

Abstract
Malformations are significant contributions to childhood mortality and disability. Their co-occurrence with intellectual disability may compound the health burden, requiring additional evaluation and management measures. Overall, malformations of greater or lesser severity occur in at least some cases of almost half of the 153 XLID syndromes. Genitourinary abnormalities are most common, but tend to contribute little or no health burden and occur in only a minority of cases of a given XLID syndrome. Some malformations (e.g., lissencephaly, hydranencephaly, long bone deficiency, renal agenesis/dysplasia) are not amenable to medical or surgical intervention; others (e.g., hydrocephaly, facial clefting, cardiac malformations, hypospadias) may be substantially corrected.
Keywords: Malformations, Birth Defects, X Chromosome, X-Linked Intellectual Disability
INTRODUCTION
Malformations or other structural anomalies occur in a number of the 153 X-linked intellectual disability (XLID) syndromes, and in a few instances they contribute to mortality or significant morbidity. Individually and in recognizable patterns, malformations provide substantive clues to diagnosis of several XLID syndromes (Table I). This is of particular importance in families with a single or few affected members.
Table I.
Malformations indicative of specific XLID syndromes
| Periventricular heterotopia | FLNA-associated XLID |
| Lissencephaly |
ARX-associated XLID CK syndrome X-linked lissencephaly |
| Hydranencephaly |
ARX-associated XLID Hydrocephaly-MASA spectrum |
| Retinal lacunae | Aicardi syndrome |
| Cleft lip | XLID cleft lip/cleft palate Telecanthus-hypospadias |
| Tracheoesophageal fistula/esophageal atresia | VACTERL-hydrocephaly |
| Imperforate anus | VACTERL-hydrocephaly Opitz FG syndrome |
| Renal agenesis | Lenz microphthalmia |
| Cystic kidneys | Oral-Facial-Digital syndrome I Simpson-Golabi-Behmel |
| Ambiguous genitalia |
ARX-associated XLID ATRX-associated XLID |
| Radial aplasia | VACTERL-hydrocephaly |
| Radioulnar synostosis | Guiffre-Tsukahara |
| Vertebral segmentation | VACTERL-hydrocephaly Christian syndrome |
Hydrocephalus, facial clefting, cardiac malformations and genitourinary anomalies are among those structural abnormalities most likely to require medical and surgical management. Malformations of the central nervous system are exceptional among the XLID syndromes and only infrequently explain the cognitive impairment or neurological manifestations.
This survey seeks to identify the structural anomalies that occur with some consistency among the XLID syndromes with emphasis on those that contribute to diagnosis and management.
MALFORMATIONS OF THE CENTRAL NERVOUS SYSTEM
Hydrocephaly (or hydranencephaly), neuronal migration abnormalities, agenesis of the corpus callosum, and cerebellar aplasia or hypoplasia are the most common and most severe of the structural anomaliesin the XLID syndromes.
Hydrocephaly
Hydrocephaly-MASA Spectrum is the prototypic XLID syndrome with a structural alteration of the central nervous system [Schrander-Stumpel et al., 1995]. Identification of mutations in L1CAM has permitted inclusion of four entities – X-Linked hydrocephaly, MASA syndrome, XLID-clasp thumb, and complicated spastic paraplegia – previously considered to be separate entities in the spectrum [Jouet et al., 1993; Vits et al., 1994; Shrander-Stumpel, 1995]. Hydrocephaly, dysgenesis of the corpus callosum, adducted thumb, unsteady gait, spastic paraplegia, and intellectual disability comprise the usual clinical findings. Hypotonia is present initially, but is replaced by spasticity by late childhood or adolescence. Hydrocephaly occurs secondary to stenosis of the aqueduct of Sylvius. The degree of intellectual development appears to correlate with the severity of hydrocephaly. Truncating mutations and mutations affecting the immunoglobulin or fibronectin domains are associated with more severe phenotypic expression and mortality [Michaelis et al., 1998; Vos et al., 2010].
VACTERL-hydrocephaly occurs as autosomal and X-linked malformation syndromes [Hilger et al., 2012]. Hydrocephaly in the company of vertebral, anorectal, tracheoesophageal, renal, and limb malformations comprise the fully-expressed phenotype. Multiple vertebral anomalies, radial ray defects, imperforate anus, and tracheoesophageal fistulas with or without esophageal atresia are typical. Infant lethality is the rule. The X-linked type accounts for a minority of cases and mutations in FANCB occur in a minority of this type.
Hydranencephaly with abnormal genitalia represents the most severe end of ARX-associated XLID [Shoubridge et al., 2010]. Hydranencephaly is accompanied by abnormalities of neuronal migration, poor development of the male genitalia, hypotonia, and seizures. Microphallus, hypospadias, undescended testes and ambiguous genitalia have been described. Death typically occurs during infancy. Mild expression has been noted in female carriers.
