

Abstract
We show, for the first time, that the tumor suppressor PTEN can have tumor promoting properties. We demonstrate that PTEN acquires these unexpected properties by enhancing gain-of-function mutant p53 (mut-p53) protein levels. We find that PTEN restoration to cells harboring mut-p53 leads to induction of G1/S cell cycle progression and cell proliferation and to inhibition of cell death. Conversely, PTEN inhibition in cells expressing wild-type PTEN and mut-p53 leads to inhibition of cell proliferation and inhibition of in vivo tumor growth. We demonstrate the dependency of the tumor promoting effects of PTEN on mut-p53 by showing that knock-down of mut-p53 expression inhibits or reverses the tumor promoting effects of PTEN. Mechanistically, we show that PTEN expression enhances mut-p53 protein levels via inhibition of mut-p53 degradation by Mdm2 and possibly also via direct protein binding. These findings describe a novel function of PTEN and have important implications for experimental and therapeutic strategies that aim at manipulating PTEN or p53 in human tumors. They suggest that the mutational status of PTEN and p53 should be considered in order to achieve favorable therapeutic outcomes. The findings also provide an explanation for the low frequency of simultaneous mutations of PTEN and p53 in human cancer.
Keywords: PTEN, p53, gain-of-function mutations, tumor suppressor, oncogene
INTRODUCTION
PTEN and p53 are the two most frequently mutated tumor suppressors in human cancer, including gliomas (1-3). A majority of malignant gliomas harbor PTEN or p53 mutations, which are considered critical events in the development and growth of these tumors (4-7). Until recently, PTEN and p53 were regarded as autonomous anti-cancer units that functioned independently of each other. However, recent evidence points to a multilevel and complex cooperation between these tumor suppressors (2). PTEN and wild-type p53 (wt-p53) can enhance each other’s tumor suppressive functions. Wt-p53 enhances PTEN gene transcription by binding to and activating the PTEN promoter (8). PTEN can protect wt-p53 from degradation through its ability to antagonize PI3-Kinase. This leads to inactivation of Mdm2, which in turn leads to inhibition of wt-p53 degradation (9, 10). PTEN also directly binds to wt-p53 leading to protein stabilization and induction of wt-p53 transcriptional activity (11). PTEN can also inhibit Mdm2 transcription by binding to the inhibitory P1 binding site (12). Recently, PTEN was found to autoregulate its expression by stabilizing wt-p53 in a phosphatase-independent manner and wt-p53 down-regulated PTEN protein stability through caspase-mediated degradation in cells with proteasome dysfunction (13).
In the present study we show, for the first time, that PTEN can have oncogenic properties by enhancing the stability of gain-of-function p53 mutants (mut-p53). Gain-of-function p53 mutants possess tumor promoting functions such as the transcriptional activation of genes that promote various malignancy parameters (14-17). We found that PTEN restoration to glioblastoma cells harboring gain-of-function p53 mutations leads to induction of cell proliferation and inhibition of cell death possibly via inhibition of mut-p53 degradation by Mdm2 and direct stabilization of mut-p53 protein. Conversely, inhibition of endogenous PTEN in glioma cells expressing mut-p53 leads to inhibition of cell proliferation and inhibition of in vivo tumor growth. This discovery describes a novel unexpected tumor promoting function of PTEN and has important implications for therapeutic strategies that aim at manipulating PTEN or p53 expression or function in human tumors. The findings also provide a potential explanation for the low frequency of simultaneous mutations of PTEN and p53 in human cancer.
MATERIALS AND METHODS
Cell culture and reagents
U373 glioma cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) (1gm/L glucose with L-glutamine) supplemented with HEPES buffer and 10% Fetal Bovine Serum (FBS). SNB19 glioma cells were grown in DMEM F12 supplemented with 10% FBS. U87 glioma cells were grown in Minimum Essential Medium Eagle medium (MEM) supplemented with sodium pyruvate, sodium bicarbonate and 10% FBS. A172 glioma cells were grown in DMEM (4.5gm/L glucose with L-glutamine) and 10% FBS. U1242 glioma cells were grown in alpha-MEM with 10% FBS. The PI3K inhibitors wortmannin and LY294002, (Calbiochem, San Diego, CA) and were added 1 hr before the cells were transfected with PTEN. Camptothecin (CPT) and trypan blue were purchased from Sigma, (St. Louis, MO). and Propidium idodide (PI), Annexin V-PE, Annexin V-FITC and BrdU from BD Pharmingen (San Diego, CA).
Vectors and transfections
Plasmids encoding wt-p53, R273H mut-p53 and Mdm2 were kindly provided by Dr. Bert Vogelstein, Johns Hopkins University. PTEN, lipid phosphatase-dead (G129E), and lipid- and protein phosphatase-dead (C124A) PTEN mutants were a kind gift from Dr. Kenneth Yamada, (NIH) (18). Adenoviruses encoding wild-type PTEN (Ad-PTEN), G129E (Ad-G129E), C124A (Ad-C124A), and control (Ad-control) were constructed by us according to Vogelstein and co-workers (19). Adenoviruses were amplified, purified and titered by the University of Pittsburgh Vector Core Facility. Viral titers were ~1010 pfu. Plasmids were transfected with Fugene-6 transfection reagent (Roche, Indianapolis, IN). Adenovirus infections were conducted at an MOI = 10.
