

Atg38 provides a physical linkage between the Vps15–Vps34 and Atg14–Vps30 subcomplexes to facilitate PI3-kinase complex I formation.
Abstract
Autophagy is a conserved eukaryotic process of protein and organelle self-degradation within the vacuole/lysosome. Autophagy is characterized by the formation of an autophagosome, for which Vps34-dervied phosphatidylinositol 3-phosphate (PI3P) is essential. In yeast, Vps34 forms two distinct protein complexes: complex I, which functions in autophagy, and complex II, which is involved in protein sorting to the vacuole. Here we identify and characterize Atg38 as a stably associated subunit of complex I. In atg38Δ cells, autophagic activity was significantly reduced and PI3-kinase complex I dissociated into the Vps15–Vps34 and Atg14–Vps30 subcomplexes. We find that Atg38 physically interacted with Atg14 and Vps34 via its N terminus. Further biochemical analyses revealed that Atg38 homodimerizes through its C terminus and that this homodimer formation is indispensable for the integrity of complex I. These data suggest that the homodimer of Atg38 functions as a physical linkage between the Vps15–Vps34 and Atg14–Vps30 subcomplexes to facilitate complex I formation.
Introduction
Macroautophagy (hereafter called autophagy) is a conserved eukaryotic process of vacuolar/lysosomal-mediated degradation of cytoplasmic proteins and organelles (Mizushima et al., 2011). Autophagy is characterized by the formation of a double-membrane structure, the autophagosome. In yeast, the autophagosome is formed as an isolation membrane that expands outwards from a nucleation site and then closes, engulfing cytoplasmic materials. The autophagosome then fuses with the vacuole, where its engulfed materials are degraded by acidic hydrolases. This conserved system plays a role not only in the recycling of proteins but also in the clearance of aberrant protein aggregates and damaged organelles.
The molecular mechanisms underlying the process of autophagy have been studied extensively in yeast (Tsukada and Ohsumi, 1993; Nakatogawa et al., 2009). Yeast genetic analyses have identified more than 35 autophagy-related genes (ATG genes). Among them, 18 ATG genes encode core components required for autophagosome formation (Atg1–10, 12–14, 16–18, 29, and 31), the majority of which localize at least partly to the preautophagosomal structure (PAS), the nucleation site from which the autophagosome originates (Suzuki et al., 2001, 2007). These 18 proteins are classified into six functional groups: Atg1 kinase and its regulators, the PI3-kinase complex, the Atg9 vesicle, the Atg2–Atg18 complex, and two ubiquitin-like conjugation systems (Nakatogawa et al., 2009).
The budding yeast has only one class III PI3-kinase, Vps34, which specifically phosphorylates position 3 of phosphatidylinositol (Schu et al., 1993). The product, phosphatidylinositol 3-phosphate (PI3P), is essential not only for autophagosome formation but also for the vacuolar protein sorting (VPS) pathway in yeast (Backer, 2008). To function in these two processes, Vps34 forms distinct complexes: PI3-kinase complex I (Vps34, Vps15, Vps30/Atg6, and Atg14), which functions in autophagy, and PI3-kinase complex II (Vps34, Vps15, Vps30, and Vps38), which is involved in the VPS pathway (Kihara et al., 2001; Obara et al., 2006). The highly specific nature of these complexes is determined by the presence of the unique proteins Atg14 and Vps38, which associate exclusively with complex I or II, respectively. Atg14 and Vps38 further play a role in bridging Vps30 and Vps34 to allow complex formation. Atg14 is required for the localization of complex I to the PAS, whereas Vps38 is responsible for the endosomal localization of complex II (Obara et al., 2006). In spite of these fundamental roles played by PI3-kinase complexes in the cell, little is known about their formation and how these complexes exert their characteristic functional specificity (Obara and Ohsumi, 2011).
In this study, we identify a yet uncharacterized protein, Atg38, in a search for proteins that tightly associate with Vps34. Immunoprecipitation analyses reveal that Atg38 co-purifies with Atg14, a complex I–specific factor, but not with Vps38, a complex II–specific factor, and is recruited to the PAS in an Atg14-dependent manner. Further biochemical and genetic analyses demonstrate that Atg38 is required for autophagy as an integral component of PI3-kinase complex I in yeast, and that Atg38 functions as a linkage connecting the Vps15–Vps34 and Vps30–Atg14 subcomplexes to facilitate complex I formation.
Results
Identification of Vps34-binding proteins
To better understand the molecular function of the PI3-kinase complexes, we initially sought to identify proteins that tightly interact with these complexes. TAP-tagged Vps34 was immunopurified from yeast cells, and co-precipitating proteins were processed for identification using liquid chromatography tandem mass spectrometry (LC-MS/MS). In addition to known components of Vps34 complexes, we identified the product of a yet uncharacterized open reading frame, YLR211c (Fig. 1 A). This gene is hereafter referred to as ATG38 to reflect its critical role in regulating autophagy as described below. Atg38 consists of 226 amino acids, with a molecular mass of 26 kD. Structurally, Atg38 is predicted to have two coiled-coil regions, one in the N-terminal half (1–120 aa) and the other in the C-terminal half (121–226 aa) of the protein (Fig. S1 A). The function of this protein has not yet been described.
Figure 1.

Atg38 is a novel component of complex I. (A) Proteins detected in Vps34-TAP immunoprecipitations by MS analysis are listed. The number of identified peptides of each protein and the peptide coverage are also shown. (B) Cells expressing TAP-tagged Atg38 and the indicated HA-tagged proteins were grown in YPD. The extracts were prepared and were immunoprecipitated by IgG-Dynabeads as described in Materials and methods. The whole-cell extract (left, “input”) and the precipitated proteins (right) were analyzed by immunoblotting with the indicated antibodies. (C) Relative abundances of proteins identified by LC-MS/MS of Atg14-TAP and Atg38-TAP purifications from YPD-grown cells. Relative abundances are defined as emPAIprey/emPAIbait and are shown as mean ± SD of two independent experiments. (D) Cell extracts were prepared from wild-type and atg14Δ cells expressing Atg38-TAP as well as HA-tagged proteins of complex I components. The IgG Dynabead-precipitated proteins (bottom), together with the whole-cell extracts (top), were immunoblotted with the indicated antibodies. (E) ATG38-2GFP ATG17-2mCherry cells in wild-type and atg14Δ were cultured at 30°C with or without rapamycin. After 180 min, cells were analyzed by fluorescence microscopy. Bar, 5 µm.
Atg38 is a stably associated partner of the PI3-kinase complex I
As Vps34 functions in two distinct complexes (complexes I and II; Kihara et al., 2001), we began our analysis by assessing which complex Atg38 exists in. To this end, we sought to determine the interactions between Atg38 and PI3-kinase complexes components. The chromosomal VPS15, VPS34, VPS30, ATG14, and VPS38 genes were individually tagged with HA epitopes in an ATG38-TAP strain. Cell extract was prepared from each strain and subjected to immunoprecipitation using IgG beads. Atg38-TAP coimmunoprecipitated with Vps34, Vps15, Vps30, and Atg14, but not Vps38, a complex II–specific component (Figs. 1 B and S1 B; Kihara et al., 2001). These results indicate that Atg38 interacts specifically with each component of complex I, which is required for autophagy, but not with complex II–specific Vps38.
We next asked whether all components of PI3-kinase complex I are able to interact with Atg38 simultaneously as a complex. We generated a yeast strain whose VPS34, VPS30, ATG14, and ATG38 genes were chromosomally tagged with the HA, myc, TAP, and FLAG tags, respectively. As the amount of complex I is very low in yeast (Kihara et al., 2001), we initially concentrated Atg14-TAP by immunoprecipitation using IgG beads before eluting with TEV protease (Fig. S1 C, input). This eluate was fractionated on a gel filtration column. Vps34, Vps30, Atg14, and Atg38 co-eluted in a fraction corresponding to ∼500 kD, which corresponds to the predicted size of complex I including Atg38 (Fig. S1 C, Fr 9). This observation provides further evidence that Atg38 tightly interacts with complex I.