AP1S2-associated XLID encompasses conditions described under three different names – X-Linked hydrocephaly with basal ganglia calcifications (Fried syndrome), Turner XLID syndrome, and MRX59 [Fried, 1972; Turner et al., 2003; Saillour et al., 2007]. Head size is variable and hydrocephaly and calcifications in the basal ganglia are not present in all cases. Hypotonia typically is present and aggressive behavior has been reported in several families.
Hydrocephaly-Cerebellar Agenesis has been afforded XLID syndrome status, but only one family has been reported [Riccardi and Marcus, 1978]. In addition to the CNS anomalies, hypotonia, areflexia or hyporeflexia, and seizures occur. All infants have died in the neonatal period. Focal glomerulosclerosis and visceral hemosiderin deposits have been noted at postmortem. The gene locus is not known and allelism with another of the XLID-hydrocephaly syndromes is possible.
In addition to these syndromes in which hydrocephaly is a defining malformation, seven other XLID syndromes include hydrocephaly or hydranencephaly as a finding, at least in some cases (Table II).
Table II.
XLID Syndromes with Central Nervous System Malformations
| Syndrome | Hydrocephaly/Hydranencephaly | Neuronal Migration Abnormality | Dysgenesis of the Corpus Callosum | Cerebellar Agenesis/Hypoplasia |
|---|---|---|---|---|
| Hydrocephalus-MASA Spectrum | + | + | ||
| VACTERL-Hydrocephaly | + | |||
| Hydranencephaly with Abnormal Genitalia (ARX-Associated XLID) | + | + | ||
| Hydrocephaly-Cerebellar Agenesis | + | + | ||
| Agenesis of the Corpus Callosum | + | |||
| Aicardi | + | + | + | |
| AP1S2-Associated XLID | + | |||
| Armfield | + | + | ||
| Bertini | + | + | ||
| Cerebro-Cerebello-Coloboma | + | + | ||
| CK | + | |||
| Christianson | + | |||
| FLNA-Associated XLID | + | |||
| Gustavson | + | + | ||
| Juberg-Marsidi-Brooks | + | + | ||
| Kang | + | + | + | |
| Lissencephaly, X-Linked | + | + | + | |
| Lissencephaly with Abnormal Genitalia (ARX-Associated XLID) | + | + | ||
| Oral-Facial-Digital I | + | + | ||
| Pettigrew | + | |||
| Schimke | + |
Abnormalities of Neuronal Migration
Disturbance in neuronal migration in the form of lissencephaly, cortical dysplasia or heterotopia occurs in seven XLID syndromes. Detection, which requires CNS imaging, provides an important diagnostic finding.
Lissencephaly, X-Linked presents as a central nervous system phenotype without malformations in other systems [Gleeson et al., 1998]. Frontal pachygyria is typical in males, but agyria, polymicrogyria, hypoplasia of the cerebellar vermis, dilated ventricles and dysgenesis of the corpus callosum may also occur. The brain is small and associated with hypotonia, spasticity, seizures and underdevelopment of the genitalia. Subcortical band heterotopia is typical in females and may be associated with mild-moderate intellectual disability, behavioral problems and seizures. Mutations in DCX are responsible [Gleeson et al., 1998; Sossay-Alaoui et al., 1998].
Lissencephaly with Abnormal Genitalia is one of a spectrum of XLID syndromes caused by mutations in ARX [Kitamura et al., 2002; Shoubridge et al., 2010]. The posterior cerebral cortex is more severely affected. Small overall brain size, dilated ventricles, and absent corpus callosum accompany the thickened smooth cortex. The abnormal genitalia may present as microphallus, undescended testes and hypospadias or be completely undifferentiated. Death usually occurs in infancy. Carrier may have dysgenesis of the corpus callosum, seizures and learning disabilities.
FLNA-Associated XLID syndromes present with variably severe skeletal findings and intellectual disability, if present, tends to be mild [Robertson, 2005]. Periventricular Nodular Heterotopia is the exception. This XLID syndrome primarily affects females, skeletal manifestations are absent, and seizures dominate the clinical picture. Truncating mutations of FLNA are generally found. Although less commonly identified in males, similar MRI findings and clinical course have been reported.
Disturbances of neuronal migration also occur in Aicardi syndrome (cortical dysplasia plus agenesis of the corpus callosum and cerebellar hypoplasia), CK syndrome (pachygyria and polymicrogyria), Kang syndrome (gyral dysplasia plus agenesis of the corpus callosum and hydrocephaly) and hydranencephaly with abnormal genitalia (thin cortex with simplified gyral pattern plus hydranencephaly) [Stevenson et al., 2012].
