

Abstract
Although histone H3 lysine 4 (H3K4) methylation is widely associated with gene activation, direct evidence for its causal role in transcription, through specific MLL family members, is scarce. Here we have purified a human MLL2 (Kmt2b) complex that is highly active in H3K4 methylation and chromatin transcription in a cell-free system. This effect requires SAM and intact H3K4, establishing a direct and causal role for MLL2-mediated H3K4 methylation in transcription. We then show that human AKAP95, a chromatin-associated protein, is physically and functionally associated with the DPY30–MLL complexes and directly enhances their methyltransferase activity. Ectopic AKAP95 stimulates expression of a chromosomal reporter in synergy with MLL1 or MLL2, whereas AKAP95 depletion impairs retinoic acid-mediated gene induction in embryonic stem cells. These results demonstrate an important role for AKAP95 in regulating histone methylation and gene expression, particularly during cell fate transitions.
Histone modifications have been closely linked to many DNA-related processes1 including transcription2. In particular, acetylation of histones H3 and H4 and methylation of H3 lysine 4, especially tri-methylation (H3K4me3), are prominent marks related to active gene expression3,4. The most notable H3K4 methyltransferases in mammals are the SET1 and MLL family complexes (hereafter MLL complexes), which, apart from some specialized subunits, contain either hSET1A, hSET1B, MLL1, MLL2, MLL3, or MLL4 as the catalytic subunit and WDR5, RbBP5, ASH2L, and DPY30 as integral core subunits that are necessary for the efficient methylation activity of the complexes4–6.
Numerous reports, based largely on chromatin immunoprecipitation (ChIP) assays, have shown a close association of H3K4 methylation with gene activation7–10, but the majority describe correlative, rather than causal, relationships. Two major lines of indirect evidence are consistent with a causal role for H3K4 methylation in gene activation. First, deletion or depletion of the catalytic or core subunits of the MLL complexes has been shown to affect H3K4 methylation and expression of specific genes5,11–16. Second, H3K4me3 can be bound, through specific domains such as PHD fingers, by effector proteins (“readers”) that are directly or indirectly involved in transcription, thus linking H3K4me3 to transcription5,17,18. Nonetheless, these lines of evidence are complicated by the following points: (i) non-catalytic activities of the methyltransferase may regulate gene expression, as exemplified by the action of MLL1 in promoting expression of a neurogenic gene (Dlx2) by recruitment of an H3K27 demethylase rather than by H3K4 methylation19; (ii) non-histone proteins can also be functional substrates for histone modifying enzymes20 including an H3K4 methyltransferase21; (iii) in principle, methylated non-histone proteins might also bind to H3K4 methyl readers and lead to observed effects and (iv) methyltransferases and methyl readers have many genomic targets, making it difficult to rigorously demonstrate, in cells, their direct effect on the specific genes of interest. Moreover, the establishment of a causal role for H3K4 methylation in transcription by histone gene mutation is also technically challenging in higher organisms due to the existence of multiple copies of individual histone genes.
To demonstrate a causal role for H3K4 methylation in transcription, we set out to purify and characterize a human MLL2 (Kmt2b, gene ID 9757) complex that was active in H3K4 methylation and chromatin-templated in vitro transcription. We then searched for novel regulators of this modification that might be associated with the MLL2 complex as well as other MLL complexes. We recently have shown that mammalian DPY30, a common subunit of all MLL complexes22, is important for efficient H3K4 methylation and transcriptional plasticity in ESC differentiation23. Using DPY30 as a bait, our current study has resulted in isolation of MLL complexes that are associated with a novel component, AKAP95.
AKAP95 is the only nuclear member of the large family of A-kinase anchoring proteins (AKAPs), which share a common function in protein kinase A (PKA) binding and spatiotemporal regulation of cellular signaling24. Among the many proposed roles of AKAP95 (refs. 25–27), which include mediation of chromatin condensation28,29, is a function in transcription regulation that is based on location in the nuclear matrix and binding to p68 RNA helicase30. However, direct evidence for a role in gene expression is lacking. Recently, AKAP95 was also found to interact with Oct4, a transcription factor crucial for ESC pluripotency31, but it is unclear whether AKAP95 is involved in ESC maintenance and differentiation. Here, following up our characterization of the MLL2 complex in chromatin-templated transcription, we have established AKAP95 as a direct and novel modulator of H3K4 methylation, a transcriptional coactivator and an important regulator of gene induction during ESC fate transitions.
RESULTS
The MLL2 complex is highly active in methylating histone H3K4
Nuclear extract derived from a 293 cell line that stably expresses FLAG-HA-tagged full-length human MLL2 (Kmt2b) (FH-MLL2) was subjected to anti-FLAG antibody-mediated immunoaffinity purification. As revealed by mass spectrometric analysis, most of the co-purified proteins were known common subunits of the MLL1 and 2 complexes and included WDR5, RbBP5, ASH2L, DPY30, and menin (Fig. 1A). An endogenous association of these proteins was confirmed by their co-immunoprecipitation with MLL2 from HeLa cell nuclear extract (Supplementary Fig. 1A).
Figure 1. An MLL2 complex that is highly active in methylating H3K4.
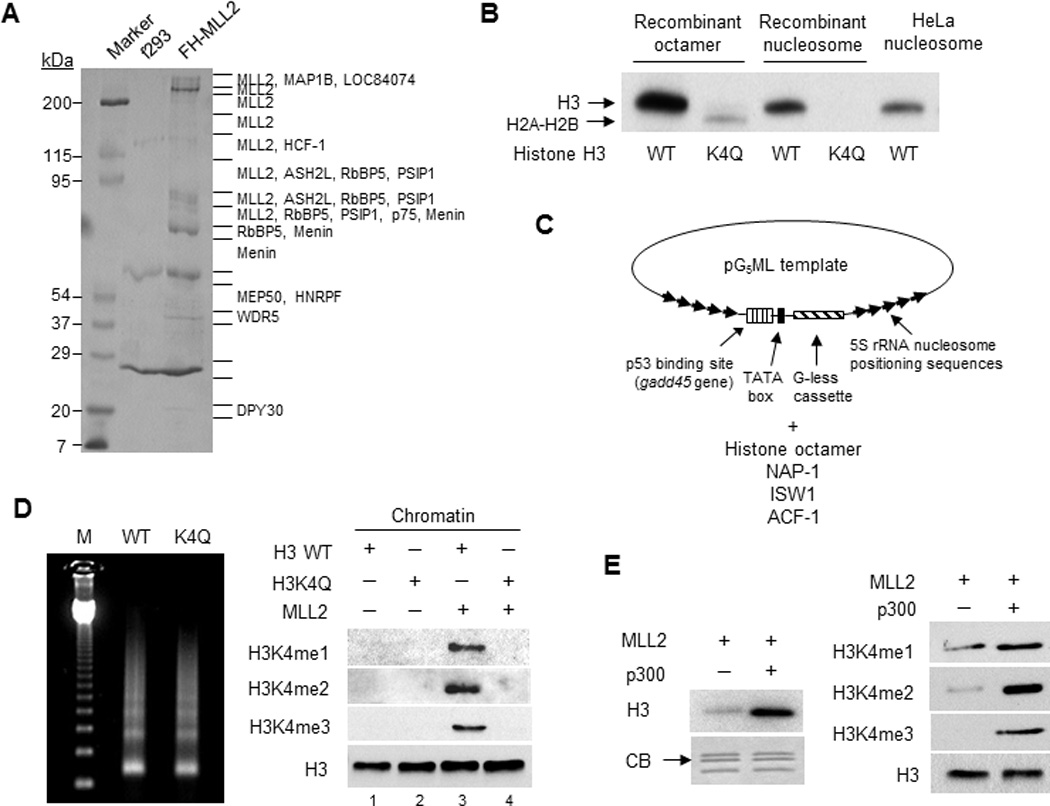
(A) Coomassie blue-stained 6–12% SDS-PAGE gel of proteins eluted from M2 agarose beads. Major proteins identified specifically from the FH-MLL2 cell line by MALDI mass spectrometry are indicated. Lines on the right of the gel indicate positions of gel excision for mass spectrometry.