To determine whether Atg38 is invariably associated with complex I, we estimated absolute protein contents in the complex. The abundance of each protein was quantified by the exponentially modified protein abundance index (emPAI), which provides an estimate of absolute abundance of proteins by quantitating the number of peptides identified by MS (Ishihama et al., 2005). To compare results from multiple purifications, we then calculated relative abundances of proteins defined as emPAIprey/emPAIbait (Fig. 1 C). Atg14-TAP proteins were purified using IgG beads before purified materials were analyzed by LC-MS/MS. Notably, we found that with the exclusion of common contaminants of proteomic analysis (Breitkreutz et al., 2010), components of complex I, including Atg38, were most abundant when analyzed by emPAI. None of these proteins were found to associate with control proteins purified by the same technique (Fig. 1 C and Table S3). Among proteins co-purifying with Atg14, Vps30 is very high in relative abundance, reflecting the fact that Atg14 forms a subcomplex with Vps30 (Noda et al., 2012). Importantly, the relative abundance of Atg38 is comparable to or even marginally higher than the abundances of Vps15 or Vps34, indicating that Atg38 is invariably associated with complex I as an integral component (Fig. 1 C and Table S3).
We next asked whether Atg38 is included in other complexes as well as in complex I. Atg38-TAP proteins were purified using IgG beads, before this purified material was analyzed by semi-quantitative LC-MS/MS as described above. Once again excluding contaminating proteins, we found that only subunits of complex I were characterized by high emPAI values, indicating increased relative abundance (Fig. 1 C and Table S3). In addition, the absence of Vps38 when Atg38-TAP eluate was assessed by emPAI analysis indicates that Atg38 does not associate with complex II at all. These data indicate that Atg38 acts exclusively as a component of complex I, supporting its specific role in autophagy through this complex.
Among the components of complex I, Atg14 is the only known protein to associate specifically with complex I, which led us to the assumption that Atg38 interacts with complex I in an Atg14-dependent manner (Kihara et al., 2001; Obara et al., 2006). To test this, we examined whether deletion of ATG14 has an effect on the interaction between Atg38 and other components of complex I. The interactions between Atg38 and Vps34, Vps15, and Vps30 observed in wild-type cells were hardly detectable in atg14Δ cells, indicating that the interaction between Atg38 and components of complex I is dependent on Atg14 (Fig. 1 D).
Treatment with rapamycin, which induces autophagy by inhibiting Tor kinase (Noda and Ohsumi, 1998), did not affect the interaction between Atg38 and other subunits of complex I, indicating that Atg38 associates stably with complex I, irrespective of whether or not autophagy is induced by rapamycin (Fig. S1 D). Thus, we conclude that Atg38 is a stable subunit of autophagy-related complex I.
Atg38 localizes to the PAS in an Atg14-dependent manner
Microscopic analyses have revealed that upon induction of autophagy, most of the Atg proteins, including Atg14, localize to a perivacuolar structure called the preautophagosomal structure (PAS) from which the autophagosome originates (Suzuki et al., 2001, 2007). As Atg38 associates with complex I, we examined whether Atg38 also localizes to the PAS. To assess this we used Atg17 as a marker of the PAS, as the localization of Atg17 to the PAS is independent of the other ATG genes (Suzuki et al., 2007). We generated yeast strains harboring a 2× GFP-tagged Atg38 and a 2× mCherry-tagged Atg17 expressed from their own promoters. Fluorescence microscopy revealed that in the absence of rapamycin, Atg38-2× GFP signals were detected on the vacuolar membrane (Fig. 1 E, top), and that in the presence of rapamycin, Atg38-2× GFP signals were detected on punctate structures in addition to the vacuolar membrane. Under these conditions, Atg38-2× GFP puncta colocalized with Atg17-2× mCherry (Fig. 1 E, middle). In atg14Δ cells, Atg38-2× GFP did not colocalize with Atg17-2× mCherry, but was diffused throughout the cytoplasm (Fig. 1 E, top). These data indicate that Atg38 localizes to the PAS in the presence of rapamycin, and further support our conclusion that Atg38 is a component of complex I through its interaction with Atg14 (Obara et al., 2006).
Autophagy is compromised in atg38Δ cells
To address the physiological function of Atg38, we next examined the effect of ATG38 deletion on autophagy, the cytoplasm-to-vacuole targeting (Cvt) pathway required for the transport of aminopeptidase I (API), and the VPS pathway required for vacuolar carboxypeptidase Y (CPY) transport. Autophagic activity was estimated by means of the alkaline phosphatase (ALP) assay, in which the hydrolytic activity of a modified variant of this enzyme (Pho8Δ60) is monitored biochemically (Noda et al., 1995). The pro-form of modified ALP, which can only be transported into the vacuole in a manner dependent on autophagy, is subsequently processed to an active form, the activity of which quantitatively indicates the progress of autophagy. In wild-type cells, ALP activity increased in response to starvation, whereas in autophagy-deficient atg14Δ cells no elevation of ALP activity was observed (Fig. 2 A). The increase of ALP activity in atg38Δ cells was ∼50% of that of wild-type cells, indicating that autophagy was significantly inhibited in the absence of Atg38. Intriguingly, we observed that atg38Δ ATG14-GFP cells showed a complete autophagy defect, although GFP fusion to Atg14 alone was not accompanied by any adverse effect on autophagy in wild-type cells (Fig. 2 A). This suggested genetic relationship lends strong support to the importance of the physical interaction between Atg14 and Atg38. We further investigated the involvement of Atg38 in autophagy using API, another cargo of autophagy. API is synthesized as a pro-form (prAPI) in the cytoplasm and is delivered to the vacuole where it is processed to a mature form (mAPI; Klionsky et al., 1992; Baba et al., 1997; Scott et al., 1997). In the presence of rapamycin, prAPI transport is mediated by autophagy. Both rapamycin-treated atg38Δ ATG14 and ATG38 ATG14-GFP cells were not characterized by any defect in API maturation (Fig. 2 B, lanes 4 and 6). However, most API was found as a pro-form in atg38Δ ATG14-GFP cells (lane 8), albeit to a lesser extent than in atg14Δ cells (lane 10). Taken together, this evidence for a genetic interaction between ATG38 and ATG14 indicates that Atg38 functions in autophagy together with Atg14, underscoring the importance of the physical interaction between Atg38 and Atg14 in autophagy.
Figure 2.

ATG38 is required for autophagy. (A) Cells expressing Pho8Δ60 were grown in YPD and shifted to SD(-N) medium for 3 h at 30°C. Lysates from each group of cells were tested for ALP activity. ALP activity is shown as mean ± SD of three independent experiments with the activity of wild-type cells normalized to 1. (B) Cells with the indicated genotypes were grown in YPD and treated with rapamycin for 3 h at 30°C. Total protein was separated by SDS-PAGE and detected by immunoblotting with anti-API. (C) Cells were grown in YPD medium for 1 h at 30°C and separated to the intracellular (I; cells) and extracellular (E; medium) fractions by centrifugation. The fractions were treated as detailed in Materials and methods and aliquots of each were separated by SDS-PAGE. The positions of the precursor (p2-CPY) and mature (m-CPY) forms of CPY are indicated.
API is transported to the vacuole via the Cvt pathway under nutrient-rich conditions. As the Cvt pathway depends on a similar mechanism to the autophagic pathway, most autophagy mutants are defective in the Cvt pathway (Harding et al., 1996). In the absence of rapamycin, atg38Δ cells showed normal maturation of API, as observed in wild-type and ATG38 ATG14-GFP cells (Fig. 2 B, lanes 1, 3, and 5), while the processing of API in atg38Δ ATG14-GFP cells (lane 7) was completely blocked as in atg14Δ cells (lane 9). The defect in the Cvt pathway observed in atg38Δ ATG14-GFP cells indicates that Atg38 also plays a role in the Cvt pathway together with Atg14.