siRNA/shRNA experiments
PTEN siRNA was purchased from Santa Cruz Biothechnology (Santa Cruz, CA). P53 siRNAs were designed following the guidelines of Ambion, Inc. and synthesized using the Silencer siRNA starter kit (Ambion Inc., Austin, TX). Random scrambled siRNAs were used as control. pSilencer plasmid vectors (Ambion Inc., Austin, TX) were constructed for stable expression of mut-p53 shRNAs, PTEN shRNA or control shRNA. ShRNAs were designed, constructed and inserted in pSilencer plasmids according to the guidelines of Ambion Inc. Two different siRNAs and shRNAs were used in all experiments. The targeted sequences for p53 were: 5′-AAACATTTTCAGACCTATGGA-3′ and 5′-AAACTACTTCCTGAAAACAAC-3′. The targeted sequences for PTEN were: 5′-AAAGAGATCGTTAGCAGAAAC-3′ and 5′-AAACATTATTGCTATGGGATT-3′. To exclude non-specific anti-viral effects, lysates of cells transfected with siRNAs/shRNAs were immunoblotted for the interferon-induced 2′-5′-oligoadenylate synthetase 3, a general marker of anti-viral responses (20). No significant antiviral response to siRNA was detected. Naked siRNAs (2 nM) were transfected using siPORT transfection reagent (Ambion Inc.). pSilencer plasmids expressing mut-p53, PTEN or control shRNAs (5 μg) were transfected with Fugene6 (Roche, Indianapolis, IN) and stable knock-down clones were selected for puromycin resistance and screened for mut-p53 or PTEN protein levels with immunoblotting as previously described (21).
Propidium Iodide Flow Cytometry
The cell cycle status was analyzed using propidium iodide (PI) flow cytometry as previously described (22). To assess the effects of PTEN on the cell cycle, the cells were infected with Ad-PTEN, Ad-G129E, Ad-C124A or Ad-control for 48 hrs. Alternatively, the cells were transfected with plasmids expressing PTEN, G129E, C124A or control together with a plasmid expressing GFP at a DNA ratio of 3:1. To analyze the dependency of the cell cycle effects on mut-p53 expression, the cells were transfected with siRNA 6 hrs prior to infection with adenoviruses as described above. All adenovirus-infected cells but only GFP-positive (transfected) plasmid- transfected cells were analyzed on a FACscan (Becton-Dickinson, Fullerton, CA).
BrdU incorporation
BrdU incorporation was used to determine if PTEN restoration induces an increase in active S phase in mut-p53 cells. U373 and U87 (control) cells were infected with Ad-PTEN or Ad-control for 36 hrs prior to treatment with 10 μM BrdU for 2 hrs. The treated cells were collected and DNA synthesis was assessed by BrdU incorporation, using a PE-anti-BrdU antibody kit following the manufacturer’s instructions (BD Biosciences) and 7AAD staining for DNA content. The cell cycle was analyzed on a FACscan.
Trypan blue and propidium iodide staining
The effects of PTEN on cell death were analyzed by PI and Trypan blue staining. Cells were infected with Ad-PTEN or Ad-control for 24 hrs as described above and subsequently treated with campthotecin (200 μM) or γ-Radiation (40 Gy) for 48 hrs. One of the latter two modalities was selected for each individual cell line in order to induce moderate cell death. The cells were then stained with trypan blue. The number of dead cells stained with trypan blue was determined by counting with a hemocytometer. To obtain morphological evidence of cell death, the cells were stained with a solution of propidium iodide (PI) in PBS (10 μg/ml). Cell suspensions were mounted on slide glasses and red (dead) cells identified by fluorescence microscopy.
Annexin-V-PE and 7AAD flow cytometry
The dependency of PTEN–mediated cell death/survival on mut-p53 expression was analyzed using Annexin-V-PE and 7AAD flow cytometry. Cells (1.106) were transfected with p53 siRNA for 6 hrs prior to transfection with plasmids expressing PTEN or control together with a plasmid expressing GFP at a DNA ratio of 3:1 for 24 hrs. Some cells were also subsequently treated with CPT (300 μM) for 48 hrs prior to evaluation of cell death. The fraction of the GFP-positive (transfected) cells was determined by flow cytometry and analyzed for cell death (Annexin V-PE/7AAD-positive cells) on a FACscan.
Growth curves
For cell proliferation experiments, 30,000 cells/well were seeded in medium containing 1% FBS. To determine the effects of PTEN on cell proliferation, the cells were infected with Ad-PTEN or Ad-control prior to cell counting. For assessing the dependency of the PTEN cell proliferative effects on mut-p53 expression, the cells were transfected with p53 siRNA 6 hrs before being infected with Ad-PTEN or Ad-control and subsequently counted. The cells were harvested by trypsinization every day for 5 days and counted with a hemocytometer and growth curves were established.
Immunoblotting and immunoprecipitation
Immunoblotting was performed as previously described using antibodies specific for PTEN, wt-p53, mut-p53, Cdk2, cyclin E, E2F-1, β-actin, α-tubulin, nucleolin (Santa Cruz Biotechnologies, Santa Cruz, CA), p27 (BD Biosciences, San Jose, CA) and Mdm2 (Calbiochem, San Diego, CA) (22). Immunoprecipitations of PTEN and p53 were performed as previously described (21). Cells were infected with plasmids expressing either, PTEN, wt-p53, R273H mut-p53 or GFP (control). After 24 hrs, lysates were immunoprecipitated with PTEN- or p53-conjugated agarose beads (Santa Cruz Biotechnologies, Santa Cruz, CA ). The beads were collected by centrifugation, washed, heated to 100°C for 5 min in Laemmli buffer and subjected to immunoblotting for p53 or PTEN as described above.