(B) Autoradiographic analysis of in vitro histone methylation (HMT) assay by MLL2C on free histone octamers or on recombinant or native (HeLa) nucleosomes with wild-type (WT) or mutated (K4Q) H3, all in the presence of [3H]-SAM.
(C) Schematic illustration of chromatin assembly protocol.
(D) In vitro HMT assay of MLL2C on assembled chromatin. Left: micrococcal nuclease (MNase) analysis of assembled chromatin with histone octamers containing wild type (WT) H3 or H3K4Q mutant. Right: Immunoblots of histone methylation on chromatin assembled with wild type H3 or the H3K4Q mutant.
(E) In vitro HMT assay of MLL2C on assembled chromatin in the absence or presence of p300. Left: an autoradiographic analysis of incorporated [3H]-SAM. CB: commassie blue staining. Right: Immunoblots with indicated antibodies.
In an in vitro histone methyltransferase (HMT) assay, the MLL2 complex (MLL2C) showed strong H3 methylation activity on free histone octamer and both recombinant and native HeLa nucleosomal substrates (Fig. 1B). No H3 methylation was detected with a recombinant histone octamer substrate carrying the H3K4Q mutation (although weak H2A and H2B methylation could be detected) or on a nucleosomal substrate carrying the H3K4Q mutation. Hence, the MLL2C methyltransferase activity is specific for H3K4. We further tested the activity of MLL2C on recombinant chromatin templates32 assembled from a plasmid and histone octamers carrying either wild type H3 or the H3K4Q mutant (Fig. 1C). Micrococcal nuclease (MNase) digestion of the two assembled chromatins produced indistinguishable DNA ladders of kinetic intermediates, indicating comparable assembly qualities for these two chromatin templates (Fig. 1D). MLL2C was shown to catalyze mono-, di-, and tri-methylation of H3K4 on chromatin assembled with wild type H3, but not on chromatin assembled with the H3K4Q mutant (Fig. 1D). Interestingly, the histone acetyltransferase (HAT) p300 strongly enhanced the MLL2C-dependent H3K4 methylation on the chromatin template, and this effect was evident for all three methylation states (Fig. 1E). Unlike the previously reported WDR5-MLL1 complex12 that associates with MOF, a MYST family HAT, no known or putative HAT was found in the purified MLL2 complex by the mass spectrometric analysis. Consistently, MLL2C failed to show any HAT activity (Supplementary Fig. 1B). Therefore, the MLL2 complex described above is robust and specific for H3K4 methylation.
H3K4 methylation stimulates chromatin transcription
To test the role of MLL2C-mediated H3K4 methylation in transcription, we used a well-established chromatin-templated in vitro transcription system32 with the same chromatin templates (Fig. 1C and 2A) used in the modification assays described above. As the template has a tandem repeat of five p53 responsive elements, we used p53 as the transcription activator and HeLa cell nuclear extract as the source of RNA polymerase II, general transcription factors and Mediator32.
Figure 2. The MLL2 complex stimulates chromatin transcription in a SAM- and H3K4-dependent manner.

(A) Schematic protocol for chromatin assembly and transcription32. SAM and acetyl-CoA were included in the assays unless specifically indicated.
(B) Autoradiographic analysis of in vitro transcription on DNA or chromatin templates. Relative transcription levels are shown in bar charts in B, C, D and F. ND, nondetectable. Refer to Supplementary Fig. 8A.
(C) Autoradiographic analysis of in vitro transcription on a chromatin template with additions of MLL2C, p300, SAM and acetyl-CoA as indicated.
(D) Autoradiographic analysis of in vitro transcription on chromatin templates assembled with histone octamers containing wild type H3 or H3K4Q mutant.
(E) Illustration of specific H3 and H4 lysine residues (red) that were mutated to arginine in E and F.
(F) Autoradiographic analysis of in vitro transcription on chromatin templates assembled with histone octamers containing wild type H3 or H3 (K14R K18R) mutant and wild type H4 or H4 (K5R K8R) mutant. p300 was not added in any of the reactions.
Addition of MLL2C had no detectable effect on p53-dependent transcription on a DNA template (Fig. 2B, lanes 1–3) or on basal transcription (in the absence of p53) on the chromatin template (Fig. 2B, lanes 4 and 5). In contrast, MLL2C showed a strong stimulatory effect on p53-dependent chromatin transcription that was comparable to the effect of p300 (Fig. 2B, lanes 7–9), a previously established robust transcription coactivator in this assay33. [Note that the p53-dependent activity observed in the absence of added p300 or MLL2C, but in the presence of acetyl-CoA and S-adenosyl methionine (SAM), likely reflects contributions of endogenous histone modifying activities (Fig. 2B, lane 7 versus lane 4)]. Furthermore, MLL2C and p300 had a synergistic effect on chromatin transcription (Fig. 2C, compare lanes 5, 8 and 9). The strong stimulatory effect of MLL2C was greatly reduced by omission of exogenous SAM from the reaction (Fig. 2C, lanes 5 and 6, and Supplementary Fig. 1C, lanes 3 and 4), indicating that MLL2C-mediated transcription enhancement depends on its methyltransferase activity. Importantly, while having a minimal effect on the transcription without exogenous MLL2C (Fig. 2D, lanes 3 and 4), the H3K4Q mutation almost completely abolished the otherwise strong stimulatory effect of MLL2C on transcription (Fig. 2D, lanes 5 and 6), indicating a direct role for MLLC-mediated methylation of H3K4, rather than possible non-histone substrates, in transcription. Notably, p300 alone had equally strong effects on transcription from the wild type H3 and H3K4Q chromatin templates (Fig. 2D, compare lanes 7 and 8), indicating retention of the overall integrity of the mutant chromatin and the lack of any contribution of an endogenous H3K4 methyltransferase activity in the nuclear extract to the p300-dependent transcription stimulation. Hence, these results unambiguously demonstrate a direct causal role for MLL2C-mediated H3K4 methylation in transcription activation, including its synergistic function with p300.
One interesting observation from our transcription experiments is the acetyl-CoA dependence of MLL2C-mediated transcription stimulation. (Fig. 2C, lanes 4 to 7 and Supplementary Fig. 1C, lanes 2 to 5). To determine whether acetylation of histones, rather than other non-histone factors, plays an important role in H3K4 methylation-mediated transcription, we performed transcription assays on chromatin templates containing either wild type histones or histones with specific lysine mutations in H3 (K14R K18R) and H4 (K5R K8R) (Fig. 2E). These lysine residues are known to be acetylated by p300 (ref. 34) and potentially other HATs. We found that mutations in H3 or H4 alone partially impaired, and simultaneous mutations in both H3 and H4 completely abolished, the transcription stimulatory effect of MLL2C (Fig. 2F, lanes 9–12). Importantly, these histone mutations barely affected the low level transcription observed in the absence of MLL2C (Fig. 2F, lanes 5–8) or the MLL2C-mediated H3K4 methylation of chromatin (Supplementary Fig. 1D), indicative of the overall integrity of chromatins containing mutated histones. Taken together, these results indicate that lysine acetylation of histone H3 and H4 is essential for manifestation of the stimulatory effect of the MLL2C-mediated H3K4 methylation on transcription.