It is conceivable that the autophagic defects in atg38Δ cells may be the result of reduced amounts of vacuolar enzymes required for the maturation of Pho8Δ60 and API in the vacuole as the transport of these proteases is mediated by the complex II–dependent VPS pathway (Herman et al., 1991; Seaman et al., 1997; Kametaka et al., 1998; Robinson et al., 1988). We therefore investigated whether atg38Δ cells show a defect in the VPS pathway. Transport of CPY, a vacuolar peptidase, is relatively complex, with the pro-form of this peptidase transiting through several organelles before delivery to the vacuole in a manner dependent on complex II (Bowers and Stevens, 2005). We therefore assessed CPY secretion, as impairment of complex II leads to mislocalization of CPY to the extracellular space. To this end, cells were washed extensively and subsequently cultured in fresh YPD medium for 1 h at 30°C. Next, cultures were separated into intracellular (I; cells) and extracellular (E; medium) fractions by centrifugation. Western blot analyses of these fractions indicated that CPY was present as a mature form (mCPY) in the intracellular fraction in wild-type, atg14Δ, and atg38Δ cells. In contrast, newly synthesized CPY was secreted as the pro-form (p2-CPY) into media from vps30Δ cells (Fig. 2 C; Kametaka et al., 1998). These data confirm that the VPS pathway is not impaired in atg38Δ cells. Thus, the reduction of autophagic activity in atg38Δ is not due to vacuolar protease deficiency, but rather reflects the important role of Atg38 in the formation of autophagosomes as a component of complex I (Fig. 2 C).
We also investigated whether Atg38 is involved in other selective autophagy pathways, first examining the requirement of Atg38 for mitophagy. We precultured cells in conditions where mitochondrial development is induced (YPL) before shifting cells to starvation conditions (SD-N; Kanki et al., 2009). Under these conditions, culture of Idh1-GFP cells, in which GFP is fused with a mitochondrial matrix protein, results in the cleavage of GFP from Idh1 as mitochondria are degraded in the vacuole by mitophagy. This cleavage can be followed by the appearance of free GFP in Western blots. Free GFP first appeared 3 h after cells were shifted to SD(-N), before reaching peak intensity after 6 h in wild-type and atg38Δ cells. Free GFP was not observed in atg32Δ cells, which lack a protein essential for mitophagic activity. These data demonstrate that deletion of ATG38 does not exert any influence on mitophagy. We additionally examined pexophagy in atg38Δ cells adopting a similar approach. Yeast cells expressing the peroxisomal protein Pex11 fused with GFP were cultured in conditions inducing peroxisomal proliferation (YM2 oleate media) before being shifted to pexophagy-inducing starvation conditions (Motley et al., 2012). This experiment indicated that Atg38 is not required for pexophagy, as free GFP, representing the vacuolar degradation of Pex11-GFP, was observed in wild-type and atg38Δ cells, but not in cells lacking the pexophagy receptor Atg36 (Fig. S1 F). Together, these results indicate that Atg38 is not implicated in the selective autophagy pathways of mitophagy and pexophagy.
Deletion of ATG38 gene decreases the association of PI3-kinase complex I with the PAS
To obtain a molecular understanding of the role of Atg38 in autophagy, we analyzed the association of complex I proteins with the PAS in atg38Δ cells in the presence of rapamycin. For this analysis, the PAS was marked by API fused with mRFP. Fluorescence microscopy revealed that deletion of ATG38 led to a decrease in colocalization of complex I to the PAS, consistent with the partial defects of autophagic activity observed in atg38Δ cells (Fig. 3, A and B). The Atg2–Atg18 complex is placed downstream of PI3-kinase in terms of the localization of Atg proteins to the PAS. Atg18 is a PI3P-binding protein and is often used as an indicator of the amount of PI3P on the PAS (Obara et al., 2008). The reduced colocalization of Atg18 to the PAS observed in atg38Δ cells (Fig. 3, A and B) was expected, as mislocalization of complex I is also observed in the absence of Atg38. These reductions were not due to changes in protein abundance, as determined by Western blotting (Fig. 3 C). These results confirm that Atg38 plays a role as a component of PI3-kinase complex I, and may indicate that the amount of PI3P produced on the PAS decreases in atg38Δ cells.
Figure 3.
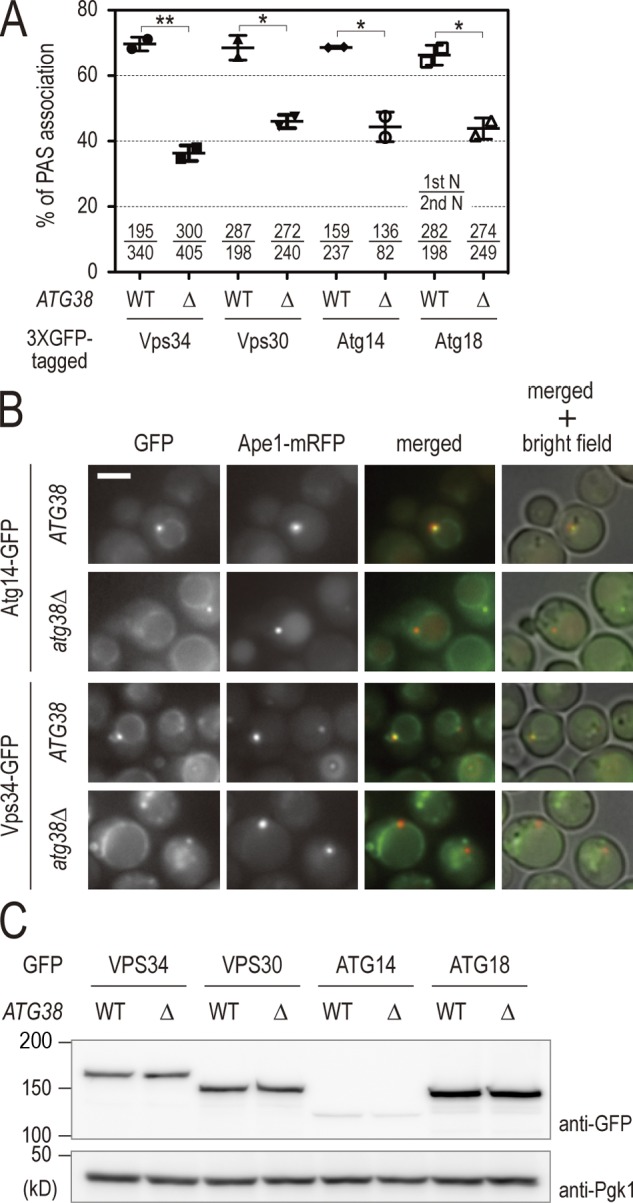
Atg38 is required for the localization of PI3-kinase and Atg18 to the PAS. (A) Wild-type and atg38Δ cells expressing the indicated GFP fusion proteins and Ape1-mRFP were cultured at 30°C in the presence of rapamycin. After 60 min, cells were analyzed by fluorescence microscopy. Of the total pool of Ape1-mRFP signal, the percentage of Ape1-mRFP colocalizing with GFP was calculated. This percentage is shown as mean ± SD of two independent experiments, with the number of cells counted indicated at the bottom of the graph. (B) Examples of wild-type and atg38Δ cells expressing either Atg14-3 × GFP or Vps34-3 × GFP in A. Bar, 5 µm. (C) Cell extracts were prepared from the cells in A, then analyzed by immunoblotting with anti-GFP and anti-Pgk1 antibodies.
Atg38 holds the Vps15–Vps34 and Atg14–Vps30 subcomplexes together
To understand the role of Atg38 in complex I, we addressed the question of whether Atg38 is involved in complex I formation. Interactions among the components of complex I in atg38Δ cells were investigated using coimmunoprecipitation analyses (Fig. 4 A). atg38Δ cells were characterized by a decrease in the amount of Vps34 and Vps15 that coimmunoprecipitated with Atg14-TAP to 24% and 23% in comparison to wild-type cells, respectively. However, we observed that most Vps30 protein still binds to Atg14 in atg38Δ cells (Fig. 4 A). These data show that the absence of Atg38 induced the radical dissociation (∼75%) of complex I into the two subcomplexes, Vps15–Vps34 and Atg14–Vps30, indicating that Atg38 is required to hold the Vps34–Vps15 and Atg14–Vps30 subcomplexes together (Kihara et al., 2001; Noda et al., 2012). Semi-quantitative LC-MS/MS analyses of purified materials from ATG14-TAP atg38Δ cells provided further evidence that these subcomplexes dissociate in the absence of Atg38 (Fig. 4 B). Importantly, in contrast to the complete autophagic defect observed in Atg14-GFP atg38Δ cells, Atg14-TAP is characterized by residual autophagy not significantly different from untagged Atg14 in an atg38Δ background (Fig. 4 C).
