

Abstract

An enantioselective α-hydroxyacetate aldol reaction that employs N-acetyl pyrroles as activated ester equivalents and generates syn 1,2-diols in good yield and diastereoselectivity is reported. This dinuclear zinc Prophenol-catalyzed transformation proceeds with high enantioselectivity with a wide variety of substrates including aryl, alyl, and alkenyl aldehydes. The resulting α,β-dihydroxy activated esters are versatile intermediates for the synthesis of a variety of carboxylic acid derivatives including amides, esters, and unsymmetrical ketones.
Enantiopure syn 1,2-diols are important pharmacophores present in bioactive polyketide natural products and drugs. A powerful method for constructing 1,2-diols in a stereocontrolled fashion is the direct α -hydroxyacetate aldol reaction, which allows assembly of the 1,2-diol functionality concomitant with formation of a new carbon–carbon bond.1 In addition, this strategy avoids the use of toxic osmium catalysts.2 While several catalytic enantioselective α-hydroxyacetyl aldol protocols have been reported, their application in synthesis has been limited due to the general requirement that the enolate partner be either an aromatic ketone3 or a hydroxyacetone derivative.4 In order to access the more synthetically versatile α,β-dihydroxyester functionality, Bayer-Villager oxidation of the α,β-dihydroxyaryl ketone products has been employed.5 Catalytic asymmetric direct glycolate aldol reactions, with aldol donors at the carboxylic acid oxidation state, are much more rare. Denmark and co-[ISP-1 check]workers6 recently reported a glycolate aldol reaction that can deliver either the syn or anti diol product in high selectivity, but this method requires the use of specialized fully protected and pre-formed silyl ketene acetal donors, which deliver the diol products in protected form. We describe herein a simplified approach that does not require the synthesis and use of enolate donors with high geometrical purity,7 but employs unprotected activated ester donors, providing free syn 1,2-diol products in high yield and enantioselectivity.
Our laboratory has pioneered the use of dinuclear zinc-Prophenol catalysts for aldol-type transformations,8 including α -hydroxyacetate aldol reactions with aryl ketones.3c In our recent efforts towards the total synthesis of laulimalide, we required a direct α -hydroxyacetate aldol reaction where the enolate donor was at the ester oxidation state.9 We found that ortho-substituted aryl ketone donor 1a provided the desired diol product (3a) in good yield and diastereoselectivity when the dinuclear zinc complex of Prophenol ligand 2a was employed as catalyst (Scheme 1). However, subsequent oxidation of 3a to the aryl ester under various conditions proceeded with poor chemoselectivity, resulting in either PMB cleavage or olefin epoxidation.8b The lower reactivity of an α -hydroxycarboxylic acid type substrate made their use seem unlikely. Nevertheless, in the course of those studies, we found that when N-(α-hydroxyacetyl)-2-ethylpyrrole (1b) was employed as the enolate partner, the desired syn 1,2-diol 3b could be obtained with good diastereoselectivity.10 In addition, the pyrrole product could easily be converted to an ester of any desired alcohol by simple esterification.
Scheme 1.

Hydroxyacetyl aldol in laulimalide synthesis.
The results of our studies towards laulimalide demonstrated that dinuclear zinc-Prophenol catalysts were capable of performing highly diastereoselective addition reactions with chiral β-siloxyaldehydes. The real challenge was whether these complexes could also facilitate highly enantioselective α -hydroxyacetate aldol reactions without the need for additional chiral directing groups in the substrate. In this report, we describe the development of a zinc-Prophenol-catalyzed enantioselective hydroxyacetate aldol reaction that employs N-acylpyrrole donors as activated ester equivalents.
Our studies towards this goal began by investigating the potential of a variety of ester equivalents to participate in the α -hydroxyacetate aldol reaction when dinuclear zinc-Prophenol complex 2a was employed as catalyst (Figure 1). While a variety of ester derivatives, including 2-acylimidazoles and acetylpyrroles, participated in the transformation, only the N-acetylpyrrole delivered promising yields and enantioselectivities. Use of an N-acylbenzoxazolinone substrate (1f) only led to decomposition. In these studies, we also confirmed that substitution of the pyrrole at the 2-position (1b vs. 1c) led to a slight increase in the enantioselectivity and diastereoselectivity of the process.
