

Abstract

An efficient and highly enantio- and diastereoselective synthesis of syn propionamide aldols is described. Formation of the Z-enolborinate via the hydroboration of 4-acryloylmorpholine with (diisopinocampheyl)borane followed by aldol reactions with representative achiral and chiral aldehydes provided syn-α-methyl-β-hydroxy morpholine carboxamides with excellent enantio- and diastereoselectivity (96–98% ee and d.r. >20:1).
Keywords: Diisopinocampheylborinate, aldol, enantioselective, diastereoselective, morpholine amide
The reductive aldol reaction of α,β-unsaturated carbonyl compounds is an important, emerging method for stereocontrolled C-C bond formation.[1] Numerous recent studies[2–6] have focused on reductive aldol reactions of enones and enoates catalyzed by transition-metal complexes. Many such reactions provide excellent levels of enantio- and diastereoselectivity in reductive aldol reactions with aromatic aldehydes, however reductive aldol reactions with aliphatic aldehydes have been generally less selective.[1,2a,b,d,i,3,4c,i,6] Catalytic enantioselective aldol reactions have also been developed that are very effective including reactions with aliphatic aldehydes.[7]
It is well established that boron enolates are exceptionally useful intermediates for asymmetric aldol reactions.[7] We reasoned that the synthetic utility of reductive aldol reactions could be enhanced by utilizing enolborinate intermediates, in view of the tight (B-O bond length 1.4–1.5 Ǻ), closed, structurally well-defined transition states that are invoked to rationalize the enhanced stereochemical control in aldol reactions of enolborinates compared to other metal enolates.[7] While examples of borane-mediated 1,4-reductions of enone and enoate Michael acceptors have been reported (including use of diisopinocampheylborane as the reducing agent),[8] with subsequent reactions with aldehydes leading to syn aldols, these processes have not yet been found to deliver the syn aldol products with synthetically useful enantioselectivity.[8b,c]
We report herein the development of a highly enantio- and diastereoselective boron-mediated reductive aldol reaction that delivers syn aldols with exceptionally high levels of stereocontrol (≥96% ee; ≥20:1 d.r). As depicted in Scheme 1, it was anticipated that hydroboration of a Michael acceptor 1 with (Ipc)2BH would proceed via transition state 2 and lead directly to a Z(O)-enolate.[8a–c] However, Z-E enolborinate equilibration through a reversible 1,3-boratropic shift was suspected to occur in prior studies of this process, thereby delivering a mixture of syn (6) and anti (7) aldol adducts from the Z(O) and E(O)-enolborinates, respectively.[8c,9] As such, two major objectives of this study became (1) the identification of a substrate that would undergo 1,4-reduction to give the Z(O)-enolborinate 3 with high kinetic (if not thermodynamic) control, and (2) identification of a chiral hydroborating reagent capable of inducing excellent enantioselectivity in the subsequent aldol reaction. We elected to pursue (diisopinocampheyl)enolborinates, which are known to be useful intermediates for enantioselective aldol reactions, albeit frequently undergoing aldol reactions with only moderate levels of enantioselectivity.[8c,10]
Scheme 1.
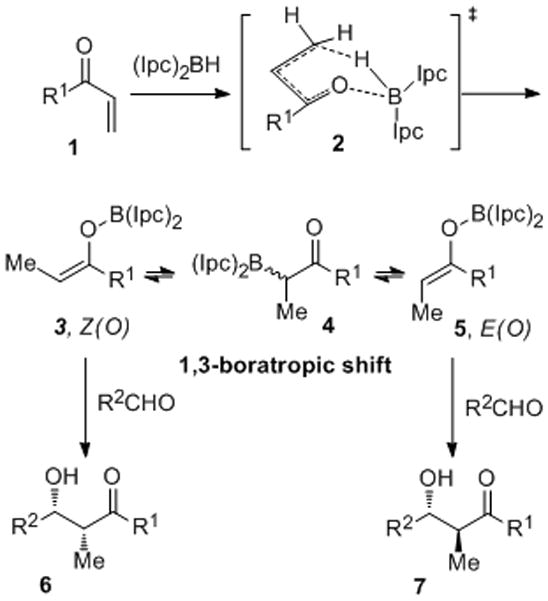
Hydroboration of α,β-unsaturated carbonyl compounds with (Ipc)2BH and subsequent aldol reactions.