Cycloheximide chase experiments
Cycloheximide chase experiments were used to assess mut-p53 degradation in response to PTEN restoration to mut-p53 cells. U373 cells were infected with Ad-PTEN or Ad-control for 24 hrs. The cells were then treated with 100 μg/ml cycloheximide. Cell lysates were collected at various time points (0-7 hrs) after cycloheximide treatment and analyzed for mut-p53 protein levels by immunoblotting as described above. Band intensities were quantified by densitometry and protein half-life was calculated.
Northern analysis
Northern analysis was performed to determine the effects of PTEN on mut-p53 mRNA. Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA). Northern analysis was performed as previously reported (23). Blots were hybridized with a full-length cDNA probe for R273H mut-p53 (a kind gift from Dr. Bert Vogelstein, Johns Hopkins University) and labeled with [32P]-dCTP using a random priming kit (Boehringer-Mannheim, Indianapolis, IN). The blots were stripped and then rehybridized with cDNA specific for GAPDH. The signal was visualized by blot exposure to film.
In vivo xenograft experiments
U1242 cells expressing PTEN and mut-p53 were stably transfected with p-Silencer vectors encoding PTEN shRNA or scrambled control shRNA. Clones were selected in puromycin and assessed for PTEN and mut-p53 protein expression levels with immunoblotting. Two control (U1242-control) and two PTEN knock-down clones (U1242-PTEN-KD) were selected for in vivo implantation. The cells (3×105) were stereotactically implanted in the striatum of immunodeficient mice (n=10). The animals were euthanized 5 weeks after tumor implantation, when three of the control animals showed signs of tumor-associated morbidity. The brains were removed and tumor maximal cross-section areas were measured using computer-assisted image analysis as previously described (22).
Statistics
All experiments were performed at least in triplicates. Numerical data were expressed as mean ± standard deviation. Two group comparisons were analyzed by two-sided Student’s t test. Multiple group comparisons were analyzed with Bonferroni/Dunn multiple comparisons tests. P values were calculated and p<0.05 was considered significant.
RESULTS
PTEN induces cell cycle progression and cell proliferation in U373 and SNB19 but inhibits cell cycle progression and cell proliferation in U87 and A172 glioblastoma cell lines
While performing PTEN gene restoration experiments to PTEN-null glioblastoma cells with the aim of inhibiting various malignancy parameters, we were surprised to find that PTEN expression consistently led to cell cycle progression in two out of four cell lines (U373 and SNB19) as measured by propidium iodide flow cytometry. PTEN restoration led to cell cycle arrest in the two other cell lines (U87 and A172) as expected from a tumor suppressor (Fig. 1A). PTEN expression reduced the G1/G0 fraction from 68.0 ± 2.4% to 53.8 ± 1.1% (n=8 ; p<0.05) in U373 and from 50.7% ± 1.1% to 42.6 ± 1.3% (n=5 ; p<0.05) in SNB19 cells. PTEN expression increased the G0/G1 fraction from 64.7 ± 1.8% to 86.7 ± 1.2% (n=8 ; p<0.05) and from 82.2 ± 0.3% to 85.0 ± 0.2% (n=4) in U87 and A172 cells, respectively. Using flow cytometry-based BrdU incorporation experiments, we confirmed that PTEN restoration induces an increase in active S phase in U373 cells and a decrease in active S phase in U87 cells (Fig. 1B). The cell cycle experiments were repeated multiple times using adenoviruses as well as plasmids to express PTEN cDNA of two different origins and consistently yielded the same results. PTEN expression in the cells was confirmed by immunoblotting and levels were comparable to those found in normal human astrocytes (not shown). PTEN lipid phosphatase function was verified by testing the ability of PTEN to inhibit the phoshphorylation of Akt in all four cell lines (not shown). Expression of PTEN lipid phosphatase mutant (G129E) and phosphatase dead mutant (C124A) did not change the cell cycle status in U373 or SNB19, indicating lipid phosphatase involvement. However, the PTEN effect on cell cycle progression could not be mimicked by treatment of the cells with the PI3K inhibitors LY294002 and wortmannin, which did not affect the cell cycle status of U373 and SNB19 (not shown). This indicates that the effects of PTEN on the cell cycle also require a component that is independent of PTEN lipid phosphatase function. The cell cycle changes described above were consistent with PTEN-induced changes in cell proliferation. PTEN significantly induced cell proliferation in U373 and SNB19 cells and inhibited proliferation in U87 and A172 cells (Fig. 1C). We also examined the effects of PTEN restoration on cell the cycle regulatory proteins p27, cdk2, cyclin E and E2F1 in U373 and U87 cells. Consistent with its effects on the cell cycle, PTEN restoration inhibited p27 and induced cdk2, cyclin E and E2F1 in U373, but induced p27 and inhibited cdk2, cyclin E and E2F1 in U87 (Fig. 1D). Therefore, PTEN restoration oppositely affected cell cycle regulatory proteins in U373 and U87 cells consistent with PTEN’s opposite effects on cell cycle and cell proliferation in these cells.
Figure 1. PTEN induces cell cycle progression and cell proliferation in U373 and SNB19 but inhibits cell cycle progression and cell proliferation in U87 and A172 glioma cell lines.