Dysgenesis of the Corpus Callosum
Dysgenesis of the corpus callosum is the most common structural anomaly of the central nervous system among all individuals and competes with hydrocephaly as the most common among the XLID syndromes (Table I). The impact of this anomaly on cognitive ability is debated. Certainly, dysgenesis or agenesis of the corpus callosum may be found incidentally among persons who appear to have normal cognitive and neurological function. It is found rather consistently in some XLID syndromes (e.g., Aicardi and Opitz FG syndromes) and less consistently in many others (Table II). In neither circumstance does it cause distinguishing manifestations in the neurological examination or neuropsychological testing profile.
Cerebellar Agenesis
Underdevelopment or absence of part or all of the cerebellum is a characteristic of several XLID syndromes (Table II). Neurological signs in the form of hypotonia, ataxia, choreoathetosis, or spasticity result. Other structural abnormalities of the nervous system are invariably present and may contribute to the neurological phenotype.
OCULAR MALFORMATIONS
Anophthalmia/microphthalmia and coloboma are the ocular malformations of note in the XLID syndromes [Stevenson et al., 2012]. Absence of the eyes or extreme microphthalmia occurs in Graham anophthalmia syndrome; less severe degrees of microphthalmia occur in Aicardi, cerebro-oculo-genital, Chassaing-Lacombe chondrodysplasia, Goltz, incontinentiapigmenti, Lenz, MIDAS, Nance-Horan, and VACTERL-hydrocephaly syndromes. Abnormalities of the anterior chamber, iris or retinal colobomas, and cataracts may accompany the microphthalmia. Retinal colobomas or lacunae occur in Aicardi and cerebro-cerebello-coloboma syndromes; iris colobomas are more typical in the other XLID syndromes with ocular anomalies.
FACIAL CLEFTING
Facial clefting has been reported in 16 XLID syndromes, although it is a consistent finding in only half of these. Cleft lip is less frequent than cleft palate (Table III). In almost all cases, malformations affecting other systems will be present (Fig 1).
Table III.
Orofacial Clefts in the XLID Syndromes
| Syndrome | Cleft Lip | Cleft Palate | ||
|---|---|---|---|---|
| Occasional | Frequent | Occasional | Frequent | |
| Armfield | + | |||
| ATRX | + | |||
| Goltz | + | + | ||
| Hall Orofacial | + | + | ||
| Lenz Microphthalmia | + | + | ||
| Oral-Facial-Digital I | P | + | ||
| Otopalatodigital I | + | |||
| Otopalatodigital II | + | |||
| Pallister W | P | + | ||
| Renpenning | + | |||
| Simpson-Golabi-Behmel | + | |||
| Snyder-Robinson | + | |||
| TARP | + | |||
| Telecanthus-Hypospadias | + | + | ||
| VACTERL-Hydrocephaly | + | |||
| XLID-Cleft Lip/Palate | + | + | ||
P=pseudocleft or incomplete cleft
Figure 1.
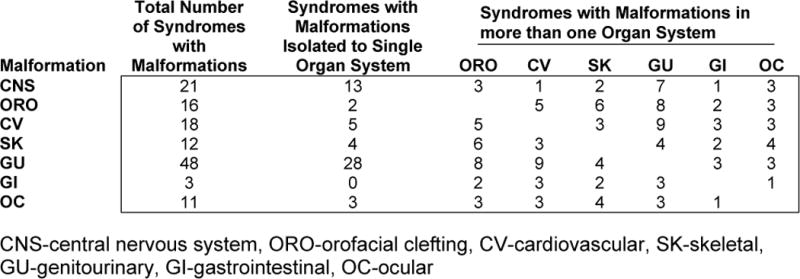
Numbers of XLID syndromes with malformations in a single organ system and in multiple organ systems.
Otopalatodigital syndrome I and Otopalatodigital syndrome II are caused by mutations in FLNA [Robertson et al., 2003]. OPD I is less severe in both physical and functional characteristics. Craniofacial findings include prominent occiput, frontal prominence with overhanging brow, broad and depressed nasal root, hypertelorism, downslanting palpebral fissures, small mouth and chin, and cleft palate. The thumbs and great toes are short and broad and the other digits blunted with bulbous tips and short nails. Other skeletal findings include irregular curvature and spacing of the digits, limited elbow movement, bowed long bones, pectus excavatum, short trunk and short stature. Conductive hearing loss accompanies these physical findings. Intellectual disability tends to be mild. Carrier females may have many of the same craniofacial and digital manifestations.
OPD II is more severe in all phenotypic aspects and may be stillborn or lethal during the first year or so. The face shows greater prominence of the forehead, flattening of the midface and nasal root, hypertelorism, small mouth, micrognathia and cleft palate. Skeletal manifestations are likewise more pronounced with short stature, underossification of the cranium, sclerotic and bowed long bones, flat vertebrae, rockerbottom feet, and irregular spacing and flexion of the blunted digits. The great toes and fibulas may be absent. Mixed hearing loss accompanies these physical findings. Many of the same findings, albeit less pronounced, may be seen in carrier females.