AKAP95 is associated with DPY30–MLL complexes
To identify novel proteins associated with MLL2C and other MLL complexes, we prepared nuclear extract from a 293 cell line that stably expresses Flag-HA-tagged human DPY30 and subjected it to an immunoaffinity purification followed by mass spectrometric analysis. In addition to the established subunits of MLL family complexes, AKAP95 was identified as one of the major DPY30-associated proteins (Fig. 3A), as further confirmed by immunoprecipitation with anti-HA antibody and immunoblotting (Supplementary Fig. 2A). Components of NURF complex35 were also found to be associated with DPY30 (Fig. 3A), but were not pursued in this study. An endogenous association of AKAP95 with DPY30 was confirmed by their co-immunoprecipitation from HeLa nuclear extract by a DPY30 antibody (Fig. 3B). As further evidence that more rigorously establishes AKAP95 as a bona fide binding partner of DPY30 in the context of MLL complexes: (i) immunoprecipitation of WDR5 or ASH2L efficiently co-precipitated AKAP95 (Fig. 3C and Supplementary Fig. 2B); (ii) epitope-tagged AKAP95 also efficiently co-precipitated ASH2L and WDR5 (Supplementary Fig. 2C); and (iii) a gel filtration chromatography analysis revealed co-elution of AKAP95 with the MLL1 and MLL2 complexes, but not with the abundant dimerized or free DPY30 (Fig. 3D).
Figure 3. AKAP95 is associated with DPY30-MLL complexes.

(A) Coomassie blue-stained 4–20% SDS-PAGE gel of proteins eluted from M2 agarose beads. Major proteins identified specifically from the FH-DPY30 cell line by MALDI mass spectrometry are indicated. Lines on the right of the gel indicate positions of gel excision for mass spectrometry.
(B) Immunoblot analysis of endogenous DPY30-MLL complexes following IP of HeLa cell nuclear extract by anti-DPY30 or normal rabbit IgG antibodies. Refer to Supplementary Fig. 8B.
(C) Immunoblot analysis of DPY30-MLL complexes following IP by M2 beads of lysates of 293T cells transiently transfected with or without FLAG-HA-WDR5 plasmid. Refer to Supplementary Fig. 8C.
(D) Immunoblot analysis of the purified FH-DPY30 complexes following fractionation by gel filtration on Superose 6. Elution volumes are indicated at the top.
AKAP95 stimulates expression of a chromosomal reporter gene
The physical interaction between AKAP95 and DPY30-MLL complexes raised the possibility of their functional relevance in regulating gene expression. In a 293T cell line (G-293T) that contains a chromosomally integrated luciferase reporter gene with upstream Gal4-binding sites23, overexpression of AKAP95 strongly stimulated reporter expression at RNA and possibly protein (indirectly indicated by the luciferase activity) levels; and this effect was critically dependent upon the presence and dose of the Gal4-VP16 activator (Fig. 4A and Supplementary Fig. 3A and 3B). Remarkably, AKAP95 showed strong synergy with either MLL1 or MLL2 in stimulating reporter gene expression (Fig. 4A and 4B). Furthermore, overexpression of either AKAP95 or MLL2 markedly enhanced the levels of H3K4me2 and H3K4me3 at the promoter region of the chromosomal Gal4-luciferase locus (Fig. 4C), strongly suggesting effects at the transcriptional level.
Figure 4. AKAP95 strongly co-activates expression of a chromosomal reporter gene.
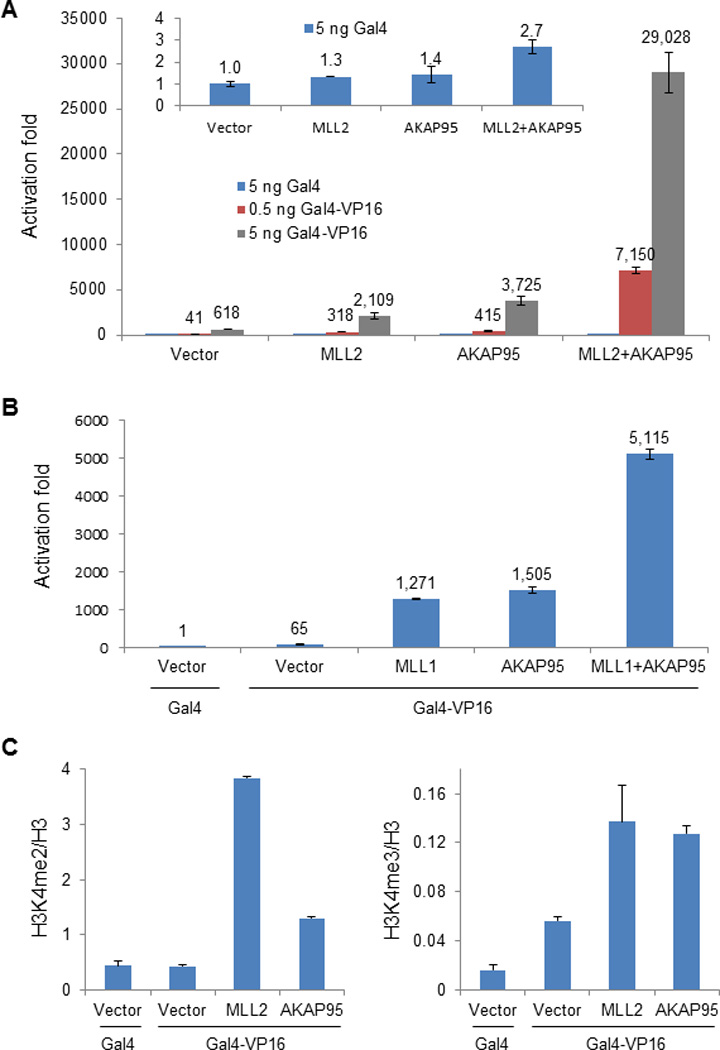
(A) Luciferase assay in G-293T cells expressing ectopic MLL2, AKAP95, or MLL2 and AKAP95 in the presence of Gal4 (blue bars, and inset) or two different doses of Gal4-VP16 (red and grey bars). In this panel and panel B, the values for activation fold are indicated above the bars and are based on the signal (arbitrarily set at 1) for cells transfected with empty vector and Gal4, and the error bars represent the range of values from two independent transfections. Representative results chosen from more than three independent experiments are shown.
(B) Luciferase assay in G-293T cells expressing ectopic MLL1, AKAP95, or MLL1 and AKAP95 in the presence of Gal4 or Gal4-VP16 as indicated.
(C) ChIP for H3K4me2 (left) or H3K4me3 (right) followed by qPCR at the Gal4-luciferase promoter region by ectopic expression of MLL2 or AKAP95. The error bars indicate s.d. from triplicate measurements from one representative experiment (out of two experiments).
Co-activation requires the DPY30-binding region on AKAP95
By serial truncations from N- and C-termini (Fig. 5A), we mapped the AKAP95 regions that are important for its transcriptional stimulatory activity in the chromosomal reporter assay. AKAP95 (1–210) and (1–340) were inactive in enhancing reporter expression (Fig. 5B). AKAP95 (1–530), despite its very low expression level, modestly enhanced the luciferase expression in a dose-dependent manner, and was thus considered active (Fig. 5C). Despite their high levels of expression, AKAP95 (341–692), AKAP95 (211–692), and AKAP95 (101–692) all failed to enhance reporter expression (Fig. 5B and 5C), indicating an essential role of the N-terminal region 1–100 in this AKAP95 function. In a further analysis, we found that the nuclear localization signal (NLS) and the two zinc fingers, but not the nuclear matrix targeting site (NMTS)30 or the RII binding domain, were important for enhancement of reporter expression (Fig. 5D and Supplementary Fig. 3C). As deletion of the RII binding region led to poor expression, we tested an AKAP95 point mutant (I582P) that was previously shown to disrupt the binding of AKAP95 to RII (ref. 36) and found it to have only a little or marginal effect on AKAP95 activity (Supplementary Fig. 3C), suggesting that binding to PKA RII is not critical for AKAP95 co-activation of reporter expression.