PTEN was transfected into four PTEN-null glioma cell lines via adenovirus infections and the cells were subsequently tested for G1/S cell cycle transition by propidium iodide flow cytometry, for active S phase changes by BrdU incorporation and for cell proliferation by cell counting. The effects of PTEN on cell cycle regulatory proteins were also assessed by immunoblotting. The results show that: A) PTEN induces cell cycle progression in two cell lines (U373 and SNB19) and inhibits cell cycle progression in the two other cell lines (U87 and A172), B) PTEN induces active S phase increase in U373 and decrease in U87. C) Similarly, PTEN induces cell proliferation in U373 and SNB19 cells and inhibits cell proliferation in U87 and A172 cells. D) PTEN has opposite effects on the levels of cell cycle regulatory proteins in U373 and U87 cells that are consistent with its opposite effects on cell cycle and cell proliferation in these cell lines. (cAdv=control, G129E=lipid phosphatase dead PTEN mutant, C124A=phosphatase dead PTEN mutant), * = p<0.05 relative to control.
PTEN inhibits cell death in U373 and SNB19 but induces cell death in U87 and A172 glioblastoma cell lines
Similar to the results obtained for cell proliferation, PTEN expression led to unexpected effects on tumor cell death and apoptosis in two out of four glioblastoma cell lines. PTEN restoration led to inhibition of radiation or chemotherapy (campthotecin) induced cell death and apoptosis in U373 and SNB19 cells but to the induction of these same parameters in U87 and A172 cells. PTEN reduced the dead cell fraction from 27.5 ± 3.4% to 8.7 ± 3.4% (n=3 ; p<0.05) in U373 cells and from 56.3 ± 1% to 23.3 ± 4% (n=3 ; p<0.05) in SNB19 cells and increased the dead cell fraction from 35.0 ± 0.3% to 43.5 ± 1.3% (n=3 ; p<0.05) in U87 cells and from 42.2 ± 3% to 53.3 ± 7% (n=3; p<0.05) in A172 cells as assessed by trypan blue staining and confirmed by propidium iodide staining (Fig. 2A,B).
Figure 2. PTEN inhibits cell death in U373 and SNB19 but induces cell death in U87 and A172 glioma cell lines.

PTEN was transfected into four PTEN-null glioma cell lines via adenovirus infections and the cells were subsequently tested for cell death by trypan blue staining (A) and propidium iodide staining (B). The results show that PTEN inhibits cell death in U373 and SNB19 cells and induces cell death in U87 and A172 cells (A,B). (Rad = Radiation, CPT = campthotecin), * = p<0.05 relative to control.
Altogether, the above results show that PTEN consistently exhibits unexpected tumor promoting characteristics in two out of four glioblastoma cell lines.
PTEN increases the levels of mut-p53 via inhibition of Mdm2-mediated degradation and possibly also via direct binding
We examined the genetic background of the four cell lines that were used in the above described experiments and noticed that U373 and SNB19 were mutated on both alleles for p53 at amino acid 273 (R273H) while U87 and A172 were wild-type for p53. These observations were based on published literature and confirmed by us through Taqman Allelic Discrimination (Supplementary data) (24-27). We hypothesized that, similar to what was previously reported for wild-type p53, PTEN might stabilize gain-of-function p53 mutants, to which the R273H mutation belongs, and thereby enhance their tumor promoting function (28-30).
To test this hypothesis, we first assessed the effects of PTEN restoration on mutant and wild-type p53 protein in U373, SNB19 and U87 cells. We found that PTEN restoration leads to an increase in wt-p53 protein levels in U87 cells and to an increase in mut-p53 protein levels in U373 and SNB19 (84% increase per densitometry on film) as assessed by immunoblotting (Fig. 3A). To determine if the PTEN-induced increase in mut-p53 protein is due to inhibition of protein degradation, we performed a cycloheximide chase of mut-p53 protein in U373 cells and calculated PTEN protein half-life. We found that PTEN restoration to U373 cells prolongs mut-p53 half-life by ~ 40% (from 3.7 hrs to 5.2 hrs) (Fig. 3B). To determine if the PTEN-induced increase in mut-p53 protein also has a transcriptional component, we analyzed the effects of PTEN restoration on mut-p53 mRNA in U373 cells by Northern blotting. We found that PTEN expression did not significantly change mut-p53 mRNA in U373 cells (Fig. 3B). Therefore, PTEN expression post-transcriptionally induces mut-p53 protein levels by inhibiting its degradation.
Figure 3. PTEN increases the levels of mut-p53 via inhibition of Mdm2-mediated degradation and possibly also via direct binding.

A) PTEN was transfected into PTEN-null glioma cells and protein levels of wild-type p53 (U87) and mutant p53 (U373 and SNB19) cells were assessed by immunoblotting. The results show that PTEN expression induces wt-p53 and mut-p53 protein levels. B) (Upper panel) PTEN was transfected into PTEN-null U373 cells and a cycloheximide chase was performed. The results show that PTEN inhibits mut-p53 protein degradation. B) (Lower panel) PTEN was transfected into PTEN-null U373 cells and mut-p53 mRNA levels were assessed by Northern blotting. The results show that PTEN does not significantly change mut-p53 mRNA levels. C) PTEN was transfected into PTEN null/mut-p53 U373 and SNB19 cells. The cells were subjected to cytoplasmic and nuclear fractionation and subsequently analyzed for Mdm2 protein levels by immunoblotting. The results show that PTEN expression inhibits Mdm2 protein levels (left panel). Mdm2 was transfected into mut-p53 U373 and SNB19 cells and the cells were assessed for mut-p53 levels by immunoblotting. The results show that Mdm2 inhibits mutant p53 levels (right panel). Together, the above shows that PTEN can induce mut-p53 levels by inhibiting its degradation by Mdm2. D) PTEN was transfected into U373 cells. The cells were subsequently immunoprecipitated (IP) for PTEN and immunoblotted (IB) for mut-p53 or immunoprecipitated for mut-p53 and immunoblotted for PTEN. The results show that PTEN co-immunoprecipitates with mut-p53.