Figure 4.

Atg38 is required to hold the Vps34–Vps15 and Atg14–Vps30 subcomplexes together. (A) Cell extracts were prepared from wild-type and atg38Δ strains expressing TAP-tagged Atg38 and the indicated HA proteins. The extracts were immunoprecipitated by IgG-Dynabeads. The whole-cell extracts (top, “input”) and the precipitates (bottom) were analyzed by immunoblotting with the indicated antibodies. Relative binding efficiency was determined as described in Materials and methods. To compare results from multiple experiments, the value for the reference sample (wild-type cells) was normalized to 1.0. Relative binding efficiency is shown as mean ± SD of three independent experiments. (B) Relative abundances of proteins identified in TAP purifications from the indicated strains were analyzed as described in Fig. 1 C and are shown as mean ± SD of two independent experiments. (C) Cells expressing Pho8Δ60 were grown in YPD and shifted to SD(-N) medium for 3 h at 30°C. Cell lysates from each group of cells were tested for ALP activity. ALP activity is shown as mean ± SD of three independent experiments with the activity of wild-type cells normalized to 1. (D) Cells expressing Vps34-6HA, Vps30-9myc, Atg38-3FLAG, and Atg14-TAP were lysed in a buffer containing 150 mM NaCl. The Atg14-TAP immunoprecipitate with IgG-Dynabeads from this lysate was split into eight fractions and washed with lysis buffer containing the indicated concentrations of NaCl. The resulting precipitates were analyzed by immunoblotting with the indicated antibodies. Relative binding efficiency was determined as described in Materials and methods, although a 150-mM NaCl sample was used as a control and normalized to 1.0. Relative binding efficiency is shown as mean ± SD of three independent experiments.
Alternatively, it is possible that Atg38 might play a role as a chaperone escorting the subcomplexes to form the mature complex I rather than a structural role in the complex as an intrinsic component. If Atg38 does act as a chaperone, the protein will not be required for the maintenance of mature complex I once formed. To determine whether this is the case, we assessed the strength of interactions among complex I components. Complex I was purified with Atg14-TAP and then washed extensively with buffers containing increasing concentrations of NaCl (Fig. 4 D). The association of Atg14 with Vps30 was found to be resistant to washes with high concentrations of salt. In contrast, Vps34 and Atg38 detached from Atg14–Vps30 in a concentration-dependent manner, but unexpectedly, Vps34 exhibited a slight recovery in Atg14 binding at high concentrations (Fig. 4 D). The similar rates of salt-induced dissociation of Atg38 and Vps34 from Atg14 are consistent with the notion that Atg38 mediates the interaction between Atg14–Vps30 and Vps15–Vps34. Collectively, these findings lend further credence to the above conclusion that Atg38 is an integral component tethering these two subcomplexes to each other, thereby facilitating the formation of complex I for autophagy.
Atg38 interacts with Atg14 through its N-terminal domain
To explore how Atg38 tethers the complex I subcomplexes, we studied the interaction between Atg38 and each component of complex I by the yeast two-hybrid system. Using medium (−LWH) and high stringency (−LWA) plates (Fig. 5 A), we found that Atg38 interacts with both Atg14 and Vps34, the interaction with Atg14 being stronger than with Vps34. Atg38 did not show any positive signal with Vps30. Furthermore, it was shown that the interaction between Atg38 and Atg14 was mediated through the N-terminal half of Atg38, but not the C-terminal half, although the interaction between Atg14 and the N-terminal half of Atg38 was weaker than the full-length protein (Fig. 4 B). These results suggest that Atg38 interacts with Atg14 through its N-terminal domain.
Figure 5.

Atg38 interacts with Atg14 and Vps34 physically via its N-terminal domain. (A) AH109 cells containing the indicated GAL4 activation domain fusions (bait) and GAL4 DNA-binding domain fusions (prey) were selected on SD-Leu-Trp plates at 30°C (−LW, left). The interaction of fusion proteins was examined by plating cells on SD-Leu-Trp-His (−LWH, middle) and SD-Leu-Trp-Ade (−LWA, right) plates at 30°C for 3 d. (B) Interactions between the indicated fusion proteins were analyzed as in A. (C) The TAP-tagged Vps34 protein complex was purified from the vps30Δ atg14Δ vps38Δ cells. The purified proteins were separated on SDS-PAGE and visualized by Coomassie staining. Recombinant GST-Atg38 (WT), GST-Atg381–120 (N), and GST-Atg38121–226 (C) were incubated with Vps15–Vps34-TAP complex bound to IgG-Dynabeads. The eluted proteins were analyzed by immunoblotting.
We next examined which domain of Atg14 is responsible for binding to Atg38. To address this question, truncated versions of Atg14 fused to 3HA-GFP were expressed in atg14Δ ATG38-TAP cells (Obara et al., 2006). Cell extracts were prepared from these cells and subjected to immunoprecipitation by IgG beads (Fig. S2 A). Atg38 coimmunoprecipitated with full-length Atg14 but not any truncated proteins of Atg14, suggesting that a rather large portion of Atg14 is necessary for tight binding to Atg38.
Atg38 interacts with Vps34 directly through its N-terminal domain
Yeast two-hybrid analyses suggested that Vps34 binds only weakly with full-length Atg38, but not with Atg381–120 or Atg38121–256 (Figs. 4 A and S2 B). We further investigated this interaction using an in vitro approach. The Vps15–Vps34-TAP complex was purified from cells in which VPS30, ATG14, and VPS38 genes had been disrupted (Fig. 5 C, left). GST-Atg38 fusion protein purified from Escherichia coli was incubated with Vps15–Vps34-TAP, followed by extensive washing. Binding of GST-Atg38 fusion protein to the Vps15–Vps34-TAP complex was then analyzed by immunoblotting (Fig. 5 C, right). The Vps15–Vps34-TAP complex associated with GST-Atg38 (WT) and GST-Atg381–120 (N), but not with GST-Atg38121–226 (C). Thus, the N-terminal half of Atg38 interacts with Vps34, as well as Atg14.
The MIT domain of Atg38 is necessary and sufficient for Atg14 binding
Using the HHpred database, a server for protein remote homologue detection and structure prediction (Söding et al., 2005), we detected high probability matches between the first 80 residues of the N terminus of Atg38 and microtubule interacting and trafficking (MIT)–containing domain proteins (Fig. S2 C; Phillips et al., 2001; Ciccarelli et al., 2003). MIT domains are known to consist of small helical modules involved in protein–protein interactions, and are conserved among Vps4, spastin, katanin, and various other proteins (Obita et al., 2007; Stuchell-Brereton et al., 2007; Kieffer et al., 2008). We asked whether the MIT domain in Atg38 is involved in the interaction with either Vps34 or Atg14. Two-hybrid analyses demonstrated that the MIT domain is necessary and sufficient for Atg14 binding, but is dispensable for binding to Vps34 (Fig. S2 D).
Atg38 forms a homodimer
We observed that Atg38 interacted with itself as shown by two-hybrid analyses, suggesting that it is able to form a homodimer or a homo-oligomer (Figs. 5 A and 6 A). Further two-hybrid analyses revealed that the self-association of Atg38 was mediated through its C-terminal domain (120–226 aa; Fig. 6 A). To determine how many molecules of Atg38 associate with each other simultaneously, the size of recombinant Atg38 protein purified from E. coli was measured by gel filtration chromatography (Fig. 6 B). We calculated that the molecular mass of the native form of Atg38 was distributed around 50–80 kD (Fig. 6 B, Fr 8–11). Judging from the predicted molecular weight of Atg38 (26.3 kD), this indicates that Atg38 most likely forms a homodimer or a trimer.
Figure 6.

Atg38 forms a homodimer. (A) Interactions between the indicated fusion proteins were analyzed as in Fig. 4 A. (B) Analysis of Atg38 by gel filtration. Recombinant Atg38 protein was fractionated over a gel filtration column. Each fraction was subjected to SDS-PAGE and visualized by Coomassie staining. (C) Analytical ultracentrifugation of Atg38. The raw absorbance distributions and the best-fit model calculated by the SEDFIT program (top). Residuals of the fit (middle), sedimentation coefficients, c(S), and distributions (bottom) of Atg38 protein.
