C, Board J, Darden T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J Chem Phys. 2000;113:10913–10927. [Google Scholar]
- 47.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 48.Zhou Y, Wang S, Zhang Y. Catalytic reaction mechanism of acetylcholinesterase determined by Born-Oppenheimer ab initio QM/MM molecular dynamics simulations. J Phys Chem B. 2010;114:8817–8825. doi: 10.1021/jp104258d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 50.Becke AD. Density-Functional Thermochemistry 3 The Role of Exact Exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 51.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 52.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER8. University of California; San Francisco: 2004. [Google Scholar]
- 53.Smith AJ, Muller R, Toscano MD, Kast P, Hellinga HW, Hilvert D, Houk KN. Structural reorganization and preorganization in enzyme active sites: comparisons of experimental and theoretically ideal active site geometries in the multistep serine esterase reaction cycle. J Am Chem Soc. 2008;130:15361–15373. doi: 10.1021/ja803213p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramalho TC, Tanos CC, Renno MN, Guimaraes AP, da Cunha EF, Kuca K. Development of new acetylcholinesterase reactivators: molecular modeling versus in vitro data. Chem Biol Interact. 2010;185:73–77. doi: 10.1016/j.cbi.2010.02.026. [DOI] [PubMed] [Google Scholar]
- 55.Chandrasekhar J, Smith SF, Jorgensen WL. Sn2 Reaction Profiles in the Gas-Phase and Aqueous-Solution. J Am Chem Soc. 1984;106:3049–3050. [Google Scholar]
- 56.Kollman P. Free-Energy Calculations - Applications to Chemical and Biochemical Phenomena. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 57.Beveridge DL, DiCapua FM. Free energy via molecular simulation: applications to chemical and biomolecular systems. Annu Rev Biophys Biophys Chem. 1989;18:431–492. doi: 10.1146/annurev.bb.18.060189.002243. [DOI] [PubMed] [Google Scholar]
- 58.Acevedo O, Jorgensen WL. Advances in Quantum and Molecular Mechanical (QM/MM) Simulations for Organic and Enzymatic Reactions. Acc Chem Res. 2010;43:142–151. doi: 10.1021/ar900171c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Acevedo O, Jorgensen WL. Cope elimination: Elucidation of solvent effects from QM/MM simulations. J Am Chem Soc. 2006;128:6141–6146. doi: 10.1021/ja057523x. [DOI] [PubMed] [Google Scholar]
- 60.Acevedo O, Jorgensen WL. Influence of inter- and intramolecular hydrogen bonding on kemp decarboxylations from QM/MM simulations. J Am Chem Soc. 2005;127:8829–8834. doi: 10.1021/ja051793y. [DOI] [PubMed] [Google Scholar]
- 61.Alexandrova AN, Rothlisberger D, Baker D, Jorgensen WL. Catalytic mechanism and performance of computationally designed enzymes for Kemp elimination. J Am Chem Soc. 2008;130:15907–15915. doi: 10.1021/ja804040s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guimaraes CR, Udier-Blagovic M, Jorgensen WL. Macrophomate synthase: QM/MM simulations address the Diels-Alder versus Michael-Aldol reaction mechanism. J Am Chem Soc. 2005;127:3577–3588. doi: 10.1021/ja043905b. [DOI] [PubMed] [Google Scholar]
- 63.Guimaraes CR, Boger DL, Jorgensen WL. Elucidation of fatty acid amide hydrolase inhibition by potent alpha-ketoheterocycle derivatives from Monte Carlo simulations. J Am Chem Soc. 2005;127:17377–17384. doi: 10.1021/ja055438j. [DOI] [PubMed] [Google Scholar]
- 64.Zhang W, Hou TJ, Qiao XB, Huai S, Xu XJ. Binding affinity of hydroxamate inhibitors of matrix metalloproteinase-2. J Mol Model. 2004;10:112–120. doi: 10.1007/s00894-004-0181-1. [DOI] [PubMed] [Google Scholar]
- 65.Udier-Blagovic M, Tirado-Rives J, Jorgensen WL. Structural and energetic analyses of the effects of the K103N mutation of HIV-1 reverse transcriptase on efavirenz analogues. J Med Chem. 2004;47:2389–2392. doi: 10.1021/jm0303507. [DOI] [PubMed] [Google Scholar]