Figure 5. Transcriptional co-activation function of AKAP95 requires the region that binds to DPY30-MLL complexes.
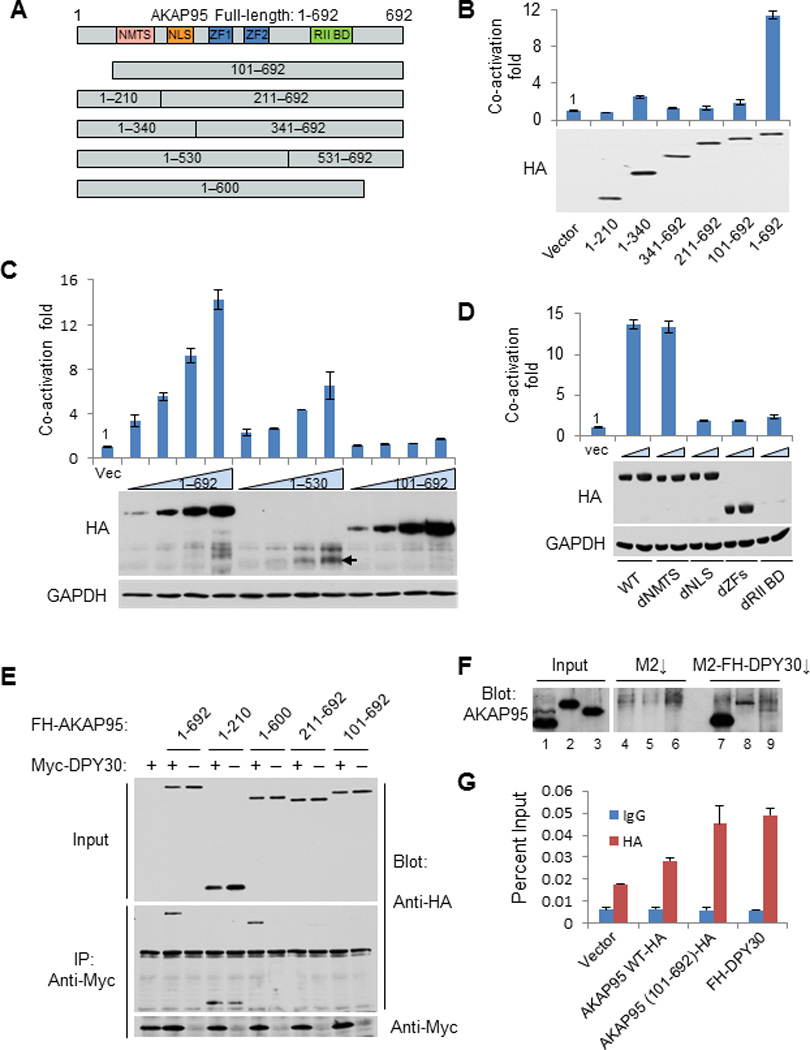
(A) Illustration of truncations of AKAP95, with putative domains marked.
(B–D) Luciferase assay (above) and immunoblot analysis (below) for G-293T cells transfected with plasmids encoding FH-full-length AKAP95 or indicated mutants, together with the Gal4-VP16 plasmid. Luciferase signals were normalized to the signal (arbitrarily set at 1) from cells transfected with empty vector plus Gal4-VP16, and the error bars represent the range of values from two independent transfections.
(E) Immunoblot analysis for anti-Myc-mediated immunoprecipitation for G-293T cells transfected with plasmids encoding full-length FH-AKAP95 or indicated truncation mutants, with or without Myc-DPY30 expressing vector as indicated.
(F) Immunoblot analysis with anti-AKAP95 antibody for the in vitro binding assay for AKAP95 and DPY30. HeLa nuclear extract (lanes 1, 4 and 7), purified GST-AKAP95 (lanes 2, 5 and 8) or GST-AKAP95 (101–692) (lanes 3, 6 and 9) was incubated with either M2 beads (lanes 4–6) or M2 bead-captured FH-DPY30 (lanes 7–9). Refer to Supplementary Fig. 8D.
(G) ChIP using control IgG or anti-HA for G-293T cells transfected with indicated plasmids in the presence of Gal4-VP16, followed by qPCR at the Gal4-luciferase promoter region. The error bars indicate s.d. from triplicate measurements from one representative experiment (out of three experiments).
We also tested the truncated AKAP95 proteins for binding to DPY30 following co-expression in G-293T cells. AKAP95 (1–210), but not AKAP95 (101–692), was sufficient for binding (Fig. 5E). Thus, the N-terminal region 1–100, which is important for enhancing reporter expression, is also critical for binding to DPY30. Although not as effective in binding to AKAP95 as endogenous DPY30 in HeLa cell nuclear extract, purified DPY30 showed considerable binding to purified AKAP95 and much weaker binding to the purified AKAP95 (101–692), thus demonstrating a direct binding that depends on the N-terminal region of AKAP95 (Fig. 5F). Importantly, truncation of this N-terminal region is unlikely to affect the overall structural integrity of AKAP95, as (i) AKAP95 (387–692), which lacks the more extensive N-terminal region, was reported to retain the full activity in mediating chromatin condensation37 and (ii) in the presence of the activator, recruitment to the reporter gene promoter was somewhat greater for AKAP95 (101–692) than for the less well expressed AKAP95 but comparable to that previously reported23 for DPY30 (Fig. 5C and 5G). The efficient enrichment of AKAP95 (101–692) also suggests that recruitment of AKAP95 to chromatin is independent of its binding to MLL complexes.
AKAP95 enhances the HMT activity of the MLL2 complex
We next tested whether AKAP95 has any direct effect on the HMT activity of MLL complexes. Recombinant Flag-HA-tagged AKAP95 and AKAP95 (101–692) proteins were expressed, purified to near homogeneity (Fig. 6A), and analyzed in the HMT assay on chromatin assembled with recombinant histone octamers and plasmid DNA. AKAP95, but not a comparable amount of AKAP95 (101–692), significantly enhanced MLL2C-mediated H3 methylation on the chromatin substrate (Fig. 6B and Supplementary Fig. 4A). Further immunoblot analyses revealed that H3K4 mono-, di- and tri-methylation were all enhanced by AKAP95 (Fig. 6C). Purified full-length and mutant AKAP95 did not possess methyltransferase activity (Fig. 6D, lanes 5 and 6; also shown in Supplementary Fig. 4B, lane 3) and were not associated with free histone H3 (Fig. 6D, lanes 8 and 9; also shown in Supplementary Fig. 4B, lane 4), a good substrate for MLL2C. These results strongly suggest that AKAP95, likely through binding to DPY30, directly stimulates the enzymatic activity of the MLL2 complex. These data are also consistent with the significant effects of AKAP95 overexpression on H3K4 methylation at the chromosomal reporter locus.
Figure 6. AKAP95 enhances the HMT activity of the MLL2 complex in vitro.

(A) Coomassie blue-stained SDS-PAGE gel for recombinant F-AKAP95 and F-AKAP95 (101–692) proteins after immunoaffinity purification from baculovirus-infected Sf9 cells on M2 beads. Refer to Supplementary Fig. 8E.