To determine the precise number of Atg38 proteins in the oligomer, we conducted analytical ultracentrifugation of Atg38 (Fig. 6 C). This sedimentation velocity experiment revealed a molecular specimen with a sedimentation coefficient of 3.0 S in solution. This indicates that Atg38 exists as a particle of 50.2 kD, which is consistent with the molecular mass of a dimer. These data therefore confirm that Atg38 forms a dimer through its C-terminal domain.
Homodimer formation of Atg38 is required for the integrity of complex I
We next asked whether homodimer formation of Atg38 is required to hold Vps34–Vps15 and Atg14–Vps30 together. If so, it might be possible to replace the endogenous homodimerization domain with an alternative pair of dimer-forming domains from another protein. To this end, we adopted the GFP entrapment strategy based on the GFP-binding protein (GBP), which efficiently binds to GFP and GFP-tagged proteins (Rothbauer et al., 2006). As a control, we generated a GBP mutant in which three amino acid residues, S33, R35, and Y37, were replaced with alanine (GBP3A) based on the structural data of the GFP–GBP complex (Kirchhofer et al., 2010). Atg9-GFP coimmunoprecipitated with Atg14–TAP–GBP, but not Atg14–TAP–GBP3A, confirming that GBP3A is unable to bind to GFP (Fig. S3 A). We then applied this artificial tethering to atg38Δ cells, in which the formation of complex I is largely defective (Fig. 4 A). We expressed the N-terminal domain of Atg38 (Atg381–120) fused to GFP and either GBP or GBP3A in atg38Δ cells and examined the interaction between Atg14 and either Vps15 or Vps34. Cell extracts were prepared from these cells and subjected to immunoprecipitation with IgG beads (Fig. 7 A). Western blotting of immunoprecipitated eluates showed that in the presence of Atg381–120-GFP and Atg381–120-GBP, Atg14-TAP coimmunoprecipitated with both Vps34 and Vps15 to a degree comparable to that in wild-type cells (89% and 94%, respectively; lanes 2 and 6). As expected, the combination of Atg381–120-GFP and Atg381–120-GBP3A failed to increase the efficiency of Atg14 binding to Vps34 and Vps15 (40% and 30%, respectively; lane 3 and 7). We confirmed this interaction using an alternative endosomal adaptor/scaffold complex consisting of MP1 and p14. (Fig. S3 B; Kurzbauer et al., 2004). These results suggest that the homodimerization of Atg38 is required to mediate the association between the Vps34–Vps15 and Atg14–Vps30 subcomplexes. This conclusion is further supported by semi-quantitative LC-MS/MS analyses of purified materials from ATG14-TAP atg38Δ atg381–120-GFP atg381–120-GBP cells (Fig. 4 B). Next, we examined whether the synthetically bound complex I, mediated by the combination of Atg381–120-GFP and Atg381–120-GBP, is able to restore the defect in autophagy in atg38Δ cells as determined by the ALP assay (Fig. S3 C). The autophagic activity of atg38Δ cells did not increase even in the presence of Atg38N-GFP and Atg38N-GBP. This implies that the C-terminal domain of Atg38 has additional, unidentified roles in autophagy besides holding Vps34–Vps15 and Atg14–Vps30 together.
Figure 7.

Homodimer formation of Atg38 is required for the integrity of complex I. (A) Cell extracts were prepared from atg38Δ strains expressing Atg14-TAP and either Vps34-6HA or Vps15-6HA with Atg381–120 fused with GFP and either GBP or GBP3A. The cell extracts were immunoprecipitated with IgG-Dynabeads. The precipitated proteins, together with the whole-cell extracts, were immunoblotted with the indicated antibodies (left). Relative binding efficiency was determined as described in Fig. 3 A and was shown as mean ± SD of three independent experiments. (B) Model for the formation of complex I.
Discussion
PI3-kinase activity plays an essential role in intracellular transport, including membrane trafficking and autophagy (Backer, 2008). Vps34, the sole class III PI3-kinase found in budding yeast, exists as two complexes known as complex I and complex II (Kametaka et al., 1998; Kihara et al., 2001). Complex I is exclusively involved in autophagy and is known to comprise the proteins Vps34, Vps15, Vps30, and Atg14. In this study, we addressed the question of how PI3-kinase complexes are formed. We identified Atg38 as a protein that associates stably with complex I, irrespective of autophagy induction (Figs. 1 A and Fig. S1 D). However, no association was apparent with Vps38, a complex II–specific protein, strongly suggesting that Atg38 associates specifically with complex I (Fig. 1 B and Fig. S1 B; Kihara et al., 2001). Supporting this observation, we found that Atg38 is present in an ∼500-kD complex containing Vps34, Vps30, and Atg14 (Fig. S1 C). Consistently, Atg38 localizes to the PAS in an Atg14-dependent manner (Fig. 1 E). Furthermore, semi-quantitative MS/MS analysis revealed that the majority of intracellular complex I contains Atg38 proteins and that Atg38 is not found in any other complex in yeast (Fig. 1 C and Table S3). Thus, we conclude that Atg38 is an integral subunit of complex I, which has most likely been overlooked thus far due to the low abundance of this complex and associated technical difficulties encountered in its analysis.
In spite of the strong evidence indicating that Atg38 associates with complex I, deletion of ATG38 does not cause a complete defect in autophagy, nor the complete mislocalization of complex I away from the PAS (Figs. 2 A and 3 A). Our subsequent investigation of the role of Atg38 in complex I formation helps to account for this apparent paradox. The absence of Atg38 induced a radical dissociation (∼75%) of complex I into the two subcomplexes, Vps15–Vps34 and Atg14–Vps30, indicating that Atg38 is required to hold these two subcomplexes together (Fig. 4, A and B). Our results from the salt-washing experiment demonstrated that the rate of salt-induced dissociation from Atg14 was similar between Vps34 and Atg38 (Fig. 4 C). On the other hand, the subcomplex partner of Atg14, Vps30, was retained by Atg14 when challenged with increasing salt concentrations. These results further support a role for Atg38 in holding these two subcomplexes together, rather than as a chaperone for complex I assembly. However, limited (∼25%) residual Vps15–Vps34 complex formation with Atg14 is observed in the absence of Atg38. This may be due to a weaker interaction between Vps30–Atg14 and Vps15–Vps34 in the absence of Atg38, or even another yet unidentified protein that is also able to facilitate this interaction (Fig. 7 B). These factors, which we were unable to identify in our investigations (Table S3), most likely account for the limited autophagy observed in atg38Δ cells.
As we determined that Atg38 is important in holding the Vps34–Vps15 and Atg14–Vps30 subcomplexes together, we next investigated the mechanistic basis of this function. Atg38 interacts with both Vps34 and Atg14 via its N-terminal domain, although whether a direct interaction between Atg14 and Atg38 exists has not been shown. Using the sensitive bioinformatic analysis tool HHpred (Söding et al., 2005), Atg38 is predicted to contain an MIT domain within its first 80 residues (Fig. S2 C). One known function of this domain is to promote protein–protein interactions, as observed in the well-studied example of Vps4. Analysis of Vps4 has revealed that its MIT domain binds target peptide motifs (Stuchell-Brereton et al., 2007; Kieffer et al., 2008), which comprise a consensus sequence that is exclusively recognized by the MIT domain, facilitating interaction between the target and Vps4. We found that the Atg38 MIT domain is necessary and sufficient for binding to Atg14, but not to Vps34. However, Atg14 lacks a typical consensus sequence required to recognize MIT domains, suggesting that the mode of interaction between Atg38 and Atg14 is different from the case of Vps4 MIT domain and its target peptides. Further structural studies will help to shed light on the molecular details of this interaction between Atg14 and the MIT domain of Atg38.