Oral-Facial-Digital Syndrome I is found almost exclusively in females. The facial findings include dystopia canthorum, flattened midface with underdevelopment of the alar cartilages, milia, and pseudocleft of the lip [Thauvin-Robinet et al., 2006]. Oral manifestations are more notable with lobation and hamartomas of the tongue, unusual hypertrophied frenula, hypodontia, malocclusion, and highly arched or irregularly cleft palate. The digits are short with proximal syndactyly and clinodactyly, especially fingers 2 and 5. Hydrocephaly, porencephaly, agenesis of the corpus callosum, adult-type polycystic kidney, alopecia or dry sparse scalp hair may occur. The syndrome is due to mutations in CXORF5.
Telecanthus-Hypospadias syndrome (Opitz BBB/G syndrome) is caused by mutations in MID1 [Quaderi et al., 1997]. The face is characterized by telecanthus, epicanthus, high broad nasal root, dysplastic or posteriorly-rotated ears and cleft lip [Stevens and Wilroy, 1988]. The forehead may be narrow with metopic ridge, and the cranium small, abnormally configured, or asymmetric. Cleft palate or uvula and other anomalies of the pharynx, larynx and trachea occur. A variety of cardiac defects have been reported, but only in a minority. Hypospadias occurs in most males and hypoplastic or cleft scrotum, undescended testes, inguinal hernia, and imperforate anus less frequently. Skeletal defects are uncommon, but short stature occurs in one-third of cases. Cognitive deficits tend to be mild. Carrier females may have telecanthus, but lack other malformations.
XLID-cleft lip/cleft palate (Siderius syndrome) typically has cleft lip and cleft palate [Siderius et al., 1999]. The face tends to be long, the nose broad, the ears cupped and the hands large. Synophrys may be present. Short attention span occurs. Behavior may be unpredictable with bouts of aggression. Mutations have been found in PHF8, a histone deacetylase [Laumonnier et al., 2005].
CARDIAC MALFORMATIONS
Structural anomalies of the cardiovascular system occur in several XLID syndromes; only in TARP syndrome do they appear to be a defining manifestation [Johnston et al., 2010]. Septal defects, valvular stenosis or incompetence and anomalies of major vessels rather than life-threatening complex cardiac malformations are typical. Septal defects are most common, occurring at least occasionally in nine XLID syndromes (Table IV). Tetralogy of Fallot, hypoplastic left heart, and other complex cardiac malformations have been reported in individual cases of alpha-thalassemia intellectual disability, Renpenning syndrome and Opitz FG syndrome. Cardiomyopathy occurs in five XLID syndromes and arrhythmias with or without cardiomyopathy in seven (Table III). Malformations in other systems commonly accompany the cardiovascular anomalies (Fig 1).
Table IV.
Cardiovascular Abnormalities Reported in XLID Syndromes
| Syndrome | ASD | VSD | Other Malformations | Valve Disease | Major Vessel Anomaly | Cardio-myopathy | Arrhyth-mias |
|---|---|---|---|---|---|---|---|
| TARP | + | + | + | ||||
| MIDAS | + | + | + | + | + | ||
| Bergia Cardiomyopathy | + | + | |||||
| Renpenning | + | + | T/F, other complex defects | + | |||
| Craniofacioskeletal (males) | + | + | + | ||||
| Myotubular Myopathy | + | + | |||||
| ATRX | + | + | T/F, D | + | TGV | ||
| Creatine Transporter Deficiency | + | + | |||||
| Duchenne Muscular Dystrophy | + | + | |||||
| Hereditary Bullous Dystrophy | + | ||||||
| Lenz Microphthalmia | + | C | |||||
| Lujan | + | ||||||
| Opitz FG | + | Hypoplastic left heart | C | ||||
| Simpson-Golabi-Behmel | |||||||
| Telecanthus-Hypospadias | + | + | + | C, + | |||
| Mucopolysaccharidosis II | + | ||||||
| n-Alpha-Acetyl Transferase Deficiency | + | ||||||
| VACTERL-Hydrocephalus | + | + | C |
C=coarctation of the aorta, T/F=Tetralogy of Fallot, TGV=Transposition of Great Vessels, D=Dextrocardia
TARP syndrome is defined by four key findings – talipes equinovarus, atrial septal defect, Robin sequence, and persistence of the left superior vena cava [Gripp et al., 2011]. Structural anomalies are, however, more widespread involving most systems. The face is distinctive with small head, upslanted palpebral fissures, low-set posteriorly-rotated ears, small jaw and cleft palate. Dysgenesis of the corpus callosum, cerebellar dysgenesis, mega cisterna magna, optic atrophy and hearing loss affect the nervous system. Cryptorchidism occurs in a majority of cases. Arrhythmias occur and contribute to early demise, generally in infancy. Mutations in RBM10, which encodes an RNA binding motif protein, are causative.