We selected commercially available (and very inexpensive)[11] 4-acryloylmorpholine (8) as substrate for the studies reported herein.[12] Morpholine amides are a safe alternative[13] to Weinreb amides but have similar modes of reactivity and comparable ease of manipulation.[14] We quickly found that excellent results were obtained when the hydroboration of 8 with (lIpc)2BH[15] was performed in Et2O at 0 °C for 2 h, followed by addition of 0.85 equiv of aldehyde at −78 °C (Scheme 2). By using this procedure, the syn-α-methyl-β-hydroxy morpholine amides 11a–f were obtained in good to excellent yields (68–91%) and with excellent enantio- and diastereoselectivities (96–98% ee, d.r. >20:1). Separation of aldols 11 from pinene-derived byproducts is trivial, essentially constituting a short column filtration, owing to the large polarity difference. The enantiofacial selectivity derived from (lIpc)2BH in these reactions is the same as in the very well studied allylboration reaction.[17]
Scheme 2.
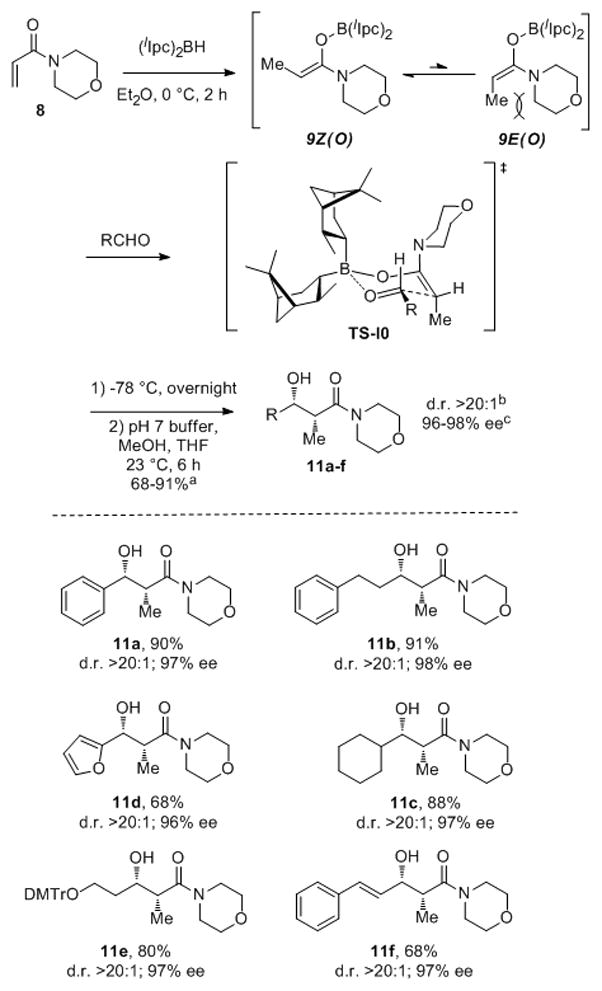
Enantioselective synthesis of syn-α-methyl-β-hydroxy morpholine amides 11 from achiral aldehydes. [a] Isolated yields of aldols 11 obtained after column chromatography. [b] Diastereomer ratios (d.r.) determined by 1H NMR analysis of crude reaction mixtures. [c] Enantiomeric excess (% ee) and absolute configurations determined by using the Mosher ester analysis.[16]
The very high selectivity realized in these reactions reflects, in part, the essentially exclusive (≥99%) formation of the Z(O)-enol diisopinocampheylborinate 9Z (which we characterized by 1D and 2D NMR experiments; see SI). Isomerization of 9Z to 9E evidently does not occur to any significant extent owing to A1,3strain between the morpholine unit and the terminal methyl substituent of the enolborinate.[18] Most remarkable, however, is the exceptional level of enantioselectivity realized in these reactions, which significantly exceeds that obtained in previous studies of enantioselective aldol reactions of (diisopinocampheyl)enolborinates.[8c,10] The relative and absolute stereochemistry determined for aldols 11 is consistent with transition state 10 being dominant in these reactions. That other aldol reactions[8c,10] using the (Ipc)2B– auxiliary proceed with significantly lower levels of enantioselectivity implies that at least one heterochirally related transition state is competitive in those cases, but significantly less so in the reactions of 9Z reported here.[19]