(B) Autoradiographic analysis of in vitro HMT activity on chromatin by MLL2C with F-AKAP95 or F-AKAP95 (101–692), all in the presence of [3H]-SAM. CB: coomassie blue staining.
(C) Immunoblot analysis of in vitro HMT activity on chromatin by MLL2C in the absence or presence of F-AKAP95.
(D) Immunoblot analysis of in vitro HMT activity on chromatin in the absence or presence of MLL2C, F-AKAP95, F-AKAP95 (101–692), SAM and chromatin as indicated.
AKAP95 regulates gene expression in ESC differentiation
Given the physical and functional interactions of AKAP95 and DPY30-MLL complexes, we tested if AKAP95 might play a role in the all-trans retinoic acid (ATRA)-mediated induction of developmental genes in human embryonic carcinoma cells (ECCs) and mouse embryonic stem cells (ESCs), as demonstrated by DPY30 (ref. 23). We effectively depleted AKAP95 from NT2 cells, a human ECC line (Fig. 7A), and then treated the cells with ATRA. Several developmental genes, including IGFBP5, HAND1, and Msx1, were strongly induced in control cells upon ATRA treatment, but induction in each case was clearly impaired by AKAP95 depletion (Fig. 7B). These results indicate an important role for AKAP95 in the ATRA-mediated gene induction in human ECCs.
Figure 7. AKAP95 modulates ATRA-mediated gene induction in human ECCs and mouse ESCs.
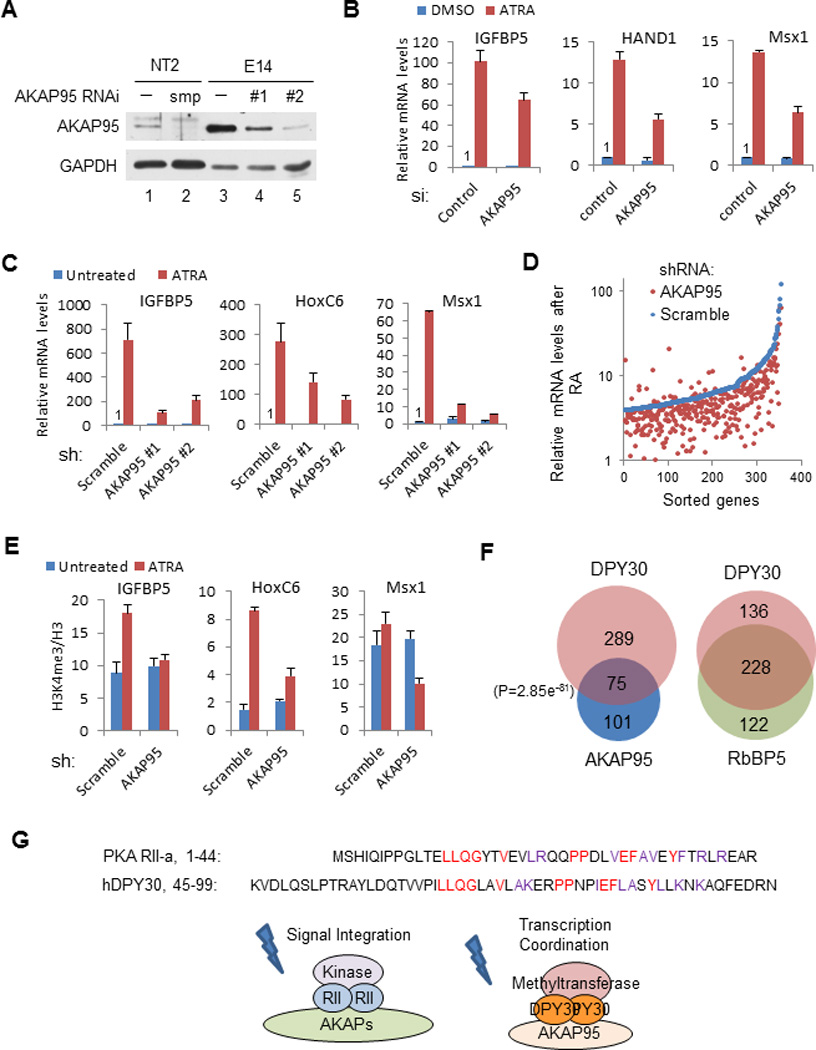
(A) Immunoblot analysis of AKAP95 depletion in cells. NT2 cells were treated with either control (−) or human AKAP95-specific Smartpool siRNA (smp), and E14 cells were infected with viruses expressing scramble shRNA (−) or two different shRNAs against mouse AKAP95 (#1 and #2).
(B) qPCR results on ATRA-mediated induction of developmental genes in control and AKAP95-depleted NT2 cells. Expression levels in DMSO-treated controls are arbitrarily set at 1. For panels B, C and E, the error bars indicate s.d. from triplicate measurements from one representative experiment (out of three experiments).
(C) qPCR results on ATRA-mediated induction of developmental genes in control and AKAP95-depleted E14 cells. Expression levels in untreated controls are arbitrarily set at 1.
(D) Microarray analysis of genes whose expression was normally induced more than 4 fold after ATRA treatment. Genes were sorted according to relative expression levels after ATRA treatment in the control cells. In panels D–F, shRNA #2 was used for AKAP95 knockdown.
(E) ChIP for H3K4me3 followed by qPCR at developmental genes in control and AKAP95-depleted E14 cells.
(F) Venn diagram analyses of genes whose post-ATRA expression levels were reduced over 2-fold by depletion of the indicated proteins in E14 cells. P value was calculated by Fisher’s exact test.
(G) Comparison of AKAP roles in signal transduction and an AKAP95 role in transcription coordination. Top: sequence relationships shared by human PKA RII and DPY30 dimerization domains.
The in vivo function of AKAP95 was further explored in the E14 murine ESC line. Depletion of AKAP95 by two different shRNAs (Fig. 7A) had little effect on ESC morphology (except that the cells appeared to be slightly more aggregated) (Supplementary Fig. 5A), proliferation rate (Supplementary Fig. 5B), or expression levels of many important pluripotency genes (Supplementary Fig. 5C). These results suggest that AKAP95 depletion does not affect self-renewal (and chromatin condensation) of mouse ESCs. AKAP95 depletion also did not affect the global H3K4me3 level, the expression of most genes by microarray analysis (H.J. et al., unpublished), or the recruitment of DPY30 to any highly expressed or bivalent genes that were tested in ESCs (Supplementary Fig. 5D). These results suggest that the co-activation function of AKAP95, similar to that of DPY30-mediated H3K4 methylation23, is obscured by potentially redundant mechanisms in the undifferentiated ESCs. However, after ATRA-mediated differentiation, and as revealed both by qPCR on selected genes (Fig. 7C) and by global expression analysis (Fig. 7D), many genes involved in early development showed strong induction in the control cells but greatly impaired induction in AKAP95-depleted cells. Consistent with a stimulatory effect of AKAP95 on the enzymatic activity of MLL complexes, the ATRA-mediated increase of H3K4me3 on these target gene loci was diminished by AKAP95 knockdown (Fig. 7E).