In addition to its binding to complex I via the N-terminal half, we found that Atg38 interacts with itself (Fig. 6 A). Gel filtration and analytical ultracentrifugation revealed that Atg38 forms a homodimer (Fig. 6, B and C). This dimerization is mediated by its C-terminal half, which is predicted to contain a coiled-coil domain. This prompted us to investigate whether Atg38 acts as a linker facilitating the dimerization of complex I. We explored this possibility by a variety of biochemical methods, including immunoprecipitation studies. Despite repeated trials, we were unable to detect dimerization of complex I (unpublished data). Our finding that the molecular mass of complex I is ∼500 kD matches the predicted size of complex I as a monomer (Fig. S1 C). Consistent with this observation, we found through an artificial dimerization system that Atg38 instead functions as a dimer within the monomeric complex I (Fig. 7 A). The defect in association between Atg14–Vps30 and Vps15–Vps34 observed in atg38Δ cells was recovered by expression of Atg38N-GFP and Atg38N-GBP, which forces the dimerization of Atg38. However, the combination of Atg38N-GFP and Atg38N-GBP3A, in which GBP cannot bind to GFP, failed to rescue proper complex I formation. These data strongly suggest that Atg38 dimerization plays an important role in mediating the interaction between Vps15–Vps34 and Atg14–Vps30.
A comparable mode of complex formation is also found at a fundamental level of bacterial life. E. coli RNA polymerase (RNAP) core enzyme is composed of two identical α subunits and one each of the β and β′ subunits. The α subunits initiate RNAP assembly by dimerizing into a platform with which the large β and β′ subunits interact (Ishihama, 1981). β and β′ subunit assembly sites are located at two different sites within the α subunit platform. Once the platform is formed, the physical location of these assembly sites determines the binding partner of each α subunit, and therefore the two subunits are considered to play different roles (Murakami et al., 1997). Using this example of the molecular function of the α subunits of E. coli RNAP as a reference, we propose a model for how complex I is formed (Fig. 7 B). In complex I formation, the two Atg38 proteins have identical sequences, but their location within complex I and their interaction within complex I most likely differ. We suspect that one Atg38 molecule interacts with Atg14 via its MIT domain, whereas the other interacts with Vps15–Vps34 through an alternative region in the N-terminal domain. The two Atg38 molecules form a dimer, resulting in a physical linkage between Vps15–Vps34 and Atg14–Vps30. Intriguingly, atg38Δ cells expressing Atg38N-GFP and Atg38N-GBP were still defective in autophagy, even though the interaction between Atg14 and Vps15–Vps34 was restored. This may indicate that the C-terminal region of Atg38 plays an additional role in autophagosome formation. However, we cannot exclude the possibility that synthetically bound complex I tethered by GFP and GBP in atg38Δ cells may not have the same conformation as that in wild-type cells, resulting in the observed inability to rescue autophagy.
As we found that Atg38 plays an important role in autophagy in yeast, we next undertook to identify homologous proteins in other organisms. Using the NIH basic local alignment search tool (BLAST) program, we identified several promising candidate Atg38 orthologues in other budding yeast species but not in higher eukaryotes such as fruit flies, worms, and mammals (unpublished data). Behrends et al. (2010) performed a proteomic screen revealing 751 interactions between 409 candidate autophagy or autophagy-interacting proteins in human cells. NRBF2 was identified as an interaction partner of hATG14, Beclin, and hVPS34, but did not bind to UVRAG, a mammalian orthologue of Vps38 (Itakura et al., 2008; Sun et al., 2008; Matsunaga et al., 2009; Zhong et al., 2009). Previously unknown to function in autophagy, NRBF2 was originally reported to be a possible gene activator protein interacting with nuclear hormone receptors, and specifically with the peroxisome proliferation activator (Yasumo et al., 2000). Behrends et al. (2010) suggest that NRBF2 is able to function as a positive regulator of autophagy by an unknown mechanism as depletion of NRBF2 results in a reduction in the number of autophagosomes. Recently, the tertiary structure of the first 86 residues of mouse NRBF2 was solved and deposited in the PDB database, showing that this region contains an MIT domain. In our HHpred search, we determined that NRBF2 comprises a region with a high degree of similarity to a portion of MIT domain found in Atg38 (Fig. S2 C). Although the significance of NRBF2 in autophagy remains to be comprehensively determined, we were also able to show that NRBF2 interacts with hAtg14 and binds to itself via its C-terminal domain (131–288 aa; unpublished data). These similarities between Atg38 and NRBF2 lend considerable credence to the notion that NRBF2 is a mammalian functional orthologue of Atg38.
In conclusion, we have found that Atg38, a hitherto uncharacterized protein, is involved in the correct formation of PI3-kinase complex I as the fifth subunit of this complex. Our data indicate that Atg38 fulfills this role by forming a physical linkage between the two subcomplexes of complex I, Vps15–Vps34 and Atg14–Vps30. Both the C and N termini of Atg38 are essential for this activity through their roles in homodimerization and subcomplex binding, respectively. We also identified candidate orthologues in several mammalian systems, raising the intriguing proposition that Atg38’s role at a structural level in complex I is conserved throughout eukaryotic organisms, underscoring the intricate role of this protein in autophagosome formation.
Materials and methods
Yeast strains and growth conditions
Yeast strains are listed in Table S1. The strains used in this study were derived from BY4741 or BJ3505 (Jones et al., 1982; Brachmann et al., 1998). Gene deletions and epitope tagging of genes at their endogenous loci were constructed by PCR-based methods and were validated by PCR (Knop et al., 1999; Janke et al., 2004). Yeast strains were grown at 30°C in yeast peptone dextrose medium with 0.1 mg/l adenine (YPAD) medium. For nitrogen starvation, SD(-N) medium (0.17% yeast nitrogen base without amino acid and ammonium sulfate supplemented with 2% glucose) was used. Synthetic complete (SC) medium (0.17% yeast nitrogen base without amino acid and ammonium sulfate, 0.5% ammonium sulfate, 2% glucose, 0.5% casamino acid, 0.002% tryptophan, 0.002% adenine, and 0.002% uracil) was used for live-cell imaging experiments. For mitophagy and pexophagy experiments, YPL medium (1% yeast extract, 2% peptone, and 2% lactate, pH 5.5) and YM2 oleate medium (0.17% yeast nitrogen base without amino acid and ammonium sulfate, 0.5% ammonium sulfate, 0.12% oleate, 0.2% Tween 40, 0.1% yeast extract, 0.1% glucose, 1% casamino acid, 0.002% tryptophan, 0.002% adenine, and 0.002% uracil) were used, respectively.
Plasmid construction
All plasmids used in this study are listed in Table S2. Yeast codon-optimized sequence of GFP binder was obtained by full gene synthesis. Mutations in GFP binder were introduced by PCR-directed mutagenesis.
Microscopic image acquisition
Cells with chromosomal gene fusions with the fluorophores mCherry and GFP were analyzed by fluorescence microscopy without fixation. Images were collected on an inverted fluorescence microscope (IX71; Olympus) equipped with a 150× total internal reflection fluorescence objective (UAPON 150×OTIRF, NA 1.45; Olympus), a CCD camera (ImagEM C9100-13; Hamamatsu Photonics), and AQUACOSMOS software (Hamamatsu Photonics). Images were processed in Adobe Photoshop. The images were not manipulated other than contrast and brightness adjustments.
Recombinant protein purification
GST-Atg38 was expressed in Escherichia coli Rosetta (DE3) by adding 0.25 mM IPTG for 3 h at 30°C and purified using glutathione-Sepharose beads (GE Healthcare) according to the manufacturer’s protocol. The recombinant proteins were eluted either with 20 mM glutathione for GST-Atg38 or with PreScission Protease (GE Healthcare) for nontagged Atg38. The nontagged protein was further purified by ion-exchange column (Mono Q HR 5/5; GE Healthcare).
In vitro binding assay
Vps15–Vps34-TAP proteins were prepared from vps30Δ atg14Δ vps38Δ yeast cells as follows. The cells were resuspended in Lysis400 buffer (50 mM Tris-HCl, pH 8, 400 mM NaCl, 10% glycerol, 1 mM dithiothreitol [DTT], 5 mM EDTA, and 1% Triton X-100) with 1 mM PMSF and Complete EDTA-free protease inhibitor cocktail (Roche) and were lysed with a glass bead homogenizer. The cleared cell extract was then incubated with magnetic beads coupled with rabbit IgG (Dynabeads; Invitrogen). Vps15–Vps34-TAP proteins immobilized on the beads were incubated with the appropriate purified GST fusion proteins in TAP-A buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 10% glycerol, 1 mM DTT, 1 mM EDTA, and 0.2% Triton X-100) for 1.5 h at 4°C. After extensive washing steps, bound proteins were eluted with SDS sample buffer and analyzed by Western blot analysis.