In order to test the utility of this reductive aldol procedure in more complex synthetic contexts, we examined the aldol reactions of 9Z (generated in situ from acrylamide 8 and (Ipc)2BH as described for the reactions in Scheme 2) in the double asymmetric manfold[20] with four chiral aldehydes, 12a, 12b,[21a] 12c,[21b] 12d (Scheme 3). The intrinsic diastereofacial preferences of these aldehydes was determined to be 1.5:1 (in favor of 13a), 1:2 (in favor of 13d), 3:1 (in favor of 13e) and 1.3:1 (in favor of 13g), respectively, by aldol reactions with the achiral enolborinate generated from 8 and dicyclohexylborane (see SI). Remarkably, the double asymmetric aldol reactions of 12a, 12b, 12c and 12d using the chiral Z-enolborinate 9Z deriving from 8 with either (lIpc)2BH or (dIpc)2BH proceeded with excellent stereocontrol (d.r. >20:1; in each case, the minor diastereomer could not be detected in any of the experiments by 1H NMR analysis of crude reaction mixtures) in both the stereochemically matched and mismatched combinations for each aldehyde substrate. The mismatched double asymmetric reaction of 2c yielding 13f (56% yield, 71% based on recovered 2c) was very slow and had not reached completion even after 48 h at −78 °C; all other experiments were complete after overnight at −78 °C. Knowing the intrinsic facial selectivity of aldehyde 12c (d.r. 3:1, see SI), the enantiofacial selectivity of the Z-enol diisopinocampheyborinate 9Z, expressed in energetic terms, must be at least 1.57 kcal/mol in order to override the intrinsic diastereofacial preference of 12c to the extent of >20:1. This corresponds to a reagent enantioselectivity of 96.5% ee, fully consistent with the data provided in Scheme 1 for reactions of 9Z with achiral aldehydes.
Scheme 3.
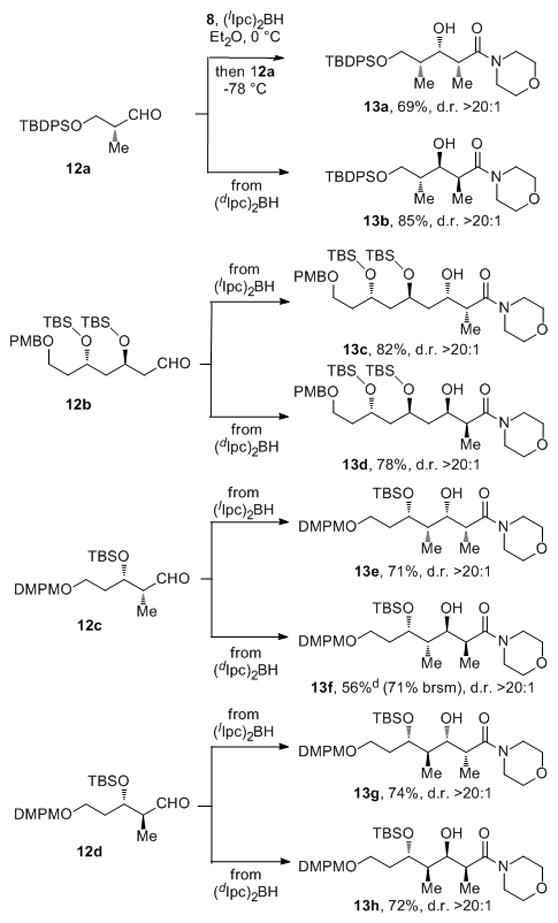
Double asymmetric aldol reactions of chiral aldehydes and the chiral Z-enolborinate generated from 8. [a] Yields of aldol adducts are for isolated products obtained by column chromatography. [b] Diastereomer ratios (d.r.) determined by 1H NMR analysis of crude reaction mixtures. [c] Absolute and relative configurations of 13a–13h determined by using Mosher ester analysis[16] and the Rychnovsky acetonide method[22] (see SI). [d] Very slow reaction, not complete after 48 h at −78 °C.