- 66.Jarmula A, Cieplak P, Les A, Rode W. Relative free energies of binding to thymidylate synthase of 2- and/or 4-thio and/or 5-fluoro analogues of dUMP. J Comput Aided Mol Des. 2003;17:699–710. doi: 10.1023/b:jcam.0000017377.07094.2e. [DOI] [PubMed] [Google Scholar]
- 67.Danciulescu C, Nick B, Wortmann FJ. Structural stability of wild type and mutated alpha-keratin fragments: molecular dynamics and free energy calculations. Biomacromolecules. 2004;5:2165–2175. doi: 10.1021/bm049788u. [DOI] [PubMed] [Google Scholar]
- 68.Funahashi J, Sugita Y, Kitao A, Yutani K. How can free energy component analysis explain the difference in protein stability caused by amino acid substitutions? Effect of three hydrophobic mutations at the 56th residue on the stability of human lysozyme. Protein Eng. 2003;16:665–671. doi: 10.1093/protein/gzg083. [DOI] [PubMed] [Google Scholar]
- 69.Zhang YK, Kua J, McCammon JA. Role of the catalytic triad and oxyanion hole in acetylcholinesterase catalysis: An ab initio QM/MM study. J Am Chem Soc. 2002;124:10572–10577. doi: 10.1021/ja020243m. [DOI] [PubMed] [Google Scholar]
- 70.Polyakov IV, Grigorenko BL, Moskovsky AA, Pentkovski VM, Nemukhin AV. Towards quantum-based modeling of enzymatic reaction pathways: Application to the acetylholinesterase catalysis. Chem Phys Lett. 2013;556:251–255. [Google Scholar]
- 71.Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience. 2002;110:627–639. doi: 10.1016/s0306-4522(01)00613-3. [DOI] [PubMed] [Google Scholar]
- 72.Masson P, Bec N, Froment MT, Nachon F, Balny C, Lockridge O, Schopfer LM. Rate-determining step of butyrylcholinesterase-catalyzed hydrolysis of benzoylcholine and benzoylthiocholine - Volumetric study of wild-type and D70G mutant behaviour. Eur J Biochem. 2004;271:1980–1990. doi: 10.1111/j.1432-1033.2004.04110.x. [DOI] [PubMed] [Google Scholar]
- 73.Mattes C, Bradley R, Slaughter E, Browne S. Cocaine and butyrylcholinesterase (BChE): Determination of enzymatic parameters. Life Sci. 1996;58:Pl257–Pl261. doi: 10.1016/0024-3205(96)00065-3. [DOI] [PubMed] [Google Scholar]
- 74.Masson P, Froment MT, Fortier PL, Visicchio JE, Bartels CF, Lockridge O. Butyrylcholinesterase-catalysed hydrolysis of aspirin, a negatively charged ester, and aspirin-related neutral esters. Bba-Protein Struct M. 1998;1387:41–52. doi: 10.1016/s0167-4838(98)00104-6. [DOI] [PubMed] [Google Scholar]
- 75.Amitay M, Shurki A. Hydrolysis of organophosphate compounds by mutant butyrylcholinesterase: A story of two histidines. Proteins. 2011;79:352–364. doi: 10.1002/prot.22864. [DOI] [PubMed] [Google Scholar]
- 76.Dvir H, Silman I, Harel M, Rosenberry TL, Sussman JL. Acetylcholinesterase: From 3D structure to function. Chem-Biol Interact. 2010;187:10–22. doi: 10.1016/j.cbi.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vellom DC, Radic Z, Li Y, Pickering NA, Camp S, Taylor P. Amino acid residues controlling acetylcholinesterase and butyrylcholinesterase specificity. Biochemistry. 1993;32:12–17. doi: 10.1021/bi00052a003. [DOI] [PubMed] [Google Scholar]
- 78.Fang L, Pan Y, Muzyka J, Zhan CG. Active site gating and substrate specificity of butyrylcholinesterase and acetylcholinesterase: Insights from molecular dynamics simulations. J Phys Chem B. 2011;115:8797–8805. doi: 10.1021/jp112030p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alvarez-Idaboy JR, Galano A, Bravo-Perez G, Ruiz ME. Rate constant dependence on the size of aldehydes in the NO(3) + aldehydes reaction. An explanation via quantum chemical calculations and CTST. J Am Chem Soc. 2001;123:8387–8395. doi: 10.1021/ja010693z. [DOI] [PubMed] [Google Scholar]