Further analyses of the microarray results for AKAP95 depletion and DPY30 or RbBP5 depletion23 in ESCs have revealed related as well as divergent relationships between AKAP95 and MLL2C core subunits. As few genes were significantly affected by either depletion before ESC differentiation, effects on the post-ATRA gene expression levels were analyzed. We found a statistically very significant overlap in the number of genes whose post-ATRA expression levels were affected over 2 fold (or 1.5 fold) by AKAP95 and DPY30 depletions, although this overlap was less significant than the overlap indicated for RbBP5 and DPY30 depletions (Fig. 7F and Supplementary Fig. 6). These results suggest that AKAP95, while not an integral core subunit of MLL complexes like DPY30 or RbBP5, is clearly functionally associated with DPY30 in co-regulating a set of common target genes. Gene ontology analyses have revealed that the genes most affected in their post-ATRA expression levels by either DPY30 (Supplementary Fig. 7A) or AKAP95 (Supplementary Fig. 7B) depletion are enriched in developmental processes, especially in neuronal differentiation. These results suggest a common role for these genes in mediating ATRA-dependent neuronal specification of mouse ESCs. While DPY30 and RbBP5 co-dependent genes are highly enriched in transcriptional regulatory functions (Supplementary Fig. 7C), AKAP95-dependent genes and AKAP95 and DPY30 co-dependent genes are highly enriched in extracellular matrix and signaling pathways (Supplementary Fig. 7B and 7D); and the genes co-affected by depletion of AKAP95, DPY30 or RbBP5 are also enriched in both extracellular matrix and in transcription regulation (Supplementary Fig. 7E). These results suggest that AKAP95 and MLL complex core subunits regulate ATRA-mediated differentiation in a coordinated yet differential manner on their targets in ESCs.
DISCUSSION
Starting with the isolation of a highly active MLL2C followed by characterization of its role in chromatin transcription in a cell-free system, we have gone further to identify and to functionally characterize a novel factor associated with DPY30-MLL complexes. Our studies establish a clear causal role for H3K4 methylation by MLL2 in gene activation and, most notably, a newly discovered modulator (AKAP95) of MLL-mediated H3K4 methylation.
MLL2 complex as a robust H3K4 methyltransferase
A catalytically active WDR5-tagged MLL1 complex was previously shown to enhance transcription on a chromatin template12, but it was not shown that this effect was through H3K4 methylation (versus a non-histone substrate) and further experimentation was impeded by the relatively weak H3 methylation activity of the complex. Although difficult to normalize according to the amount of the catalytic subunit, the currently described MLL2C was empirically much more active in our analyses than the isolated MLL1 (ref. 12) and MLL3/4 (ref. 38) complexes in methylating all levels of substrates (H.J. et al., unpublished observations), potentially due either to an enhanced occupancy of the catalytic subunit in the isolated complex or to an intrinsic difference between MLL2 and other MLL family members. As discussed later, the robust activity of the MLL2C may be a key factor in helping to reveal an effect of H3K4 methylation on transcription. Compared to the previously purified WDR5-MLL1 complex that was associated with MOF12, the lack of any detectable HAT in the purified MLL2 complex could result from the different tagging and purification strategies or from MLL1 and MLL2 sequence variations that result in differential binding capabilities.
Direct co-activation of transcription by H3K4 methylation
The strict requirement for SAM and an intact H3K4 residue in the chromatin template for the stimulatory effect of the MLL2C in transcription unambiguously demonstrate a causal role for H3K4 methylation per se in activator-dependent transcription. This conclusion is corroborated by our cell-based studies showing that overexpression of either MLL1 or MLL2 strongly stimulates chromosomal reporter gene expression23. Although a previous report failed to see an effect of H3K4 methylation on transcription of a chromatin template39, this may have reflected either a low efficiency of methylation in the completely defined in vitro system that was employed or, more likely, the absence in this system of transcription-related effector proteins that recognize H3K4 methylation. This contrasts with the present analysis in which the recombinant chromatin template was robustly methylated by the MLL2C and transcribed in a nuclear extract that should contain previously described effectors for H3K4 methyl marks18. Consistent with this interpretation, recent studies have shown effects of an H3K4 tri-methyl analog on transcription in a HeLa nuclear extract-based assay, through CHD1 (ref. 40) and TAF3 (ref. 41) as effectors.
Our robust cell-free H3K4 methylation and transcription system now offers an ideal platform for analysis of factors and mechanisms underlying H3K4 methylation-dependent transcription, including factors that generate or regulate the methylation and factors (effectors) that read the H3K4 methyl marks. This could involve either an unbiased biochemical fractionation and factor purification or systematic analyses of factors already shown to enhance H3K4 methylation or to recognize the H3K4me3 mark. Indeed, our initial results suggest an important role for histone lysine acetylation in MLL2C- and H3K4 methylation-dependent transcription that needs to be further explored. Furthermore, our new findings of an enhancement of MLL2C-mediated H3K4 mono-, di-, and tri-methylation by p300 (Fig. 1E) and synergy between MLL2C and p300 in transcription (Fig. 2C) establish a novel link between enzymes responsible for “writing” two prevalent histone marks associated with gene activation. These results complement those of recent studies showing an enhancement of SET1C-mediated H3K4 methylation and associated p53-dependent transcription by p300 (ref. 42).
Association of AKAP95 with DPY30-MLL complexes
As DPY30 is shared among all MLL complexes, it is possible, but has not been rigorously demonstrated, that AKAP95 may also be commonly associated with all MLL complexes. Our discovery of AKAP95 as a bona fide partner and regulator of DPY30-MLL complexes has provided a novel example of a protein factor, other than the well-established core subunits, that directly modifies the enzymatic activity of the H3K4 methyltransferases. Most of the known AKAPs bind specifically to PKA through the dimerization domain of the RII subunit43. Interestingly, the sequence of the DPY30 dimerization domain shares a striking homology to the RII dimerization domain (Fig. 7G). Hence, it might be expected that a nuclear AKAP would actually associate with DPY30, although the regions on AKAP95 responsible for binding to DPY30 and RII seem to be different. The molecular mechanism by which AKAP95 directly regulates the catalytic activity of the MLL2 complex is not clear and merits further biophysical studies. It will also be interesting to examine whether AKAP95 directly enhances H3K4 methylation mediated by other MLL complexes.
A role for AKAP95 in gene expression
A role for AKAP95 in gene regulation has been established by in vitro and in vivo results in this work. How does AKAP95 enhance chromosomal transcription? Primarily three AKAP95 regions, including the NLS, the two zinc fingers, and the N-terminal region 1–100, have been found to be important for this activity. The NLS requirement suggests that AKAP95 affects transcription in the nucleus. The exact role of AKAP95 zinc fingers in regulating transcription is not clear, but they might facilitate association of AKAP95 with chromatin or transcription-related proteins, similar to their reported roles in mediating chromosome condensation through chromatin binding and condensin recruitment37. The AKAP95 N-terminal region is required for AKAP95-enhanced transcription, and is also critical for AKAP95 binding to DPY30 and for stimulation of the methylation activity of the DPY30-MLL2 complex. Our data, therefore, are consistent with a model in which AKAP95 enhances gene expression through binding to, and modulation of, the activity of DPY30-MLL complexes, but do not exclude other possible mechanisms. The related yet divergent relationship between AKAP95 and MLL complexes is underscored by their impact on ATRA-mediated gene induction in ESCs. Lineage specification for ESCs involves signaling pathways (including extracellular ligand-cellular receptor interactions) eventually transduced to instruct nuclear transcription regulation, and our analyses suggest that AKAP95 and MLL core subunits may preferentially focus on different parts of the system, in addition to co-regulating many common targets, to coordinate the ESC fate transition. Our findings allow us to envision a role for AKAP95 in coordinating transcription by serving as a platform for the docking and integration of H3K4 methyltransferases together with other potential transcription-related factors -- analogous to the function of other AKAPs in organizing kinase and signaling molecules for a spatiotemporal regulation of signal transduction (Fig. 7G). In this regard, it is particularly interesting that the DPY30 dimerization domain bears a striking homology to the otherwise unrelated PKA RII dimerization domain, with each domain mediating an association between an AKAP protein and an enzymatic activity specific for a distinct chemical modification (methylation versus phosphorylation) (Fig. 7G). It thus appears that nature has adopted similar strategies for two important but distinct biological processes.