This method for synthesis of syn-α-methyl-β-hydroxy morpholine carboxamides 11 and 13 is a highly attractive and highly competitive alternative to existing methods for the enantioselective synthesis of syn aldols.[1–7,23] It also sheds light on the great potential of boron-mediated reductive aldol reactions, despite the less than stellar history associated with prior studies of (diisopinocampheyl)enolborinates in enantioselective aldol transformations of achiral substrates.[8c,10]
The aldol reactions of 9Z described here are performed under exceptionally mild and simple conditions, with no added bases. The results summarized in Schemes 2 and 3 demonstrate that standard (e.g., TBDPS, PMB, DMPM) as well as potentially sensitive protecting groups such as dimethoxytrityl (DMTr, see 11e) are fully compatible. The diastereo- and enantioselectivity of this procedure rivals that of the very best technology currently available.[1–7,23] The morpholine amide unit in the aldol products exhibits Weinreb amide-like ease of manipulation in subsequent steps.[13,14] Our procedure requires only two steps, starting with the straightforward synthesis of diisopinocampheylborane.[15] Strikingly, the cost of raw materials required for the synthesis of enolborinate 9Z (including the synthesis of diisopinocampheylborane) is less than $0.25 per mmol scale aldol reaction (2012 Sigma-Aldrich prices for bulk quantities of reagents).[11] Integrating over cost, reagent accessibility, selectivity (both enantio- and diastereoselectivity), substrate scope and generality, and the ease of manipulation of the morpholine amide aldol products,[14] we submit that the new reductive aldol procedure described here is the least expensive[24] and ranks high among the most highly enantio- and diastereoselective, substrate-general methods for synthesis of syn aldols compared to all other currently available procedures.
In summary, we have developed a highly enantioselective synthesis of syn-α-methyl-β-hydroxy morpholine amides 11 and 13 from achiral and chiral aldehydes, respectively, via the hydroboration of 4-acryloylmorpholine (8) with diisopinocampheylborane. This reaction produces the Z-(diisopinocampheyl)enolborinate 9Z with excellent selectivity, which undergoes highly enantioselective aldol reactions with achiral aldehydes (96–98% ee, Scheme 2) and equally highly diastereoselective double asymmetric reactions with a range of chiral aldehydes (Scheme 3). The exceptional enantioselectivity of this process is also noteworthy, especially given that the vast majority of literature examples of aldol reactions of (diisopinocampheyl)enolborinates generally proceed with lower levels of enantioselectivity, which suggests that transition state control in the aldol reactions reported herein is more precise than with the previously studied aldol reactions of (diisopinocampheyl)enolborinates.[8c,10] Extensions of this methodology to other aldol substrates, as well as applications in the synthesis of natural products are currently under investigation and will be reported in due course.
Experimental Section
At 0 °C, to a suspension of (l or dIpc)2BH (weighed in a glovebox, 72 mg, 0.25 mmol) or (Cy)2BH (weighed in a glovebox, 45 mg, 0.25 mmol) in Et2O (1.0 mL) was added 4-acryloylmorpholine (8) (35 μL, 0.275 mmol). The solution was stirred 2 h at 0 °C at which time it became homogeneous. The resulting mixture was cooled to −78 °C, aldehyde (0.213 mmol) was added, and the solution was stirred overnight at −78 °C. Aqueous pH 7 buffer solution (0.5 mL), MeOH (0.5 mL) and THF (0.5 mL) were added and the reaction was stirred for 6 h at room temperature. The aqueous phase was extracted three times with CH2Cl2 (10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification of the crude product by flash chromatography through a short plug of silica gel (1:1 CH2Cl2-ethyl acetate) provided the corresponding β–hydroxymorpholine amide 11 or 13.