Figure 1.

Variation of activated ester structure.
The synthesis of our desired α-hydroxyester equivalent 1b is easily accomplished in a two-step process from commercially available 2-ethylpyrrole11 and benzyloxyacetyl chloride (Scheme 2). Treatment of 2-ethylpyrrole with n-butyllithium at low temperature, followed by addition of the pyrrole anion to the benzyloxyacetyl chloride provided the desired N-acylpyrrole 4 in respectable yield on scales up to 77 mmol. Simple cleavage of the benzyl group with boron trichloride gave the α -hydroxyacetylpyrrole 1b in excellent yield.
Scheme 2.
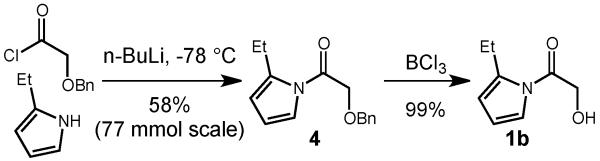
Synthesis of N-(α-hydroxyacetyl)-2-ethylpyrrole.
Having developed a large-scale synthesis of our desired activated ester derivative, we next sought to optimize the aldol transformation in terms of yield and enantioselectivity (Table 1). We found that use of two equivalents of the aldehyde was optimal to obtain the 1,2-diol product in good yield and improved enantioselectivity (entries 1–3). Increasing the equivalents of aldehyde to more than two equivalents did not provide any additional benefits. The effects of solvent (entry 4), temperature (entry 5) and concentration (entry 6) were explored and led to slight improvements in enantioselectivity, but the reaction still suffered from low conversion. Additives that have been shown to improve catalysts turnover also failed to increase the conversion (entries 7–8).3c Finally, by increasing the catalyst loading (entries 9–10) and conducting the reaction at 0 °C for 48 h we were able to isolate the diol product in excellent diastereoselectivity, enantioselectivity, and yield. We next looked at the effect of catalyst structure on the enantioselectivity of the process and found that ligand 2b provided the product in improved yield and selectivity (entry 11). Modification of the aryl substituents on the diaryl prolinol in 2a, however, did not significantly affect the selectivity of the reaction (see supporting information).
Table 1.
Optimization studies.
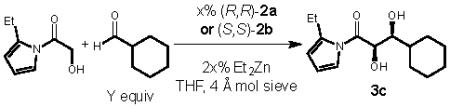
| entrya | Y | temp. | Yield (%) | dr (syn/anti) | ee (%) |
|---|---|---|---|---|---|
| 1 | 0.75 | r.t. | 55 | 5:1 | 68 |
| 2 | 1.5 | r.t | 63 | 4:1 | 84 |
| 3 | 2.0 | r.t. | 72 | 5:1 | 83 |
| 4b | 2.0 | r.t. | 53 | 3:1 | 85 |
| 5 | 2.0 | 0 °C | 48 | 3.5:1 | 95 |
| 6c | 2.0 | 0 °C | 53 | 5:1 | 94 |
| 7d | 2.0 | 0 °C | 47 | 4.7:1 | 93 |
| 8e | 2.0 | 0 °C | 43 | 3.9:1 | 96 |
| 9cf | 2.0 | 0 °C | 42 | 7.7:1 | 92 |
| 10c,g | 2.0 | 0 °C | 67 | 7.7:1 | 90 |
| 11c,h | 2.0 | 0 °C | 77 | 11:1 | −92 |
Unless otherwise noted, reactions performed using 15% (R,R)-2a, 30% Et2Zn and 0.2 mmol 1b in THF (0.2M).
Toluene as solvent.
Run at 0.4 M in THF.
Triphenylphosphine (30 mol%) used as additive.
Methyl 2,2-dimethyl-3-hydroxypropionate (30%) used as additive.
5% (R,R)-2a, and 10% diethylzinc.
20% (R,R)-2a, and 40% diethylzinc.
With (S,S)-2b.