METHODS
Cloning, mutagenesis, plasmids and expression vectors
Human MLL2 cDNA was cloned on the basis of the KIAA0304 sequence (Kazusa DNA Research Institute, Japan) and inserted into pcDNA5/FRT/TO (pc5, Invitrogen) with an N-terminal Flag-HA tag. cDNA for human AKAP95 was generated by reverse transcription using 293 cells as the source of RNA. AKAP95 mutants were generated by PCR-based deletion or site-directed mutagenesis. Detailed information for some of these mutants is as follows: dNMTS: 1–110 + 141–692; dNLS: 1–287 + 306–692; dZF: 1–389 + 504–692; dRII: 1–571 + 590–692.
Cell lines, complex purifications and co-immunoprecipitation
The FH-MLL2 and FH-DPY30 cell lines were made by co-transfecting Flp-In-293 cells (f293 cells, Invitrogen) with pOG44 plasmid and either an FH-MLL2-pc5 vector or an FH-DPY30-pc5 vector. Nuclear extracts were obtained from these cell lines by a modified Dignam procedure44, incubated with M2 agarose beads in BC300, 0.1% NP40 at 4°C for 6 hr and extensively washed with BC300, 0.1% NP40 followed by BC100, 0.1% NP40. The complexes were eluted with 0.4 mg/ml FLAG peptide in BC100, 0.1% NP40. DPY30-associated proteins were fractionated on Superose 6 (Smart System; Pharmacia) in buffer BC200 containing 0.05% NP40. Co-immunoprecipitation of endogenous protein association was carried out in BC200, 0.1% NP40.
Antibodies
Anti-DPY30 antibody was obtained from C. Hughes (Rockefeller University) and E. McIntush (Bethyl Laboratories). Anti-WDR5 was kindly provided by J. Wysocka (Stanford University). Anti-MLL1 was developed previously in our laboratory2. Anti-MLL2-N was generated using a recombinant epitope in the N-terminal fragment of human MLL2. Anti-CFP1 was generated using a recombinant epitope in human CFP1. Other antibodies were obtained commercially as follows: anti-MLL2-C, ASH2L, Menin, and RbBP5 (Bethyl Laboratories, A300-113A, A300-107A, A300-105A, and A300-109A, respectively); anti-H3K4me2 and H3K4me3 (Upstate Biotechnology, 07–030 and 07–473 respectively); anti-HA [12CA5, for immunoblotting] (Roche, 11583816001); anti-HA [for ChIP assay] (Abcam, ab9110); anti-H3 (Abcam, ab1791); anti-GAPDH (Chemicon, MAB374); anti-FLAG [M2 beads] (Sigma, A2220); anti-MBP (New England BioLabs, E8032S); anti-AKAP95, hSNF2, p68, and myc (Santa Cruz Biotechnology, sc-10766, sc-13054, sc-32858, and sc-40, respectively).
Recombinant proteins and in vitro binding assay
A FLAG-tagged AKAP95 or AKAP95 (101–692) cDNA was inserted into the pFastBac1 vector (Invitrogen) for expression in Sf9 cells. Baculoviruses were generated according to the Bac-to-Bac Baculovirus Expression System (Invitrogen) protocols. Glutathione S-transferase (GST)-tagged AKAP95 or AKAP95 (101–692) was expressed from the pGEX vector in bacteria and purified on glutathione-Sepharose. For the in vitro binding assay, HeLa nuclear extract and purified GST-AKAP95 or GST-AKAP95 (101–692) were incubated with either M2 beads or M2 bead-captured FH-DPY30 protein purified from bacteria in BC200 and 0.1% NP40. Bead-captured proteins were extensively washed with BC200 and 0.1% NP40, resolved by SDS-PAGE and detected by immunoblotting with anti-AKAP95 antibody.
Histone modification and transcription assays
Histone modification assays on non-chromatin substrates were carried out essentially as described12. Chromatin assembly, chromatin-based histone modification assays, and transcription assays were performed as previously described32,45. Chromatin composed of 250 to 350 ng of DNA template and a similar amount of histone octamer was used in each histone methylation assay. When present, about 0.5 µg of AKAP95 or AKAP95 (101–692) was used in each methylation reaction. Chromatin composed of 40 ng of DNA template and a similar amount of histone octamer, 40 ng of p53 and about 100 ng of each coactivator (p300 and/or MLL2-C in the MLL2 complex) were used, along with nuclear extract (50 µg of protein) in each transcription assay. SAM and acetyl-CoA were present in all transcription reactions unless otherwise indicated. Relative transcription levels were quantified by Quantity One software.
RNA Interference
Lentiviral constructs expressing short hairpin RNA (shRNA) sequences (listed in Supplementary Table 1) were purchased from OpenBiosystems. Negative control constructs containing a scrambled shRNA (Addgene plasmid 1864) were purchased from Addgene. Viral particles were produced by following the recommended protocols (Addgene). Two days after infection of ESCs with viruses, puromycin was added at 2 µg/ml to select for stably-infected cells. For siRNA-mediated knockdown in NT2 cells, chemically synthesized siRNAs of the indicated sequences (listed in Supplementary Table 1) were purchased from Dharmacon. An on-target plus non-targeting pool was used as the negative control. NT2 cells were transfected with siRNA duplexes using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
RT-PCR, ChIP, and qPCR
Total RNAs were extracted using the RNeasy kit (Qiagen) and reverse-transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen) with an oligo dT20 primer. Histone or histone modification ChIP assays followed the fast ChIP protocol46, but with the lysis method and washing protocols from the Chromatin Immunoprecipitation Assay Kit (Upstate Biotechnology). ChIP for HA-tagged proteins followed the same protocol except that the lysis method was as follows: cell pellets were first lysed in L1 buffer (50 mM Tris [pH8.0], 2 mM EDTA, 0.1% NP40, 10% glycerol, 1 mM DTT, and protease inhibitors) on ice for 8 min, which was followed by centrifugation at 1,300 × g for 5 min. Collected nuclei were then lysed in IP buffer (ChIP dilution buffer from the Upstate ChIP Assay Kit with additional 0.1% SDS and protease inhibitors). Cell lysates were passed through 27G1/2 needles before sonication. qPCR was performed in triplicate with SYBR Advangtage qPCR Premix (Clontech) on a 7300 Real-Time PCR System (Applied Biosystems). Fold differences in gene expression levels were calculated according to the 2−ΔΔCt method47 and normalized against GAPDH. Levels of histone modifications were presented as the value of 2−ΔCt in which ΔCt is the difference of Ct values of the histone modification immunoprecipitation and the H3 immunoprecipitation for a given primer pair. Primers are listed in Supplementary Table 2.
ES Cell Culture and Gene Induction
The mouse ES cell line E14TG2A (E14) was cultured on 0.1% gelatin-coated tissue-culture plates in complete ES growth medium (knockout DMEM [Invitrogen] supplemented with 15% ESC-certified fetal bovine serum [Omega Scientific], 2 mM L-glutamine, 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, and recombinant LIF) without irradiated murine embryonic fibroblasts. Stably-infected ESCs were maintained in complete ESC growth medium with 2 µg/ml puromycin (Invivogen). To monitor ESC proliferation, cells were subjected to proliferation assays using the Aqueous One Solution Cell Proliferation Assay system (Promega) by following the manufacturer’s protocols. For ATRA-mediated gene induction, ESCs were plated on gelatinized 6-well tissue culture dishes at 30,000 cells per well in complete growth medium. Cells were washed and incubated in differentiation medium for 4 days and then in differentiation medium with 1 µM ATRA for 4 additional days.