GENITOURINARY MALFORMATIONS
Almost one-third of the XLID syndromes will have genitourinary anomalies at least in a minority of cases (Table V). In many instances, the anomalies are trivial without associated morbidity; in a few instances the anomalies require medical or surgical management. Incompletely differentiated genitalia represents the most severe end of the spectrum of genital anomalies, shawl scrotum and undescended testes the least severe. Hydronephrosis is the most severe co-occurring urinary system anomaly.
Table V.
Abnormalities of the Genitourinary System in XLID Syndromes
| Anomalies of Genitalia | Anomalies of Urinary System | ||||||
|---|---|---|---|---|---|---|---|
| Syndrome | Undifferentiated | Hypospadias | Minor Anomalies | Macroorchidism/ Microorchidism | Renal Dysplasia | Hydro-nephrosis | Renal Agenesis |
| Aarskog | S, UT | ||||||
| Ahmad | MP | Mi | |||||
| Alpha-Thalassemia Intellectual Disability | + | + | UT, MP | Mi | |||
| Armfield | UT | ||||||
| ARX-Associated XLID | + | + | MP, UT | ||||
| Atkin-Flaitz | Ma | ||||||
| Börjesson-Forssman-Lehman | MP | Mi | |||||
| Branchial Arch | UT | ||||||
| Cerebro-Oculo-Genital | + | MP, UT | |||||
| Cornelia de Lange | Mi | ||||||
| Craniofacioskeletal | + | UT | + | ||||
| Dyskeratosis Congenita | MP | Mi | |||||
| Fragile X | Ma | ||||||
| Hall Orofacial | UT | + | |||||
| Hereditary Bullous Dystrophy | UT | Mi | |||||
| Juberg-Marsidi-Brooks | MP | Mi | |||||
| Lenz Microphthalmia | + | UT | + | + | |||
| Lissencephaly, X-Linked | MP | Mi | |||||
| Lujan | Ma | ||||||
| Martin-Probst | MP | Mi | |||||
| MEHMO | MP, UT | ||||||
| Myotubular Myopathy | UT | ||||||
| Opitz FG | + | UT | |||||
| Oral-Facial-Digital I | + | ||||||
| Pettigrew | Mi | ||||||
| PPMX | Ma | ||||||
| Prieto | UT | ||||||
| Renpenning | + | Mi | Horseshoe kidney | ||||
| Shashi | Ma | ||||||
| Simpson-Golabi-Behmel | UT | + | + | ||||
| Smith-Fineman-Myers | UT | ||||||
| Telecanthus-Hypospadias | + | UT, S | |||||
| Urban | MP, UT | Ma | |||||
| VACTERL-Hydrocephalus | MP | Mi | Horseshoe kidney | ||||
| Vasquez | MP | Mi | |||||
| Wilson-Turner | MP, UT | Mi | |||||
| Wittwer | E | UT | + | ||||
| XLID-Cleft Lip/Palate | Mi | ||||||
| XLID-Hypogamma-globulinemia | UT | ||||||
| XLID-Hypogonadism-Tremor | Mi | ||||||
| XLID-Hypospadias | + | ||||||
| XLID-Ichthyosis-Hypogonadism | + | MP, UT | Mi | ||||
| XLID-Macrocephaly-Macroorchidism | Ma | ||||||
| XLID-Microcephaly-Testicular Failure | + | UT | Mi | ||||
| XLID-Panhypopituitarism | MP | Mi | |||||
| XLID-Precocious Puberty | Ma | ||||||
| XLID-Psoriasis | UT | ||||||
| Young-Hughes | MP | Mi | |||||
E=epispadias, S=shawl scrotum or cleft scrotum, MP=microphallus, UT=undescended testes, Mi=small testes, Ma=macroorchidism
Lissencephaly with abnormal genitalia and hydranencephaly with abnormal genitalia are allelic conditions caused by mutations in ARX [Kitamura et al., 2002; Shoubridge et al., 2010]. The genitalia may be undifferentiated or be less severely affected with microphallus, hypospadias and undescended testes. Microcephaly generally accompanies the brain malformations. Profound developmental failure, hypotonia and seizures dominate the clinical presentation and death occurs in the neonatal period or later in infancy.
Alpha-thalassemia intellectual disability commonly has some form of genital abnormality [Gibbons et al., 2008]. Undifferentiated genitalia is the most severe but least common anomaly. Genital hypoplasia, hypospadias and undescended testes are said to occur in most cases. This may represent a biased view since males with more typical phenotype are more likely to be diagnosed. For example, only 20% of males with the p.R37X mutation in ATRX have genital anomalies [Basehore et al., unpublished].