With optimal conditions for the α-hydroxyacetate aldol reaction in hand, we next set out to determine the scope of aldehydes that can participate in this transformation (Table 2). A range of branched and linear alkyl aldehydes reacted in good yield, d.r. and enantioselectivity (entries 1-7). This result is particularly important because other glycolate aldol methodologies have only proved particularly effective for aryl and mono-substituted alkyl aldehydes. In fact, α -branched aldehydes like cylcohexanecarboxaldehyde (Table 2, entry 1) often provide none of the desired 1,2-diol product when previously reported methods are employed.6b In our zinc-Prophenol-catalyzed aldol addition, the reaction proceeds with highest selectivity when aldehydes are disubstituted at the α -carbon (entries 1–3). Aldehydes with a tetrasubstituted alpha center also react with excellent diastereoselectivity, and only slightly reduced ee (entries 8–10). On the other hand, aromatic aldehydes (entries 11–12) and α,β-unsaturated aldehydes (entry 13) reacted in high enantioselectivity when the transformation was performed at slightly lower temperatures. These promising results demonstrate the potential utility of this transformation for a broad range of aldehyde substrates and further development of the catalytic system to improve the diastereoselectivity with certain substrates is on-going in our laboratory. Of further note, this transformation tolerates a variety of functional groups in addition to the free hydroxyl group of the starting material and the free 1,2-diol of the product, including ketals (entries 7, 9), olefins (entry 9), and silyl ethers (entry 10).
Table 2.
Substrate scope.
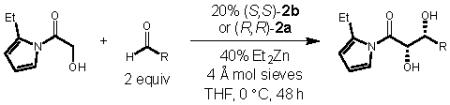
| entrya | R | yield (%)b |
dr (syn/anti)c |
ee (%)d | |
|---|---|---|---|---|---|
| 1 | Cyclohexyl | 3c | 77 | 11:1 | 92 |
| 2 | i-Pr | 3d | 79 | 7.1:1 | 90 |
| 3 | CH(CH2CH3)2 | 3e | 67 | 5.2:1 | 88 |
| 4 | CH2CH(CH3)2 | 3f | 86 | 3.7:1 | 87 |
| 5 | n-Butyl | 3g | 56 | 3.9:1 | 87 |
| 6 | CH2CH2Ph | 3h | 72 | 2.6:1 | 84 |
| 7 |
|
3i | 84 | 3.6:1 | 88 |
| 8e | t-Butyl | 3j | 51 | 15:1 | −75 |
| 9e |
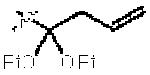
|
3k | 66 | 19:1 | −74 |
| 10e |
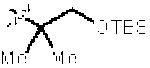
|
3l | 57 | 16:1 | −72 |
| 11e,f | C6H5 | 3m | 65 | 1.7:1 | −92 |
| 12f | p-ClC6H4 | 3n | 59 | 1.8:1 | 91 |
| 13f |
|
3o | 59 | 1.6:1 | 87 |
Reactions performed using 0.2 mmol 1b, 2.0 equiv aldehyde, 0.025 g 4 Å molecular sieves, 20% (S, S)-2b or (R,R)-2a, and 40% Et2Zn in THF (0.4 M) at 0 °C for 48 h.
Isolated Yield.
Determined by 1H NMR of crude reaction mixture.
Determined by chiral HPLC analysis
20 mol% (R,R)-3a was used.
Run for 72 h at −15 °C
Confirming our initial goals of this project, the acetylpyrrole proved to be a highly versatile activated ester equivalent for the synthesis of a variety of carboxylic acid derivatives, ketones, or alcohols (Scheme 3). The free 1,2-diol could be easily converted to the methyl ester (5a), benzyl ester (5b), or benzyl amide (5c) in high yield by simply treating the diol with the appropriate nucleophile. Conversion of the diol to the protected acetonide (6) also proceeded in excellent yield, and subsequent reduction to the alcohol (7) with LiAlH4 or conversion to the non-symmetrical ketone (8) by treatment with a Grignard reagent then DBU12 proceeded in 99% and 88% yield respectively.
Scheme 3.

Derivatization of N-acylpyrrole products.