We then asked if PTEN-induced changes of mut-p53 levels are mediated by changes in the levels and cellular localization of Mdm2 and/or by direct binding and stabilization of the mutants by PTEN protein. To assess the involvement of Mdm2 in PTEN-induced mut-p53 protein levels, we first studied the effect of PTEN expression on the cytoplasmic and nuclear levels of Mdm2. PTEN restoration to mut-p53 U373 and SNB19 cells led to a reduction of the levels of nuclear Mdm2 as assessed by immunoblotting after cell fractionation (Fig. 3C, left panel). This shows that PTEN regulates the levels and distribution of Mdm2 in these cells. We then studied the effects of Mdm2 on mut-p53 levels by transfecting U373 and SNB19 cells with an Mdm2 cDNA expression vector and analyzing mut-p53 levels by immunoblotting. We found that Mdm2 expression leads to the decrease of nuclear mut-p53 levels in U373 and SNB19 cells (Fig. 3C, right panel). This demonstrates that mut-p53 is regulated by Mdm2 in these cells. Together, the results described above show that PTEN expression could increase the levels of mut-p53 via inactivation of Mdm2. To determine if PTEN can also regulate mut-p53 levels by direct binding, we transfected U373 cells with PTEN prior to immunoprecipitation of either PTEN or mut-p53. Immunoprecipitates were immunoblotted for mut-p53 (PTEN) or PTEN (mut-p53). We found that PTEN protein binds to mut-p53 protein in the cells (Fig. 3D). Therefore, similar to what has been recently shown for wt-p53, PTEN protein might also increase the levels of mut-p53 via protein binding and stabilization.
PTEN-induced cell cycle progression, cell proliferation and cell survival are dependent on mut-p53 expression
We then asked if PTEN-induced cell cycle progression, cell proliferation and cell survival are dependent on mut-p53 expression. To answer this question, we inhibited mut-p53 expression in U373 with p53 siRNA and studied the effects of PTEN restoration on cell cycle, cell proliferation and apoptosis in this setting. R273H mut-p53 was inhibited by either transient expression of siRNA or stable expression of shRNA encoded in pSilencer plasmids. P53 siRNA/shRNA expression in U373 cells led to substantial inhibition of mut-p53 protein as assessed by immunoblotting (Fig. 4A). U373 cells transfected with siRNA/shRNA were then infected with Ad-PTEN or Ad-control. The cell cycle status and apoptosis were determined by flow cytometry as described above and cell proliferation was determined by cell counting.
Figure 4. PTEN-induced cell cycle progression, cell proliferation and cytoprotection are dependent on gain-of-function mut-p53 expression.

R273H mut-p53 expression was inhibited in U373 cells by transfection with siRNA or shRNA prior to restoration of PTEN. Control cells were transfected with scrambled siRNA/shRNA (scRNA). Expression inhibition was confirmed by immunoblotting (upper right panel). The effects of PTEN on cell cycle progression (A) cell proliferation (B) and cytoprotection (C) were subsequently assessed in this setting using propidium iodide flow cytometry, cell counting or annexin V flow cytometry, respectively. The results show that inhibition of mut-p53 expression leads to partial inhibition of PTEN-mediated induction of cell cycle progression, complete inhibition of PTEN-induced cell proliferation and complete reversal of PTEN-induced cytoprotection. * = p<0.05 relative to control.
Inhibition of basal levels of R273H mut-p53 in U373 cells led to cell cycle arrest with the G0/G1 fraction increasing from 57.4 ± 8.8% in control cells to 82.4 ± 0.1% (n=5 ; p<0.05) in mut-p53-inhibited cells (Fig. 4A). This demonstrates that R273H is a gain-of-function p53 mutant that induces cell cycle progression. PTEN expression reduced the G0/G1 fraction by 50.5% in control cells (p<0.05) and by a statistically insignificant (p>0.05) 32% in mut-p53-inhibited cells (Fig. 4A). PTEN induction of cell cycle progression was therefore inhibited by 45% in the setting of partially inhibited mut-p53 expression.
Similarly, inhibition of mut-p53 expression in U373 cells led to complete inhibition of PTEN-induced cell proliferation. Cells transfected with control-siRNA and Ad-control grew ~3-fold while cells transfected with control-siRNA and Ad-PTEN grew ~ 5-fold in 5 days (n=6 ; p<0.01). Cells transfected with p53 siRNA and Ad-control grew only ~2-fold, confirming that mut-p53 in U373 is gain-of-function. Cells transfected with mut-p53-siRNA and Ad-PTEN grew less than 2-fold after 5 days (n=6, p>0.05 relative to control) (Fig. 4B). Therefore, inhibition of mut-p53 expression completely abrogated the cell proliferative effect of PTEN.
Inhibition of mut-p53 expression also led to inhibition of PTEN-induced cytoprotection as measured by Annexin V flow cytometry. In U373 cells transfected with control siRNA, PTEN reduced cell death from 5.8 ± 0.1% to 2.9 ± 0.2% (n=3, p< 0.01). Inhibition of mut-p53 expression with siRNA led to induction of cell death from 5.8 ± 0.1% to 11.4 ± 2.1% (p<0.01) (Fig. 4C). This further demonstrates that R273H is a gain-of-function p53 mutant which elicits cytoprotective effects. In the setting of siRNA-inhibited mut-p53, PTEN increased cell death from 11.4 ± 2.1 to 22.8 ± 0.1%. (p<0.01) (Fig. 4C). Therefore, inhibition of mut-p53 led to a complete reversal of the effects of PTEN on cell death. These results demonstrate that the PTEN-induced cytoprotective effects are dependent on gain-of-function mut-p53 expression. Altogether, the above experiments consistently show that the tumor promoting properties of PTEN are dependent on mut-p53 expression.