Coimmunoprecipitation
Cells were resuspended in Lysis150 buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 10% glycerol, 1 mM DTT, 5 mM EDTA, and 1% Triton X-100) with 1 mM PMSF and Complete EDTA-free protease inhibitor cocktail (Roche), and were lysed with a glass bead homogenizer. TAP-tagged proteins were precipitated with magnetic beads coupled with rabbit IgG (Dynabeads; Invitrogen). The beads were washed three times with Lysis150 buffer. Bound proteins were eluted in HU buffer (200 mM Na-phosphate, pH 6.8, 8 M urea, 5% SDS, 0.1 mM EDTA, 0.005% Bromophenol blue, and 15 mg/ml DTT) by heating for 15 min at 65°C. Proteins were resolved by SDS-PAGE and analyzed by standard Western blotting techniques. Western blots were quantified with Image Gauge software (Fujifilm). To calibrate binding efficiency, the intensity of precipitated HA protein was divided by that of TAP protein in the same lane. To compare results from multiple experiments, data were normalized to results of wild-type control.
Analytical ultracentrifugation
Before analysis by analytical ultracentrifugation, nontagged Atg38 was purified by Mono Q chromatography and dialyzed against buffer containing 50 mM Tris-HCl, pH 7.5, and 150 mM NaCl. The dialysate was used as a reference solution. Sedimentation velocity experiments were performed on an analytical ultracentrifuge (Optima XL-I; Beckman Coulter) in a 4-hole An60Ti rotor or 8-hole An50Ti rotor (both from Beckman Coulter) at 20°C with standard double-sector centerpieces and quartz windows. Concentration profiles of samples were monitored by absorbance at 280 nm. The sedimentation velocity experiments were performed at A280 of 0.8 for Atg38 at a rotor speed of 45,000 rpm. Scans were recorded rapidly without intervals between successive scans. The sedimentation coefficient distribution function, c(S), was obtained using the SEDFIT program (Schuck, 2000; Schuck et al., 2002). The molecular mass distribution, c(M), was obtained by converting c(S) on the assumption that the frictional ratio f/f0 was common to all the molecular species as implemented in SEDFIT. The partial specific volume 0.7373 cm3/g for Atg38 was based on the amino acid composition of the proteins and together with the partial specific volume, solvent viscosity, and solvent density were calculated by the Sednterp program (Laue et al., 1992).
Mass spectrometry analysis
Cells expressing TAP-tagged proteins were resuspended in Lysis150 buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 10% glycerol, 1 mM DTT, and 1% Triton X-100) supplemented with 1 mM PMSF, Complete EDTA-free protease inhibitor cocktail (Roche), 5 mM EDTA, 50 mM NaF, 2 mM Na3VO4, 10 mM Na4P2O7, and 60 mM β-glycerophosphate, and were lysed with a glass bead homogenizer. TAP-tagged proteins were immunoprecipitated using rabbit IgG (Sigma-Aldrich) conjugated to M270 epoxy Dynabeads. Bound proteins were eluted by 0.1 M glycine at pH 2.5. The eluate was reduced with 10 mM DTT and alkylated with 55 mM iodoacetamide, followed by trypsin digestion. The peptides were desalted with reversed phaseStageTips (Rappsilber et al., 2007). NanoLC-MS/MS analysis was conducted using an TripleTOF 5600 System (AB Sciex) equipped with an Ultimate 3000 pump (Dionex), HTC-PAL autosampler (CTC Analytics), and a self-pulled analytical column needle packed with ReproSil-Pur C18-AQ materials (3 µm; Dr. Maisch GmbH). Peak lists were created using Mass Navigator v1.2 (Mitsui Knowledge Industry) based on the recorded fragmentation spectra. Peptides and proteins were identified by means of automated database searching using Mascot v2.3 (Matrix Science) against UniProt/Swiss-Prot (ver. 2011_06) with a precursor mass tolerance of 20 ppm, a fragment ion mass tolerance of 0.1 D, and strict trypsin specificity allowing for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification, and methionine oxidation. Peptides were initially rejected if the Mascot score was below the 95% confidence limit based on the “identity” score of each peptide and their length was less than seven amino acid residues. Further, we verified all MS/MS spectra by manual inspection. Protein abundance in the immunoprecipitated samples was measured based on exponentially modified protein abundance index (emPAI; Ishihama et al., 2005) using an open-source web tool, emPAI Calc (Shinoda et al., 2010).
Gel filtration
Cells were lysed with a glass bead homogenizer in Lysis150 buffer with 1 mM PMSF and Complete EDTA-free protease inhibitor cocktail (Roche). After the removal of cell debris by centrifugation at 20,000 g for 5 min, the sample was centrifuged at 100,000 g for 30 min. TAP-tagged Atg14 proteins were precipitated with magnetic beads coupled with rabbit IgG (Dynabeads; Invitrogen). The beads were washed three times with Lysis150 buffer. Bound proteins were eluted with TEV protease (Life Technologies) in TAP-A buffer without Triton X-100. The eluate was separated by size exclusion chromatography on a Superdex 200 column (GE Healthcare).
Statistical analyses
Significance of differences was determined using an unpaired two-tailed Student’s t test. *, P < 0.05; **, P < 0.01 were considered statistically significant.
Online supplemental material
Fig. S1 shows the identification of Atg38 as a subunit of complex I. Fig. S2 shows the requirement of the Atg38 MIT domain for the interaction between Atg14 and Atg38. Fig. S3 shows functionality of an artificial homodimer of Atg38. Table S1 lists yeast strains used in this study. Table S2 lists plasmids used in this study. Table S3 shows lists of proteins identified by LC-MS/MS in Figs. 1 C and 4 B. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201304123/DC1.
Supplementary Material
Acknowledgments
We thank E. Asai and Y. Nakamura for excellent technical support. p14 and MP1 were gifts from S. Nada.