The utility of the α-hydroxyacetate aldol reaction was further demonstrated in our current efforts toward the total synthesis of cytotoxic natural product peloruside A (9). In our initial route, the α,β-dihydroxyester was installed via an olefination/dihydroxylation sequence starting from commercially available ester 10 (Scheme 4). Reduction of 10 to the aldehyde, followed by Horner-Wadsworth-Emmons olefination provided α ,β-unsaturated ester 11. Osmium-catalyzed dihydroxylation of the olefin then provided our desired syn 1,2-diol 12 in good yield and enantioselectivity under optimized conditions. Employing our Prophenol-catalyzed reaction, we were able to construct the same functionality in a single atom economic transformation that avoids the olefination reaction and precludes the use of toxic osmium salts. Variation of the acetal protecting group on the aldehyde allowed us to improve the diastereoselectivity of the reaction and obtain the diols (3p–r) in good yield and enantioselectivity.
Scheme 4.

Diol synthesis towards peloruside A.
In conclusion, we have demonstrated that dinuclear zinc-Prophenol complexes are capable of facilitating highly stereoselective α -hydroxyacetate aldol reactions with N-acylpyrroles as activated ester equivalents. This process generates completely unprotected syn 1,2-diols in good yield, diastereoselectivity and enantioselectivity.13 In addition, α-branched alkyl aldehydes, which are often unreactive or proceed with poor selectivity in other glycolate aldol systems, perform remarkably well under our reaction conditions. The resulting N-acylpyrrole products are versatile activated ester intermediates for a variety of functionalization reactions, including conversion to ester, amide, ketone, or alcohol derivatives. The broad substrate scope and mild reaction conditions make this procedure a valuable reaction for the synthesis of enantioenriched 1,2-diol structures and further studies to improve the selectivity with certain substrates is ongoing in our laboratory.
Supplementary Material
Acknowledgment
Funding was provided by the National Institutes of Health (GM033049). We thank the NSF for their generous support of our programs. D.J.M. wishes to thank the NIH for a postdoctoral fellowship (F32 GM093467)
Footnotes
Supporting Information Available: Detailed experimental details, compound characterization data, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Mukaiyama T, Shiina I, Uchiro H, Kobayashi S. Bull. Che. Soc. Jpn. 1994;67:1708–1716. (b) For a review of direct aldol reactions, which includes α-hydroxyacetate aldols, see: Trost BM, Brindle CS. Chem. Soc. Rev. 2010;39:1600–1632. doi: 10.1039/b923537j. (c) For a recent review on additions to imines, which includes α-hydroxycarbonyl enolate donors, see: Kobayashi S, Mori Y, Fossey JS, Salter MM. Chem. Rev. 2011;111:2626–2704. doi: 10.1021/cr100204f.
- (2).For selected reviews of osmium-catalyzed syn dihydroxylations, see: Tse MK, Schroeder K, Beller M. In: Modern Oxidation Methods. 2nd edition Bäckvall J-E, editor. Wiley-VCH; Weinheim: 2010. pp. 1–36. Zaitsev AB, Adolfsson H. Synthesis. 2006:1725–1756. Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547.
- (3).(a) Yoshikawa N, Kumagai N, Matsunaga S, Moll G, Ohshima T, Suzuki T, Shibasaki M. J. Am. Chem. Soc. 2001;123:2466–2467. doi: 10.1021/ja015580u. [DOI] [PubMed] [Google Scholar]; (b) Kumagai N, Matsunaga S, Kinoshita T, Harada S, Okada S, Sakamoto S, Yamaguchi K, Shibasaki M. J. Am. Chem. Soc. 2003;125:2169–2178. doi: 10.1021/ja028926p. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Ito H, Silcoff ER. J. Am. Chem. Soc. 2001;123:3367–3368. doi: 10.1021/ja003871h. [DOI] [PubMed] [Google Scholar]
- (4).Notz W, List B. J. Am. Chem. Soc. 2000;122:7386–7387. For selected recent examples, see the following and references therein: Ma G, Bartoszewicz A, Ibrahem I, Córdova A. Adv. Synth. Catal. 2011;353:3114–3122. Wu C, Fu X, Li S. Tetrahedron: Asymmetry. 2011;22:1063–1073. Chen X-H, Luo S-W, Tang Z, Cun L-F, Mi A-Q, Jiang Y-Z, Gong L-Z. Chem. Eur. J. 2007;13:689–701. doi: 10.1002/chem.200600801. Ramasastry SSV, Zhang H, Tanaka F, Barbas CF., III J. Am. Chem. Soc. 2007;129:288–289. doi: 10.1021/ja0677012.