Supplementary Material
Footnotes
We thank the National Institutes of Health (GM038436) for support of this research. We also sincerely thank Professor Glenn Micalizio and Dr. Daniel Canterbury for fruitful discussions and comments on this work.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Guo HC, Ma JA. Angew Chem Int Ed. 2006;45:354–366. doi: 10.1002/anie.200500195. [DOI] [PubMed] [Google Scholar]; b) Nishiyama H, Shiomi T. Top Curr Chem. 2007;279:105–137. [Google Scholar]; c) Han SB, Hassan A, Krische MJ. Synthesis. 2008;17:2669–2679. doi: 10.1055/s-2008-1067220. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Garner SA, Han SB, Krische MJ. Metal Catalyzed Reductive Aldol Coupling. In: Andersson P, Munslow I, editors. Modern Reduction Methods. Wiley-VCH; Weinheim: 2008. pp. 387–408. [Google Scholar]
- 2.Rhodium catalyzed asymmetric reductive aldol reactions: Taylor SJ, Duffey MO, Morken JP. J Am Chem Soc. 2000;122:4528–4529.Russell AE, Fuller NO, Taylor SJ, Aurriset P, Morken JP. Org Lett. 2004;6:2309–2312. doi: 10.1021/ol049591e.Bocknack BM, Wang L-C, Krische MJ. Proc Natl Acad Sci. 2004;101:5421–5424. doi: 10.1073/pnas.0307120101.Fuller NO, Morken JP. Synlett. 2005:1459–1461.Nishiyama H, Shiomi T, Tsuchiya Y, Matsuda I. J Am Chem Soc. 2005;127:6972–6973. doi: 10.1021/ja050698m.Jung C-K, Krische MJ. J Am Chem Soc. 2006;128:17051–17056. doi: 10.1021/ja066198q.Han SB, Krische MJ. Org Lett. 2006;8:5657–5660. doi: 10.1021/ol0624023.Ito J, Shiomi T, Nishiyama H. Adv Synth Catal. 2006;348:1235–1240.Shiomi T, Ito J, Yamamoto Y, Nishiyama H. Eur J Org Chem. 2006:5594–5600.Shiomi T, Nishiyama H. Org Lett. 2007;9:1651–1654. doi: 10.1021/ol070251d.Hashimoto T, Shiomi T, Ito J, Nishiyama H. Tetrahedron. 2007;63:12883–12887.Hashimoto T, Ito J, Nishiyama H. Tetrahedron. 2008;64:9408–9412.Bee C, Han SB, Hassan A, Iida H, Krische MJ. J Am Chem Soc. 2008;130:2746–2747. doi: 10.1021/ja710862u.Shiomi T, Adachi T, Ito J, Nishiyama H. Org Lett. 2009;11:1011–1014. doi: 10.1021/ol802939u.Sugiura M, Sato N, Sonoda Y, Kotani S, Nakajima M. Chem–Asian J. 2010;5:478–481. doi: 10.1002/asia.200900450.
- 3.Iridium catalyzed asymmetric reductive aldol reactions: Zhao CX, Duffey MO, Taylor SJ, Morken JP. Org Lett. 2001;3:1829–1831. doi: 10.1021/ol015859f.
- 4.Copper catalyzed asymmetric reductive aldol reactions: Lam H, Murray GJ, Firth JD. Org Lett. 2005;7:5743–5746. doi: 10.1021/ol052599j.Lam H, Joensuu PM. Org Lett. 2005;7:4225–4228. doi: 10.1021/ol051649h.Chuzel O, Deschamp J, Chausteur C, Riant O. Org Lett. 2006;8:5943–5946. doi: 10.1021/ol062398v.Deschamp J, Chuzel O, Hannedouche J, Riant O. Angew Chem Int Ed. 2006;45:1292–1297. doi: 10.1002/anie.200503791.Zhao D, Oisaki K, Kanai M, Shibasaki M. Tetrahedron Lett. 2006;47:1403–1407.Zhao D, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2006;128:14440–14441. doi: 10.1021/ja0652565.Lipshutz BH, Amorelli B, Unger JB. J Am Chem Soc. 2008;130:14378–14379. doi: 10.1021/ja8045475.Deschamp J, Riant O. Org Lett. 2009;11:1217–1220. doi: 10.1021/ol802879f.Kato M, Oki H, Ogata K, Fukuzawa S. Synlett. 2009:1299–1302.Ou J, Wong WT, Chiu P. Tetrahedron. 2012;68:3450–3456.
- 5.Cobalt catalyzed asymmetric reductive aldol reaction: Lumby RJ, Joensuu PM, Lam HW. Tetrahedron. 2008;64:7729–7740.
- 6.Tertiary amine as a hydride donor for asymmetric reductive aldol reactions: Osakama K, Sugiura M, Nakajima M, Kotani S. Tetrahedron Lett. 2012;53:4199–4201.