GASTROINTESTINAL MALFORMATIONS
Gastrointestinal malformations are rarely found among the XLID syndromes. Tracheoesophageal fistula and esophageal atresia are sentinel findings in VACTERL-Hydrocephaly syndrome (Table I). Imperforate anus or other anorectal malformations occur in VACTERL-Hydrocephaly, Opitz FG and Telecanthus-Hypospadias syndromes.
SKELETAL MALFORMATIONS AND DYSPLASIAS
Malformations involving the chondroosseous structures are distinctly rare among the XLID syndromes. The skeletal features can be divided into two main classifications: 1) anatomical, defined as primary malformations and 2) functional, defined as secondary and include most cases of joint contractures and other positional deformities. These functional anomalies are typically secondary to poor fetal or postnatal movement and sometimes accompanying primary central nervous system malformations which lead to spasticity, contractures, and even scoliosis. The primary anatomic abnormalities, which provide diagnostic clues to the specific syndrome and its etiology, can be further divided into generalized skeletal dysplasias and specific focal skeletal malformations (Tables VI and VII).
Table VI.
XLID Syndromes with Skeletal Malformations
| Syndrome (gene) | Long bones | Digits | Associated features |
|---|---|---|---|
| Giuffre-Tsukahara | Radioulnar synostosis | Fifth finger clinodactyly | Microcephaly in males and females |
| Goltz (PORCN) | Asymmetry, longitudinal striations of metaphyses | Reduction defects with missing digital rays | Dermal hypoplasia, ectodermal dysplasia, cleft lip/palate, renal anomalies |
| Lenz Microphthalmia (BCOR) | – | Duplicated and/or hypoplastic thumbs, syndactyly, camptodactyly | Microphthalmia and anterior chamber defects, cleft lip/palate, urogenital anomalies |
| Opitz FG (MED12) | – | Broad flat thumbs, rarely duplicated | Macrocephaly, dysplastic corpus callosum, cardiac defects, imperforate anus, constipation, hypotonia |
| Oral-Facial-Digital (OFD1) | – | Asymmetrically shortened fingers, syndactyly | Cleft lip, lingual hamartomas, accessory frenulae, CNS anomalies |
| Otopalatodigital II (FLNA) | Fibular deficiency, bowing of long bones | Short digits, broad thumbs and great toes, variable absence of thumbs, polydactyly and syndactyly | Prominent forehead, hypertelorism, flat midface, downslanting palpebral fissures, small mouth, micrognathia, cleft palate, spondylobrachydactyly, poor calvarial ossification, abnormally formed carpals and tarsals, narrow thorax |
| VACTERL-Hydrocephaly (FANCB) | Radial deficiency | Absent thumbs | Early lethality, hydrocephalus, vertebral, tracheoesphageal, cardiac, renal, and anal anomalies |
Table VII.
XLID Syndromes with Generalized Skeletal Dysplasias
| Syndrome (gene) | Type of Skeletal Dysplasia | Other Skeletal Features | Associated Features |
|---|---|---|---|
| Chassaing-Lacombe Chondrodysplasia (HDAC6) | Spondylometaphyseal | Platyspondyly, metaphyseal flaring, 11 ribs, poor mineralization, brachydactyly | Hydrocephaly, microphthalmia, male lethality, females affected |
| Christian | Spondylobrachydactyly | Prominent metopic ridge, hemivertebrae, scoliosis, sacral hypoplasia, brachydactyly with short middle phalanges | Abducens palsy, downslanting palpebral fissures, glucose intolerance |
| Otopalatodigital I (FLNA) | Spondylobrachydactyly | Short trunk with pectus excavatum, brachydactyly, short and broad thumbs and halluces, abnormal carpals and tarsals | Pugilistic facies, downslanting palpebral fissures, cleft palate, large anterior fontanel with delayed closure |
| Otopalatodigital II (FLNA) | Spondylobrachydactyly (more severe, but similar skeletal findings to those seen in OPD1) | Short trunk with pectus excavatum, brachydactyly, short and broad thumbs and halluces, abnormal carpals and tarsals, bowed long bones, hypoplastic/absent fibulae | Prominent forehead, downslanting palpebral fissures, large anterior fontanel with delayed closure, hypertelorism, small mouth, cleft palate, micrognathia |
| Roifman | Spondyloepiphyseal | Wavy and irregular vertebral endplates, generalized epiphyseal changes, short tapered digits | Microcephaly, downslanting palpebral fissures, long philtrum, thin upper lip, pigmentary retinopathy, immune deficiency with frequent infections |
| XLID-SEMD | Spondyloepimetaphyseal | Progressive dysplasia of spine, metaphyses, and epiphyses, chest deformation | Coarse facies, progressive mental deterioration |
Long bone deficiencies occur only in VACTERL-hydrocephaly (radial aplasia) and otopalatodigitalsyndrome II (fibular aplasia) [Robertson et al., 2003; Stevenson and Hunter, 2013]. Oligodactyly occurs in VACTERL-Hydrocephaly (absent thumbs) and Goltz syndrome (absence of any digits). Polydactyly occurs in Lenz Microphthalmia and rarely in Opitz FG, otopalatodigital II and Simpson-Golabi-Behmel syndromes. Some cases of Aarskog, oral-facial-digital II, and otopalatodigital II syndromes have incomplete cutaneous syndactyly. Brachydactyly of greatest severity is present in oral-facial-digital syndrome I. Less severe shortening of the digits occurs in alpha-thalassemia intellectual disability, Aarskog, Atkin-Flaitz, Simpson-Golabi-Behmel, hereditary bullous dystrophy, and the Otopalatodigital syndromes [Stevenson et al., 2012]. Broad thumbs typically occur in Opitz FG and otopalatodigital I syndromes. Carpal and tarsal coalitions may be seen in otopalatodigitalsyndrome II and radioulnarsynostosis in Giuffre-Tsukahara syndrome. Abnormalities of vertebral segmentation are usually present in VACTERL-hydrocephaly and Christian syndrome; less commonly in Aicardi and Goltz syndromes. Most of these syndromes are readily diagnosed on the basis of clinical findings (Table VI). The clinical diagnosis may be confirmed in most cases with molecular testing.