- (5).For an enantioselective direct aldol reaction with ester derivatives, see: Misaki T, Takimoto G, Sugimura T. J. Am. Chem. Soc. 2010;132:6286–6287. doi: 10.1021/ja101216x. and references therein.
- (6).(a) Denmark SE, Chung W.-j. Angew. Chem. Int. Ed. 2008;47:1890–1892. doi: 10.1002/anie.200705499. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Chung W.-j. J. Org. Chem. 2008;73:4582–4595. doi: 10.1021/jo8006539. [DOI] [PubMed] [Google Scholar]
- (7).For leading references on glycolate aldol reactions, see: Gennari C. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon Press; New York: 1991. Chapter 2.4. Cameron JC, Paterson I. Org. React. 1997;51:1. Carreira EM. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 3. Springer; Heidelberg: 1999. Chapter 29.1. Mahrwald R, editor. Modern Aldol Reactions. Wiley-VCH; Weinheim, Germany: 2004. Nelson SG. Tetrahedron: Asymmetry. 1998;9:357–389. Machajewski TD, Wong C-H. Angew. Chem. Int. Ed. 2000;39:1352–1374. doi: 10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j.
- (8).For selected examples, see: Trost BM, Hirano K. Angew. Chem. Int. Ed. 2012;51:6480–6483. doi: 10.1002/anie.201201116. Trost BM, Hirano K. Org. Lett. 2012;14:2446–2449. doi: 10.1021/ol300577y. Trost BM, Malhotra S, Fried BA. J. Am. Chem. Soc. 2009;131:1674–1675. doi: 10.1021/ja809181m. Trost BM, Lupton DW. Org. Lett. 2007;9:2023–2026. doi: 10.1021/ol070618e. Trost BM, Jaratjaroonphong J, Reutrakul V. J. Am. Chem. Soc. 2006;128:2778–2779. doi: 10.1021/ja057498v. Trost BM, Shin S, Sclafani JA. J. Am. Chem. Soc. 2005;127:8602–8603. doi: 10.1021/ja051526s. Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660–2661. doi: 10.1021/ja038666r. Trost BM, Terrell LR. J. Am. Chem. Soc. 2003;125:338–339. doi: 10.1021/ja028782e. Trost BM, Yeh VSC. Angew. Chem. Int. Ed. 2002;41:861–863. doi: 10.1002/1521-3773(20020301)41:5<861::aid-anie861>3.0.co;2-v.
- (9).(a) Trost BM, Amans D, Seganish WM, Chung CK. J. Am. Chem. Soc. 2009;131:17087–17089. doi: 10.1021/ja907924j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Seganish WM, Chung CK, Amans D. Chem. Eur. J. 2012;18:2948–2960. doi: 10.1002/chem.201102898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trost BM, Amans D, Seganish WM, Chung CK. Chem. Eur. J. 2012;18:2961–2971. doi: 10.1002/chem.201102899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For a repot of N-(α-hydroxyacetyl)pyrrole as an aldol donor for asymmetric mannich type reactions, see: Harada S, Handa S, Matsunaga S, Shibasaki M. Angew. Chem. Int. Ed. 2005;44:4365–4368. doi: 10.1002/anie.200501180.
- (11).For a large-scale synthesis of 2-ethyl pyrrole see: Alonso Garrido DO, Buldain G, Frydman B. J. Org. Chem. 1984;49:2619–2622.
- (12).Evans DA, Borg G, Scheidt KA. Angew. Chem. Int. Ed. 2002;41:3188–3191. doi: 10.1002/1521-3773(20020902)41:17<3188::AID-ANIE3188>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- (13).The absolute configuration of the diol products was confirmed by comparison of the optical rotation of 5a to reported values and then by analogy. See: Neisius NM, Plietker B. J. Org. Chem. 2008;73:3218–3227. doi: 10.1021/jo800145x.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.