Inhibition of endogenous PTEN in wt-PTEN/mut-p53 U1242 glioma cells leads to inhibition of cell proliferation
To ascertain the tumor promoting effects of endogenous PTEN in the setting of gain-of-function mut-p53, we inhibited endogenous PTEN expression in cells that express PTEN and gain-of-function mut-p53 and studied the effects of this inhibition on tumor cell proliferation. We used U1242 glioblastoma cell lines that express PTEN and have gain-of-function mutations of p53 at codon 175 (R175H) (27, 30, 31). We used siRNA to inhibit PTEN expression in these cells and analyzed them for proliferation by cell counting. Consistent with the results described in the previous sections, inhibition of PTEN expression in these mut-p53 cells led to inhibition of cell proliferation. While control siRNA transfected cells U1242 glioma cells grew ~30-fold in 5 days, PTEN-siRNA transfected cells grew only ~10 fold (Fig. 5A). This provides evidence that endogenous PTEN has tumor promoting effects in the setting of gain-of-function p53 mutations.
Figure 5. Inhibition of endogenous PTEN expression in wt-PTEN/mut-p53 glioma cells leads to inhibition of cell proliferation and in vivo tumor growth.

A) Endogenous PTEN was inhibited in wt-PTEN/mut-p53 U1242 cells by siRNA transfection and the cells were assessed for proliferation by cell counting. The results show that inhibition of PTEN expression in these mut-p53 cells leads to inhibition of cell proliferation. B) PTEN was inhibited in U1242 cells by stable transfection with p-Silencer plasmids encoding PTEN-shRNA. Control U1242 cells were transfected with p-Silencer plasmids encoding scrambled control-shRNA. Clones were selected for puromycin resistance and assessed for PTEN and mut-p53 protein levels by immunoblotting (right panel). Two PTEN-knock-down and two control clones were selected and implanted intracranially (3×105 cells) in immunodeficient mice (n=10 for each clone). The mice were sacrificed 5 weeks post-implantation and the brains were cryosectioned and H&E stained. Tumor maximal crossectional areas were measured with computer-assisted image analysis. The results show that endogenous PTEN inhibition in mut-p53 U1242 cells leads to inhibition of tumor growth (left panel: representative tumors ; middle panel: quantification of tumor sizes). * = p<0.05 relative to control.
Inhibition of PTEN in wt-PTEN/mut-p53 U1242 cells leads to inhibition of in vivo tumor growth
To determine if the tumor promoting characteristics of PTEN lead to in vivo tumor growth, we inhibited PTEN expression in wt-PTEN/mut-p53 U1242 cells and assessed the effect of this inhibition on in vivo tumor growth. These cells were also chosen because they are very tumorigenic in vivo as opposed to U373 which do not form tumors in animals. We generated PTEN knock-down U1242 clones by stable transfection with plasmids encoding PTEN shRNA and control clones by transfection with plasmids encoding scrambled shRNA. We verified PTEN knock-down as well as the ensuing mut-p53 inhibition in the clones by immunoblotting (Fig. 5B, right panel). We implanted two PTEN knock-down clones and two control clones in the brains of immunodeficient mice, euthanized the mice after 5 weeks and measured tumor sizes. We found that inhibition of PTEN expression in U1242 cells that harbor a gain-of-function mutation of p53 at codon 175 leads to a significant inhibition of in vivo xenograft growth. While control clones had a crossectional area of 155.6 × 103 ± 6.9 × 103 μm2, PTEN knock-down clones had a crossectional area of 43.9 × 103 ± 11.1 × 103 μm2 (n=10 ; p<0.01) (Fig. 5B). Therefore, inhibition of PTEN expression in glioblastoma cells harboring gain-of-function mut-p53 leads to inhibition of in vivo tumor growth, indicating that PTEN has tumor promoting properties in vivo in the setting of mut-p53.
DISCUSSION
We demonstrated, for the first time, that PTEN can have tumor promoting properties in cells that harbor gain-of-function p53 mutations. We found that PTEN induces cell cycle progression, cell proliferation, cell survival and in vivo tumor growth in mut-p53 glioma cells but exerts the opposite effects in wt-p53 cells. PTEN increased the levels of mut-p53 protein by inhibiting its degradation possibly via inhibition of PI3K/Mdm2 and physical binding. We showed that the unexpected effects of PTEN are dependent on mut-p53 expression.
We first found that PTEN induces cell proliferation and cell survival of glioma cells that have gain-of-function p53 mutations. Since these findings were unexpected and seemingly contrary to established knowledge, we carefully and thoroughly excluded potential procedural and other non-specific errors. The following steps were taken: 1) To exclude adenovirus-induced non-specific effects, all experiments showing that PTEN has tumor promoting effects were repeated using plasmid-based transfections coupled with GFP selection. This approach yielded similar results to adenovirus-based experiments. 2) To ensure the integrity and functionality of expressed PTEN protein, the ability of PTEN to dephosphorylate Akt was demonstrated. Additionally, PTEN cDNA of two different independent sources were used and yielded the same results. 3) Complementary techniques were used to assess cell proliferation (PI flow cytometry, BrdU incorporation and growth curves) and cell death (trypan blue staining, PI staining and Annexin V flow cytometry). 4) To avoid unphysiologically high expression levels of restored PTEN protein, adenoviral titers used were adjusted to yield PTEN expression levels comparable to the ones in primary normal human astrocytes. Additionally, proliferation assays and in vivo tumor growth experiments were conducted after inhibition of endogenous PTEN in cells with mut-p53 and yielded results consistent with the ones obtained in PTEN restoration experiments. 5) All experiments were repeated multiple times by at least two independent investigators and consistently yielded comparable results. Also, the fact that PTEN consistently behaved as a tumor suppressor in U87 and A172 wt-p53 cells and as a tumor promoter in U373, SNB19 and U1242 mut-p53 cells under the same experimental conditions is an additional indication that the findings are real pathophysiological events.