The X-linked skeletal dysplasias with intellectual disability include Chassaing-Lacombe chondrodysplasia, FLNA-Associated XLID (otopalatodigital Syndromes I and II), Roifman syndrome and XLID-spondyloepimetaphyseal dysplasia (Table VII). Chassaing-Lacombe chondrodysplasia is lethal in males as are most cases of otopalatodigital syndrome II. The nonskeletal findings are useful in differentiating the few XLID syndromes with chondroosseous dysplasias (Table VI). The responsible genes are known for Chassaing-Lacombe Chondrodysplasia (HDAC6) and the Otopalatodigital Syndromes (FLNA) [Stevenson et al., 2012; Robertson et al., 2003].
SUMMARY
Major malformations occur in 2–5% of all infants and comprise the most common cause of death during infancy. The prevalence of malformations of varying severity among the XLID syndromes exceeds this rate, being found at least in some cases of almost half (72) of the 153 XLID syndromes. Malformations of the central nervous system occur in 14% (21/153), facial clefting in 10% (16/153), cardiovascular malformations or cardiomyopathy in 12% (18/153), genitourinary anomalies in 31% (48/153) and skeletal malformations or dysplasias in 8% (12/153). Malformations play a significant health role in over half of the 18 XLID syndromes with infant or early childhood lethality.
Acknowledgments
This work was supported in part by NIH grants (NS073854 and HD26202) to CES; MH57840 to RES), the South Carolina Department of Disabilities and Special Needs, and the Greenwood Genetic Center Foundation.
References
- Fried K. X-linked mental retardation and/or hydrocephalus. Clin Genet. 1972;3:258–263. [PubMed] [Google Scholar]
- Gibbons RJ, Wada T, Fisher CA, Malik N, Mitson MJ, Steensma DP, Fryer A, Goudie DR, Krantz ID, Traeger-Synodinos J. Mutations in the chromatin-associated protein ATRX. Hum Mutat. 2008;29:796–802. doi: 10.1002/humu.20734. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, Biesecker LG. Long term survival in TARP syndrome and confirmation of RBM10 as the disease causing gene. Am J Med Genet A. 2011;155A:2516–2520. doi: 10.1002/ajmg.a.34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger A, Schramm C, Draaken M, Mughal SS, Dworschak G, Bartels E, Hoffmann P, Nöthen MM, Reutter H, Ludwig M. Familial occurrence of the VATER/VACTERL association. Pediatr Surg Int. 2012;28:725–729. doi: 10.1007/s00383-012-3073-y. [DOI] [PubMed] [Google Scholar]
- Jouet M, Rosenthal A, MacFarlane J, Kenwrick S, Donnai D. A missense mutation confirms the L1 defect in X-linked hydrocephalus (HSAS) Nat Genet. 1993;4:331. doi: 10.1038/ng0893-331. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo S, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, Morohashi K. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, Schwartz CE, Lespinasse J, Van Esch H, Lacombe D, Goizet C, Phan-DinhTuy F, van Bokhoven H, Fryns JP, Chelly J, Ropers HH, Moraine C, Hamel BC, Briault S. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J Med Genet. 2005;42:780–786. doi: 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis RC, Due YZ, Schwartz CE. The site of a missense mutation in the extracellular Ig or FN domains of L1CAM influences infant mortality and the severity of X linked hydrocephaly. J Med Genet. 1998;35:901–904. doi: 10.1136/jmg.35.11.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 1997;17:285–291. doi: 10.1038/ng1197-285. [DOI] [PubMed] [Google Scholar]
- Riccardi VM, Marcus ES. Congenital hydrocephalus and cerebellar agenesis. Clin Genet. 1978;13:443–447. doi: 10.1111/j.1399-0004.1978.tb04143.x. [DOI] [PubMed] [Google Scholar]
- Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R, Orstavik KH, Quarrell OW, Schwartz CE, Shears DJ, Suri M, Kendrick-Jones J, Wilkie AO, OPD-spectrum Disorders Clinical Collaborative Group Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet. 2003;33:487–491. doi: 10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- Robertson SP. Filamin A: phenotypic diversity. Curr Opin Genet Dev. 2005;15:301–307. doi: 10.1016/j.gde.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Saillour Y, Zanni G, Des Portes V, Heron D, Guibaud L, Iba-Zizen MT, Pedespan JL, Poirier K, Castelnau L, Julien C, Franconnet C, Bonthron D, Porteous ME, Chelly J, Bienvenu T. Mutations in the AP1S2 gene encoding the sigma 2 subunit of the adaptor protein 1 complex are associated with syndromic X-linked mental retardation with hydrocephalus and calcifications in basal ganglia. J Med Genet. 2007;44:739–744. doi: 10.1136/jmg.2007.051334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrander-Stumpel C, Höweler C, Jones M, Sommer A, Stevens C, Tinschert S, Israel J, Fryns JP. Spectrum of X-linked hydrocephalus (HSAS), MASA syndrome, and complicated spastic paraplegia (SPG1): Clinical review with six additional families. Am J Med Genet. 1995;57:107–116. doi: 10.1002/ajmg.1320570122. [DOI] [PubMed] [Google Scholar]
- Shoubridge C, Fullston T, Gécz J. ARX spectrum disorders making inroads into the molecular pathology. Hum Mutat. 2010;31:889–900. doi: 10.1002/humu.21288. [DOI] [PubMed] [Google Scholar]
- Siderius LE, Hamel BCJ, van Bokhoven H, de Jager F, van den Helm B, Kremer H, Heineman-de Boer JA, Ropers HH, Mariman EC. X-linked mental retardation associated with cleft lip/cleft palate maps to Xp11.3-q21.3. Am J Med Genet. 1999;85:216–220. [PubMed] [Google Scholar]
- Sossey-Alaoui K, Hartung AJ, Guerrini R, Manchester DK, Posar A, Puche-Mira A, Andermann E, Dobyns WB, Srivastava AK. Human doublecortin (DCX) and the homologous gene in mouse encode a putative Ca2+-dependent signaling protein which is mutated in human X-linked neuronal migration defects. Hum Mol Genet. 1998;7:1327–1332. doi: 10.1093/hmg/7.8.1327. [DOI] [PubMed] [Google Scholar]
- Stevens CA, Wilroy RS. The telecanthus-hypospadias syndrome. J Med Genet. 1988;25:536–542. doi: 10.1136/jmg.25.8.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson RE, Hunter AGW. Considering the Embryopathogenesis of VACTERL Association. Mol Syndromol. 2013;4:7–15. doi: 10.1159/000346192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson RE, Schwartz CE, Rogers RC. Atlas of X-Linked Intellectual Disability Syndromes. New York: Oxford Univ Press; 2012. p. 344. [Google Scholar]
- Thauvin-Robinet C, Cosseé M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, Goldenberg A, Mortier G, Héron D, Sagot P, Bouvier AM, Huet F, Cusin V, Donzel A, Devys D, Teyssier JR, Faivre L. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43:54–61. doi: 10.1136/jmg.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner G, Gedeon A, Kerr B, Bennett R, Mulley J, Partington M. Syndromic form of X-linked mental retardation with marked hypotonia in early life, severe mental handicap, and difficult adult behavior maps to Xp22. Am J Med Genet A. 2003;117A:245–250. doi: 10.1002/ajmg.a.10005. [DOI] [PubMed] [Google Scholar]
- Vits L, Van Camp G, Coucke P, Fransen E, De Boulle K, Reyniers E, Korn B, Poustka A, Wilson G, Schrander-Stumpel C, Winter RM, Schwartz C, Willems PJ. MASA syndrome is due to mutations in the neural cell adhesion gene L1CAM. Nat Genet. 1994;7:408–413. doi: 10.1038/ng0794-408. [DOI] [PubMed] [Google Scholar]
- Vos YJ, de Walle HE, Bos KK, Stegeman JA, Ten Berge AM, Bruining M, van Maarle MC, Elting MW, den Hollander NS, Hamel B, Fortuna AM, Sunde LE, Stolte-Dijkstra I, Schrander-Stumpel CT, Hofstra RM. Genotype-phenotype correlation in L1 syndrome: a guide for genetic counseling and mutation analysis. J Med Genet. 2010;47:169–175. doi: 10.1136/jmg.2009.071688. [DOI] [PubMed] [Google Scholar]