- 7.Selected reviews of enantioselective aldol reactions: Heathcock CH. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. 2 . Pergamon Press; New York: 1991. pp. 181–238.Kim BM, Williams SF, Masamune S. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon Press; New York: 1991. pp. 239–275.Cowden CJ, Paterson I. Org React. 1997;51:1–200.Mahrwald R. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; Weinheim: 2004. Denmark SE, Fiujimori S. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; Weinheim: 2004. pp. 229–326.Shibasaki M, Matsunaga S, Kumagai N. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; Weinheim: 2004. pp. 197–227.Johnson JS, Nicewicz DA. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; Weinheim: 2004. pp. 69–103.Bisai V, Bisai A, Singh VK. Tetrahedron. 2012;68:4541–4580.
- 8.a) Evans DA, Fu GC. J Org Chem. 1990;55:5678–5680. [Google Scholar]; b) Boldrini GP, Mancini F, Tagliavini E, Trombini C, Umani-Ronchi A. J Chem Soc, Chem Commun. 1990:1680–1681. [Google Scholar]; c) Boldrini GP, Bortolotti M, Mancini F, Tagliavini E, Trombini C, Umani-Ronchi A. J Org Chem. 1991;56:5820–5826. [Google Scholar]; d) Matsumoto Y, Hayashi T. Synlett. 1991:349–350. [Google Scholar]; e) Ghosh AK, Kass J, Anderson DD, Xu X, Marian C. Org Lett. 2008;10:4811–4814. doi: 10.1021/ol801971t. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Huddleston RR, Cauble DF, Krische MJ. J Org Chem. 2003;68:11–14. doi: 10.1021/jo020629f. [DOI] [PubMed] [Google Scholar]
- 9.a) Masamune S, Mori S, van Horn D, Brooks SW. Tetrahedron Lett. 1979;20:1665–1668. [Google Scholar]; b) Duffy JL, Yoon TP, Evans DA. Tetrahedron Lett. 1995;36:9245–9248. [Google Scholar]; c) Abiko A, Inoue T, Masamune S. J Am Chem Soc. 2002;124:10759–10764. doi: 10.1021/ja0260014. [DOI] [PubMed] [Google Scholar]
- 10.a) Paterson I, Goodman JM, Lister MA, Schumann RC, McClure CK, Norcross RD. Tetrahedron. 1990;46:4663–4684. [Google Scholar]; b) Ramachandran PV, Pratihar D. Org Lett. 2009;11:1467–1470. doi: 10.1021/ol802850w. Ramachandran reports the synthesis of syn aldols with ≥94:6 d.r. and 90–98% ee from the Ipc2BOTf mediated aldol reactions of methyl propionate. However, after repeated attempts, the best selectivity we have achieved is 2:1 d.r. and 79% ee for the syn aldol from the reaction of methyl propionate and cinnamaldehyde using Ramachandran’s procedure. [DOI] [PubMed] [Google Scholar]
- 11.Cost of reagents used in the synthesis of (Ipc)2BH and enolborinate 9Z (2012 Sigma Aldrich prices): N-acryloyl morpholine ($168 per 250 mL, or $0.008 per mmol); (+)-pinene ($72 per Kg, or $0.01 per mmol); (−)-pinene is less expensive than the (+)-enantiomer; borane-dimethyl sulfide ($550 for 800 mL of 10.0 M solution; or $0.07 per mmol).
- 12.The Weinreb amide of acrylic acid failed to undergo the reductive aldol reaction, presumably owing to chelation of boron by the N-methoxy group after the 1,4-reduction.