We then showed that PTEN expression regulates the levels of mut-p53 protein. PTEN appeared to regulate gain-of-function mut-p53 levels via PI3K dependent and independent mechanisms as indicated by the fact that phosphatase-dead PTEN mutants have no effect on cell proliferation and cell death in mut-p53 cells combined with the fact that the PTEN effect cannot be mimicked by pharmacological inhibition of PI3K. We found that PTEN enhances mut-p53 levels at least partly through its effects on PI3K-regulated Mdm2. We demonstrated the involvement of Mdm2 by showing that PTEN regulates nuclear Mdm2 levels in mut-p53 cells and that Mdm2 regulates mut-p53 levels in these same cells. We also examined the dependency of mut-p53 induction by PTEN on Mdm2. We inhibited Mdm2 binding to p53 with Nutlin-3 and assessed the effects of PTEN on mut-p53 protein in this setting. We found that Nutlin-3 and PTEN additively increase mut-p53 protein levels (see supplementary data). These additive effects are probably due to incomplete inhibition of Mdm2 by Nutlin-3 as has been reported in mut-p53 cells not subjected to chemotherapy and/or to partial dependence of PTEN-induced mut-p53 on Mdm2 inhibition (32). The latter is consistent with our data suggesting that PTEN might stabilize mut-p53 protein through physical binding. These findings are altogether consistent with what was reported on the regulation of wt-p53 by PTEN (9-11).
We next demonstrated that the tumor promoting effects of PTEN are dependent on mut-p53 expression. We found that siRNA-mediated down-regulation of mut-p53 expression partially inhibits the PTEN effects on cell cycle progression, completely inhibits the effects of PTEN on cell proliferation and completely reverses the PTEN effects on cytoprotection. The p53 mutations R273H and R175H present in the cells that were used in the present study have been previously characterized as gain-of-function mutations (28-30, 33, 34). About 80% of p53 gene mutations are missense mutations that occur within the DNA binding region of the protein (35). Some of these mutants acquire oncogenic activities per se (16, 17). These mutants function by activating the transcription of oncogenes such as c-myc, c-fos, EGFR, IGF1-R, NF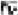 B and others (15, 36). By increasing the protein levels of these mutants, PTEN could acquire tumor promoting functions. While PTEN can acquire tumor promoting properties in the setting of gain-of-function p53 mutations, PTEN probably still exerts tumor suppressive effects in mut-p53 cells through p53-independent mechanisms. The overall effects of PTEN could therefore result from the balance of mut-p53-mediated tumor promoting effects and mut-p53-independent tumor suppressive effects.
B and others (15, 36). By increasing the protein levels of these mutants, PTEN could acquire tumor promoting functions. While PTEN can acquire tumor promoting properties in the setting of gain-of-function p53 mutations, PTEN probably still exerts tumor suppressive effects in mut-p53 cells through p53-independent mechanisms. The overall effects of PTEN could therefore result from the balance of mut-p53-mediated tumor promoting effects and mut-p53-independent tumor suppressive effects.
We describe a novel function for PTEN and provide new insights into the interactions between PTEN and p53. Our findings provide a potential explanation for the low frequency of simultaneous occurrence of PTEN and p53 mutations in human cancer (4, 37, 38). Loss of PTEN in cells harboring gain-of-function p53 mutations would confer growth disadvantage to these cells as compared to wt-PTEN/mut-p53 cells. Our findings also have implications for therapeutic approaches that aim at manipulating PTEN and p53 expression or function in tumors. These approaches would have to take into consideration the mutational status of PTEN and p53 to avoid unwanted harmful effects.
Supplementary Material
ACKNOWLEDGMENTS
Supported by NIH grant RO1 NS045209 (RA) and the Jean Maxwell / American Brain Tumor Association Fellowship (YL).