Gene Expression Microarray Analysis
Total RNAs were submitted to the Genomics Resource Center at the Rockefeller University for labeling and hybridization to the Illumina 8-sample BeadChip. Signals were normalized against the median in each sample. For expression analysis after ATRA treatment, genes were sorted based on the effect of ATRA treatment in the control samples.
Reporter Assay
The reporter assay was performed as described23. Briefly, the assay system is based on a Gal4-293T cell line (G-293T) that was made by stable transfection of 293T cells with a Gal4 UAS × 5-SV40-firefly luciferase reporter construct described previously48. G-293T cells were plated in a 24-well dish at 35,000 cells per well on day 0 and transfected with 0.2 ng of pGL4.75 [hRluc/CMV] (Promega), 5 ng (unless otherwise indicated) of plasmid expressing Gal4 or Gal4-VP16 and 200 ng (unless otherwise indicated) of control vector or constructs expressing indicated co-activator genes (MLL1, MLL2 or AKAP95) on day 1 using TransIT-LT1 (Mirus Bio LLC). Luciferase assays were performed on day 3 using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. The firefly luciferase light signals were normalized to the Renilla luciferase light signals, which served as an internal control for transfection efficiency. Figures show the mean and standard deviation of duplicate samples in representative experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kazusa DNA Research Institute (Japan) for providing the cDNA KIAA0304. We thank J. Kim [Rockefeller University (RU)] for histone H3 and H4 with mutations in acetylation sites, E. McIntush (Bethyl laboratories) and C. Hughes (RU) for DPY30 antibody, Q. Yang (RU) for multiple valuable reagents, J. Wysocka (Stanford University) for WDR5 antibody, A. Goldberg and D. Allis (RU) for LIF, and Z. Fu and Z. Yan (RU) for excellent technical assistance. H. Jiang was supported by a fellowship from the Leukemia and Lymphoma Society and X. Lu was a recipient of the C. H. Li Memorial Scholar Award. This work was supported by grants from the NIH (CA129325 and DK071900) and the Ellison Medical Foundation (AG-SS-2665-11) to R.G. Roeder and by a Leukemia and Lymphoma Society SCOR grant (7132-08).
Footnotes
Accession codes
The microarray data have been deposited in the GEO database with the accession code GSE48128.
AUTHOR CONTRIBUTIONS
H. J. and X. L. conceived the project, designed and performed the experiments, analyzed the data and wrote the paper. M. S. and Y. D. performed experiments. Z.T. generated the K4Q mutant octamer. R.G.R conceived the project, analyzed the data, wrote the paper, supervised the project and had overall responsibility for the joint research.
References
- 1.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 4.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 6.Shilatifard A. The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos-Rosa H, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 8.Schneider R, et al. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein BE, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Dou Y, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 12.Dou Y, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 13.Hughes CM, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 14.Milne TA, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 15.Wysocka J, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 16.Demers C, et al. Activator-mediated recruitment of the MLL2 methyltransferase complex to the beta-globin locus. Mol Cell. 2007;27:573–584. doi: 10.1016/j.molcel.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vermeulen M, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 18.Vermeulen M, Timmers HT. Grasping trimethylation of histone H3 at lysine 4. Epigenomics. 2010;2:395–406. doi: 10.2217/epi.10.11. [DOI] [PubMed] [Google Scholar]
- 19.Lim DA, et al. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature. 2009;458:529–533. doi: 10.1038/nature07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rathert P, Dhayalan A, Ma H, Jeltsch A. Specificity of protein lysine methyltransferases and methods for detection of lysine methylation of non-histone proteins. Mol Biosyst. 2008;4:1186–1190. doi: 10.1039/b811673c. [DOI] [PubMed] [Google Scholar]
- 21.Zhang K, et al. The Set1 methyltransferase opposes Ipl1 aurora kinase functions in chromosome segregation. Cell. 2005;122:723–734. doi: 10.1016/j.cell.2005.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho YW, et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang H, et al. Role for Dpy-30 in ES Cell-Fate Specification by Regulation of H3K4 Methylation within Bivalent Domains. Cell. 2011;144:513–525. doi: 10.1016/j.cell.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 25.Jungmann RA, Kiryukhina O. Cyclic AMP and AKAP-mediated targeting of protein kinase A regulates lactate dehydrogenase subunit A mRNA stability. J Biol Chem. 2005;280:25170–25177. doi: 10.1074/jbc.M502514200. [DOI] [PubMed] [Google Scholar]
- 26.Eide T, et al. Protein kinase A-anchoring protein AKAP95 interacts with MCM2, a regulator of DNA replication. J Biol Chem. 2003;278:26750–26756. doi: 10.1074/jbc.M300765200. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, et al. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006;20:2566–2579. doi: 10.1101/gad.1455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collas P, Le Guellec K, Tasken K. The A-kinase-anchoring protein AKAP95 is a multivalent protein with a key role in chromatin condensation at mitosis. J Cell Biol. 1999;147:1167–1180. doi: 10.1083/jcb.147.6.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bomar J, Moreira P, Balise JJ, Collas P. Differential regulation of maternal and paternal chromosome condensation in mitotic zygotes. J Cell Sci. 2002;115:2931–2940. doi: 10.1242/jcs.115.14.2931. [DOI] [PubMed] [Google Scholar]
- 30.Akileswaran L, Taraska JW, Sayer JA, Gettemy JM, Coghlan VM. A-kinase-anchoring protein AKAP95 is targeted to the nuclear matrix and associates with p68 RNA helicase. J Biol Chem. 2001;276:17448–17454. doi: 10.1074/jbc.M101171200. [DOI] [PubMed] [Google Scholar]
- 31.van den Berg DL, et al. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell. 2010;6:369–381. doi: 10.1016/j.stem.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.An W, Roeder RG. Reconstitution and transcriptional analysis of chromatin in vitro. Methods Enzymol. 2004;377:460–474. doi: 10.1016/S0076-6879(03)77030-X. [DOI] [PubMed] [Google Scholar]
- 33.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 34.An W, Palhan VB, Karymov MA, Leuba SH, Roeder RG. Selective requirements for histone H3 and H4 N termini in p300-dependent transcriptional activation from chromatin. Mol Cell. 2002;9:811–821. doi: 10.1016/s1097-2765(02)00497-5. [DOI] [PubMed] [Google Scholar]
- 35.Tsukiyama T, Wu C. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell. 1995;83:1011–1020. doi: 10.1016/0092-8674(95)90216-3. [DOI] [PubMed] [Google Scholar]
- 36.Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- 37.Eide T, et al. Distinct but overlapping domains of AKAP95 are implicated in chromosome condensation and condensin targeting. EMBO Rep. 2002;3:426–432. doi: 10.1093/embo-reports/kvf089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goo YH, et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol. 2003;23:140–149. doi: 10.1128/MCB.23.1.140-149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavri R, et al. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125:703–717. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 40.Lin JJ, et al. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev. 2011;25:2198–2209. doi: 10.1101/gad.17554711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lauberth SM, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Z, et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell. 2013;154:297–310. doi: 10.1016/j.cell.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for Methods
- 44.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guermah M, Kim J, Roeder RG. Transcription of in vitro assembled chromatin templates in a highly purified RNA polymerase II system. Methods. 2009;48:353–360. doi: 10.1016/j.ymeth.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Kalkum M, Chait BT, Roeder RG. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9:611–623. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.