- 13.a) Jackson MM, Leverett C, Toczko JF, Roberts JC. J Org Chem. 2002;67:5032–5035. doi: 10.1021/jo025682i. [DOI] [PubMed] [Google Scholar]; b) Peters R, Waldmeier P, Joncour A. Org Process Res Dev. 2005;9:508–512. [Google Scholar]
- 14.a) Martin R, Romea P, Tey C, Urpi F, Vilarrasa J. Synlett. 1997;49:1414–1416. [Google Scholar]; b) Concellón JM, Rodríguez-Solla H, Méjica C, Blanco EG. Org Lett. 2007;9:2981–2984. doi: 10.1021/ol070896d. [DOI] [PubMed] [Google Scholar]; c) Dhoro F, Kristensen TE, Stockmann V, Yap GPA, Tius MA. J Am Chem Soc. 2007;129:7256–7257. doi: 10.1021/ja0718873. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Concellón JM, Rodríguez-Solla H, Díaz P. J Org Chem. 2007;72:7974–7979. doi: 10.1021/jo701417z. [DOI] [PubMed] [Google Scholar]; e) Lin KW, Tsai C-H, Hsieh I-L, Yan TH. Org Lett. 2008;10:1927–1930. doi: 10.1021/ol8004326. [DOI] [PubMed] [Google Scholar]; f) Concellón JM, Rodríguez-Solla H, del Amo V, Díaz P. Synthesis. 2009:2634–2645. [Google Scholar]; g) Rye C, Barker D. Synlett. 2009:3315–3319. [Google Scholar]
- 15.Brown HC, Singaram B. J Org Chem. 1984;49:945–947. [Google Scholar]
- 16.a) Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519. [Google Scholar]; b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 17.a) Jadhav PK, Bhat KS, Perumal PT, Brown HC. J Org Chem. 1986;51:432–439. [Google Scholar]; b) Brown HC, Bhat KS, Randad RS. J Org Chem. 1989;54:1570–1576. [Google Scholar]; c) Racherla US, Brown HC. J Org Chem. 1991;56:401–404. [Google Scholar]
- 18.Hoffmann RW. Chem Rev. 1989;89:1841–1860. [Google Scholar]
- 19.Computational studies of aldol reactions of enolborinate intermediates: Bernardi A, Capelli AM, Gennari C, Goodman JM, Paterson I. J Org Chem. 1990;55:3576–3581.Li Y, Paddon-Row MN, Houk KN. J Org Chem. 1990;55:481–493.Bernardi F, Robb MA, Suzzi-Valli G, Tagliavini E, Tromboni C, Umani-Ronchi A. J Org Chem. 1991;56:6472–6475.
- 20.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed. 1985;24:1–30. [Google Scholar]
- 21.a) Nuhant P, Kister J, Lira R, Sorg A, Roush WR. Tetrahedron. 2011;67:6497–6512. doi: 10.1016/j.tet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lira R, Roush WR. Org Lett. 2007;9:4315–4318. doi: 10.1021/ol7018746. [DOI] [PubMed] [Google Scholar]
- 22.Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945–948. [Google Scholar]
- 23.a) Evans DA, Bartroli J, Shih TL. J Am Chem Soc. 1981;103:2127–2129. [Google Scholar]; b) Crimmins MT, King BW, Tabet AE. J Am Chem Soc. 1997;119:7883–7884. [Google Scholar]; c) Crimmins MT, Chaudhary K. Org Lett. 2000;2:775–777. doi: 10.1021/ol9913901. [DOI] [PubMed] [Google Scholar]
- 24.By comparison, the cost of the valine-derived N-propionyl oxazolidinone (e.g., Evans’ aldol reagent)[23] is $20/mmol (2012 Sigma-Aldrich). The current cost of the chiral oxazolidinone (use of which requires an N-acylation step prior to the aldol reaction) is $6.80/mmol. The cost of the parent (S)-valinol (the less expensive of the two valinol enantiomers), common to both the Evans and Crimmin’s aldol methods, is $1.90/mmol, but two additional synthetic steps are required to generate the reagents used in the aldol rexperiments. Virtually all of the catalytic enantioselective methods[1–7] currently available use of expensive transition metal catalysts and/or expensive chiral ligands (many of which require multi-step synthesis if not commercially available). For example, Rh(COD)2OTf and [Rh(COD)Cl]2,, two of the least expensive and most accessible Rh(I) catalyst starting materials used in catalytic enantioselective reductive aldol reactions,[2] cost $62–$86 per mmol, respectively (5% Rh(I) loading is used in many of the published examples, therefore the Rh(I) cost is $3–$5 for a 1 mmol aldol experiment). (R)-BINAP, one of the least expensive and widely available chiral phosphine ligands, costs $80/mmol; hence the cost of this ligand when used at the 5 mol% level in a catalytic enantioselective reductive aldol experiment is approximately $5 per mmol aldol experiment.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.