LITERATURE
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Trotman LC, Pandolfi PP. PTEN and p53: who will get the upper hand? Cancer Cell. 2003;3:97–9. doi: 10.1016/s1535-6108(03)00022-9. [DOI] [PubMed] [Google Scholar]
- 3.Kim RH, Mak TW. Tumours and tremors: how PTEN regulation underlies both. Br J Cancer. 2006;94:620–4. doi: 10.1038/sj.bjc.6602994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–9. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 5.Kato H, Kato S, Kumabe T, et al. Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin Cancer Res. 2000;6:3937–43. [PubMed] [Google Scholar]
- 6.Knobbe CB, Merlo A, Reifenberger G. Pten signaling in gliomas. Neuro-oncol. 2002;4:196–211. [PMC free article] [PubMed] [Google Scholar]
- 7.Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum- sensitive G1 growth arrest in glioma cells. Cancer Res. 1998;58:5002–8. [PubMed] [Google Scholar]
- 8.Stambolic V, MacPherson D, Sas D, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–25. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 9.Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem. 2002;277:5484–9. doi: 10.1074/jbc.M108302200. [DOI] [PubMed] [Google Scholar]
- 10.Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27:462–7. doi: 10.1016/s0968-0004(02)02166-7. [DOI] [PubMed] [Google Scholar]
- 11.Freeman DJ, Li AG, Wei G, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 12.Chang CJ, Freeman DJ, Wu H. PTEN regulates Mdm2 expression through the P1 promoter. J Biol Chem. 2004;279:29841–8. doi: 10.1074/jbc.M401488200. [DOI] [PubMed] [Google Scholar]
- 13.Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66:736–42. doi: 10.1158/0008-5472.CAN-05-1557. [DOI] [PubMed] [Google Scholar]
- 14.Cadwell C, Zambetti GP. The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of-function on cell growth. Gene. 2001;277:15–30. doi: 10.1016/s0378-1119(01)00696-5. [DOI] [PubMed] [Google Scholar]
- 15.Kim E, Deppert W. Transcriptional activities of mutant p53: when mutations are more than a loss. J Cell Biochem. 2004;93:878–86. doi: 10.1002/jcb.20271. [DOI] [PubMed] [Google Scholar]
- 16.Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene. 2007;26:2202–11. doi: 10.1038/sj.onc.1210294. [DOI] [PubMed] [Google Scholar]
- 17.Strano S, Dell’orso S, Mongiovi AM, et al. Mutant p53 proteins: Between loss and gain of function. Head Neck. 2007;29:488–96. doi: 10.1002/hed.20531. [DOI] [PubMed] [Google Scholar]
- 18.Gu J, Tamura M, Yamada KM. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J Cell Biol. 1998;143:1375–83. doi: 10.1083/jcb.143.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He T-C, Zhou S, DaCosta LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl Acad Sci USA. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marie I, Rebouillat D, Hovanessian AG. The expression of both domains of the 69/71 kDa 2′,5′ oligoadenylate synthetase generates a catalytically active enzyme and mediates an anti-viral response. Eur J Biochem. 1999;262:155–65. doi: 10.1046/j.1432-1327.1999.00361.x. [DOI] [PubMed] [Google Scholar]
- 21.Abounader R, Ranganathan S, Lal B, et al. Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c- met expression. J Natl Cancer Inst. 1999;91:1548–56. doi: 10.1093/jnci/91.18.1548. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Lal B, Kwon S, et al. The scatter factor/hepatocyte growth factor: c-met pathway in human embryonal central nervous system tumor malignancy. Cancer Res. 2005;65:9355–62. doi: 10.1158/0008-5472.CAN-05-1946. [DOI] [PubMed] [Google Scholar]
- 23.Abounader R, Ranganathan S, Kim BY, Nichols C, Laterra J. Signaling pathways in the induction of c-met receptor expression by its ligand scatter factor/hepatocyte growth factor in human glioblastoma. J Neurochem. 2001;76:1497–508. doi: 10.1046/j.1471-4159.2001.00158.x. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor PM, Jackman J, Bae I, et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997;57:4285–300. [PubMed] [Google Scholar]
- 25.Wang Y, Zhu S, Cloughesy TF, Liau LM, Mischel PS. p53 disruption profoundly alters the response of human glioblastoma cells to DNA topoisomerase I inhibition. Oncogene. 2004;23:1283–90. doi: 10.1038/sj.onc.1207244. [DOI] [PubMed] [Google Scholar]
- 26.Ishii N, Maier D, Merlo A, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–79. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhrbom L, Nister M, Westermark B. Induction of senescence in human malignant glioma cells by p16INK4A. Oncogene. 1997;15:505–14. doi: 10.1038/sj.onc.1201227. [DOI] [PubMed] [Google Scholar]
- 28.Dittmer D, Pati S, Zambetti G, et al. Gain of function mutations in p53. Nat Genet. 1993;4:42–6. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 29.Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000;60:6788–93. [PubMed] [Google Scholar]
- 30.Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Chuang YY, Chen Q, Brown JP, Sedivy JM, Liber HL. Radiation-induced mutations at the autosomal thymidine kinase locus are not elevated in p53-null cells. Cancer Res. 1999;59:3073–6. [PubMed] [Google Scholar]
- 32.Ambrosini G, Sambol EB, Carvajal D, Vassilev LT, Singer S, Schwartz GK. Mouse double minute antagonist Nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene. 2007;26:3473–81. doi: 10.1038/sj.onc.1210136. [DOI] [PubMed] [Google Scholar]
- 33.Wong RP, Tsang WP, Chau PY, Co NN, Tsang TY, Kwok TT. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol Cancer Ther. 2007;6:1054–61. doi: 10.1158/1535-7163.MCT-06-0336. [DOI] [PubMed] [Google Scholar]
- 34.Scian MJ, Stagliano KE, Anderson MA, et al. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol Cell Biol. 2005;25:10097–110. doi: 10.1128/MCB.25.22.10097-10110.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soussi T, Lozano G. p53 mutation heterogeneity in cancer. Biochem Biophys Res Commun. 2005;331:834–42. doi: 10.1016/j.bbrc.2005.03.190. [DOI] [PubMed] [Google Scholar]
- 36.Weisz L, Damalas A, Liontos M, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67:2396–401. doi: 10.1158/0008-5472.CAN-06-2425. [DOI] [PubMed] [Google Scholar]
- 37.Soyoola EO, Pattillo RA. PTEN/MMAC1 mutations correlate inversely with an altered p53 tumor suppressor gene in gynecologic tumors. Am J Obstet Gynecol. 2003;188:S33–6. doi: 10.1067/mob.2003.243. [DOI] [PubMed] [Google Scholar]
- 38.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32:355–7. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.