This work was supported by Grants-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Abbreviations used in this paper:
- ALP
- alkaline phosphatase
- API
- aminopeptidase I
- ATG
- autophagy related
- CPY
- carboxypeptidase Y
- emPAI
- exponentially modified protein abundance index
- GBP
- GFP-binding protein
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- MIT
- microtubule interacting and trafficking
- PAS
- preautophagosomal structure
- PI3-kinase
- phosphatidylinositol 3-kinase
- VPS
- vacuolar protein sorting
References
- Baba M., Osumi M., Scott S.V., Klionsky D.J., Ohsumi Y. 1997. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol. 139:1687–1695 10.1083/jcb.139.7.1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backer J.M. 2008. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J. 410:1–17 10.1042/BJ20071427 [DOI] [PubMed] [Google Scholar]
- Behrends C., Sowa M.E., Gygi S.P., Harper J.W. 2010. Network organization of the human autophagy system. Nature. 466:68–76 10.1038/nature09204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K., Stevens T.H. 2005. Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta. 1744:438–454 10.1016/j.bbamcr.2005.04.004 [DOI] [PubMed] [Google Scholar]
- Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14:115–132 [DOI] [PubMed] [Google Scholar]
- Breitkreutz A., Choi H., Sharom J.R., Boucher L., Neduva V., Larsen B., Lin Z.Y., Breitkreutz B.J., Stark C., Liu G., et al. 2010. A global protein kinase and phosphatase interaction network in yeast. Science. 328:1043–1046 10.1126/science.1176495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli F.D., Proukakis C., Patel H., Cross H., Azam S., Patton M.A., Bork P., Crosby A.H. 2003. The identification of a conserved domain in both spartin and spastin, mutated in hereditary spastic paraplegia. Genomics. 81:437–441 10.1016/S0888-7543(03)00011-9 [DOI] [PubMed] [Google Scholar]
- Harding T.M., Hefner-Gravink A., Thumm M., Klionsky D.J. 1996. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 271:17621–17624 10.1074/jbc.271.30.17621 [DOI] [PubMed] [Google Scholar]
- Herman P.K., Stack J.H., DeModena J.A., Emr S.D. 1991. A novel protein kinase homolog essential for protein sorting to the yeast lysosome-like vacuole. Cell. 64:425–437 10.1016/0092-8674(91)90650-N [DOI] [PubMed] [Google Scholar]
- Ishihama A. 1981. Subunit of assembly of Escherichia coli RNA polymerase. Adv. Biophys. 14:1–35 [PubMed] [Google Scholar]
- Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. 2005. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics. 4:1265–1272 10.1074/mcp.M500061-MCP200 [DOI] [PubMed] [Google Scholar]
- Itakura E., Kishi C., Inoue K., Mizushima N. 2008. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell. 19:5360–5372 10.1091/mbc.E08-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C., Magiera M.M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., Knop M. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 21:947–962 10.1002/yea.1142 [DOI] [PubMed] [Google Scholar]
- Jones E.W., Zubenko G.S., Parker R.R. 1982. PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics. 102:665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kametaka S., Okano T., Ohsumi M., Ohsumi Y. 1998. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 273:22284–22291 10.1074/jbc.273.35.22284 [DOI] [PubMed] [Google Scholar]
- Kanki T., Wang K., Cao Y., Baba M., Klionsky D.J. 2009. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell. 17:98–109 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer C., Skalicky J.J., Morita E., De Domenico I., Ward D.M., Kaplan J., Sundquist W.I. 2008. Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell. 15:62–73 10.1016/j.devcel.2008.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Kihara A., Noda T., Ishihara N., Ohsumi Y. 2001. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 152:519–530 10.1083/jcb.152.3.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhofer A., Helma J., Schmidthals K., Frauer C., Cui S., Karcher A., Pellis M., Muyldermans S., Casas-Delucchi C.S., Cardoso M.C., et al. 2010. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 17:133–138 10.1038/nsmb.1727 [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Cueva R., Yaver D.S. 1992. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J. Cell Biol. 119:287–299 10.1083/jcb.119.2.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., Schiebel E. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 15(10B):963–972 [DOI] [PubMed] [Google Scholar]
- Kurzbauer R., Teis D., de Araujo M.E., Maurer-Stroh S., Eisenhaber F., Bourenkov G.P., Bartunik H.D., Hekman M., Rapp U.R., Huber L.A., Clausen T. 2004. Crystal structure of the p14/MP1 scaffolding complex: how a twin couple attaches mitogen-activated protein kinase signaling to late endosomes. Proc. Natl. Acad. Sci. USA. 101:10984–10989 10.1073/pnas.0403435101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue T.M., Shah B.D., Ridgeway T.M., Pelletier S.L. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. Analytical ultracentrifugation in biochemistry and polymer science. Harding S.E., Rowe A.J., Horton J.C., Royal Society of Chemistry, Cambridge, UK: 90–125 [Google Scholar]
- Matsunaga K., Saitoh T., Tabata K., Omori H., Satoh T., Kurotori N., Maejima I., Shirahama-Noda K., Ichimura T., Isobe T., et al. 2009. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11:385–396 10.1038/ncb1846 [DOI] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T., Ohsumi Y. 2011. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27:107–132 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- Motley A.M., Nuttall J.M., Hettema E.H. 2012. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 31:2852–2868 10.1038/emboj.2012.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K., Kimura M., Owens J.T., Meares C.F., Ishihama A. 1997. The two alpha subunits of Escherichia coli RNA polymerase are asymmetrically arranged and contact different halves of the DNA upstream element. Proc. Natl. Acad. Sci. USA. 94:1709–1714 10.1073/pnas.94.5.1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H., Suzuki K., Kamada Y., Ohsumi Y. 2009. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10:458–467 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- Noda N.N., Kobayashi T., Adachi W., Fujioka Y., Ohsumi Y., Inagaki F. 2012. Structure of the novel C-terminal domain of vacuolar protein sorting 30/autophagy-related protein 6 and its specific role in autophagy. J. Biol. Chem. 287:16256–16266 10.1074/jbc.M112.348250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T., Ohsumi Y. 1998. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273:3963–3966 10.1074/jbc.273.7.3963 [DOI] [PubMed] [Google Scholar]
- Noda T., Matsuura A., Wada Y., Ohsumi Y. 1995. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 210:126–132 10.1006/bbrc.1995.1636 [DOI] [PubMed] [Google Scholar]
- Obara K., Ohsumi Y. 2011. PtdIns 3-Kinase Orchestrates Autophagosome Formation in Yeast. J. Lipids. 2011:498768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara K., Sekito T., Ohsumi Y. 2006. Assortment of phosphatidylinositol 3-kinase complexes—Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol. Biol. Cell. 17:1527–1539 10.1091/mbc.E05-09-0841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara K., Sekito T., Niimi K., Ohsumi Y. 2008. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem. 283:23972–23980 10.1074/jbc.M803180200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obita T., Saksena S., Ghazi-Tabatabai S., Gill D.J., Perisic O., Emr S.D., Williams R.L. 2007. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 449:735–739 10.1038/nature06171 [DOI] [PubMed] [Google Scholar]
- Phillips S.A., Barr V.A., Haft D.H., Taylor S.I., Haft C.R. 2001. Identification and characterization of SNX15, a novel sorting nexin involved in protein trafficking. J. Biol. Chem. 276:5074–5084 10.1074/jbc.M004671200 [DOI] [PubMed] [Google Scholar]
- Rappsilber J., Mann M., Ishihama Y. 2007. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2:1896–1906 10.1038/nprot.2007.261 [DOI] [PubMed] [Google Scholar]
- Robinson J.S., Klionsky D.J., Banta L.M., Emr S.D. 1988. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 8:4936–4948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbauer U., Zolghadr K., Tillib S., Nowak D., Schermelleh L., Gahl A., Backmann N., Conrath K., Muyldermans S., Cardoso M.C., Leonhardt H. 2006. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods. 3:887–889 10.1038/nmeth953 [DOI] [PubMed] [Google Scholar]
- Schu P.V., Takegawa K., Fry M.J., Stack J.H., Waterfield M.D., Emr S.D. 1993. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 260:88–91 10.1126/science.8385367 [DOI] [PubMed] [Google Scholar]
- Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78:1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P., Perugini M.A., Gonzales N.R., Howlett G.J., Schubert D. 2002. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82:1096–1111 10.1016/S0006-3495(02)75469-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S.V., Baba M., Ohsumi Y., Klionsky D.J. 1997. Aminopeptidase I is targeted to the vacuole by a nonclassical vesicular mechanism. J. Cell Biol. 138:37–44 10.1083/jcb.138.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman M.N., Marcusson E.G., Cereghino J.L., Emr S.D. 1997. Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J. Cell Biol. 137:79–92 10.1083/jcb.137.1.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda K., Tomita M., Ishihama Y. 2010. emPAI Calc—for the estimation of protein abundance from large-scale identification data by liquid chromatography-tandem mass spectrometry. Bioinformatics. 26:576–577 10.1093/bioinformatics/btp700 [DOI] [PubMed] [Google Scholar]
- Söding J., Biegert A., Lupas A.N. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33(Web Server issue):W244-8 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuchell-Brereton M.D., Skalicky J.J., Kieffer C., Karren M.A., Ghaffarian S., Sundquist W.I. 2007. ESCRT-III recognition by VPS4 ATPases. Nature. 449:740–744 10.1038/nature06172 [DOI] [PubMed] [Google Scholar]
- Sun Q., Fan W., Chen K., Ding X., Chen S., Zhong Q. 2008. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 105:19211–19216 10.1073/pnas.0810452105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K., Kirisako T., Kamada Y., Mizushima N., Noda T., Ohsumi Y. 2001. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20:5971–5981 10.1093/emboj/20.21.5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K., Kubota Y., Sekito T., Ohsumi Y. 2007. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 12:209–218 10.1111/j.1365-2443.2007.01050.x [DOI] [PubMed] [Google Scholar]
- Tsukada M., Ohsumi Y. 1993. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333:169–174 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- Yasumo H., Masuda N., Furusawa T., Tsukamoto T., Sadano H., Osumi T. 2000. Nuclear receptor binding factor-2 (NRBF-2), a possible gene activator protein interacting with nuclear hormone receptors. Biochim. Biophys. Acta. 1490:189–197 10.1016/S0167-4781(99)00244-4 [DOI] [PubMed] [Google Scholar]
- Zhong Y., Wang Q.J., Li X., Yan Y., Backer J.M., Chait B.T., Heintz N., Yue Z. 2009. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11:468–476 10.1038/ncb1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.