

Abstract
Pneumococcal conjugate vaccines (PCVs) have been introduced worldwide. However, few developing countries have high-quality surveillance systems available for monitoring vaccine impact. We evaluated whether data from nasopharyngeal carriage studies can be used to accurately monitor post-PCV changes in the incidence of invasive pneumococcal disease (IPD) among children under 5 years of age. For various dates during 1991–2010, data on nasopharyngeal pneumococcal carriage and on IPD before and after administration of 7-valent PCV (PCV7) were available from England and Wales, the Netherlands, the Navajo and White Mountain Apache American Indian populations, and the US states of Massachusetts and Alaska. We estimated the change in carriage prevalence for each serotype in each study and then either calculated the average change (inverse variance-weighted) among vaccine and nonvaccine serotypes (model 1) or used mixed-effects models to estimate the change for each serotype individually, pooling serotype data within or between studies (models 2 and 3). We then multiplied these values by the proportion of IPD caused by each serotype during the pre-PCV7 period to obtain an estimate of post-PCV7 disease incidence. Model 1 accurately captured overall changes in IPD incidence following PCV7 introduction for most studies, while the more detailed models, models 2 and 3, were less accurate. Carriage data can be used in this simple model to estimate post-PCV changes in IPD incidence.
Keywords: carriage; conjugate vaccine, pneumococcal; pneumococcus; surveillance; vaccine effectiveness; vaccines
Pneumococcal conjugate vaccines (PCVs) for children have successfully reduced the burden of invasive pneumococcal disease (IPD) in diverse epidemiologic settings (1–3). Declines in overall IPD incidence are the net result of large reductions in the incidence of targeted vaccine serotypes (hereafter called vaccine types (VTs)) and smaller increases in the incidence of nonvaccine serotypes (nonvaccine types (NVTs)). PCVs are now being introduced into developing countries and are expected to confer a significant benefit in these high-incidence populations as well. However, given global variations in the prevaccine serotype distribution and the potential for increases in the incidence of NVTs (i.e., serotype replacement), it is imperative to measure the impact of PCVs on IPD incidence. This information is needed to inform policy decisions regarding sustaining or modifying PCV immunization programs.
Few developing countries have population-based surveillance systems available for monitoring IPD incidence. Several years’ worth of data from a mature, stable surveillance system are required to accurately estimate baseline IPD rates (3), and these surveillance systems are expensive to maintain. Therefore, there has been interest in using pneumococcal nasopharyngeal carriage data as a proxy measure for monitoring the impact of PCVs (4–6). Unlike IPD, carriage of pneumococcus is common among children, and carriage data are less expensive and technically easier to collect in an unbiased way than population-based IPD data. Carriage is a prerequisite for disease, and PCVs protect against VT carriage, so changes in the IPD incidence of a serotype should be predicted by changes in the carriage prevalence of that serotype, as long as virulence (cases of IPD per carriage episode) does not change (4–8).
Despite the relative ease of obtaining carriage samples, there are challenges in interpreting such data. Because serotypes of pneumococci colonizing the nasopharynx differ in their capacity to cause IPD, there needs to be some measure of how much IPD would be expected given the carriage prevalence of specific serotypes, either by calculating invasiveness (ratio of cases of IPD to carriers) (6, 8, 9) or by directly including information on prevaccine IPD incidence.
In this study, we considered whether changes in carriage prevalence following the introduction of 7-valent PCV (PCV7) could be used to estimate the overall impact of the vaccines on IPD incidence in several populations. We found that estimates that accounted for changes in individual serotypes were highly inaccurate. However, a simpler model that grouped together VTs and NVTs can provide an accurate estimate of the overall change in IPD following PCV introduction. We discuss how such a method could be implemented in settings that lack high-quality IPD data.
MATERIALS AND METHODS
Data sources
Carriage data were gathered from published and unpublished sources from the US state of Massachusetts (10) (2001–2009), the Navajo and White Mountain Apache (WMA) American Indian populations (11–13) (1998–2008), the US state of Alaska (14, 15) (1998–2008), the Netherlands (16) (2005–2010), and England and Wales (17) (2001–2009) (see Web Table 1, available at http://aje.oxfordjournals.org/). For the Navajo/WMA and Netherlands populations, prevaccine colonization data were from the control groups of randomized controlled trials conducted in these populations. IPD incidence data were obtained from published and unpublished sources for the Massachusetts (18) (2001–2009), Navajo/WMA (19) (1995–2007), Netherlands (20) (2004–2010), Alaska (14, 21) (1991–2008), and England and Wales (3) (2000–2009) populations (Web Table 1). The Alaska data were subdivided into rural and urban communities, based on previous findings (21). Data included years before and after the introduction of PCV7 but did not include time periods when 10-valent PCV (PCV10) or 13-valent PCV (PCV13) was in use. For studies with multiple post-PCV periods, separate estimates were obtained for each period. The age groups and years included for each study are summarized in Web Table 1. All data were drawn from children under age 5 years.
Serotypes 6A and 6C were grouped together, because not all studies distinguished between them (22). For Massachusetts carriage and IPD data and the Navajo/WMA IPD data, missing serotype information was imputed based on the assumption that isolates not serotyped had the same serotype distribution as those typed within a given pre- or post-PCV sampling period.
Each of the study protocols was approved by the relevant institutional review board, and for the Navajo/WMA studies, protocols were approved by the respective tribes.
Definition of the raw serotype-specific carriage prevalence ratio (post-PCV/pre-PCV)
Prevalence was defined as the number of positive nasopharyngeal swabs for a given serotype divided by the total number of swabs collected during a given sampling period. The serotype-specific carriage prevalence ratio (PRi) was defined as the carriage prevalence of a serotype following vaccine introduction divided by the carriage prevalence before introduction. For serotypes with 0 isolates before or after PCV7, a continuity correction of 0.5 was added to the numerator and denominator of the prevalence calculation to allow for a nonzero or noninfinite estimate of PRi. The variance of the raw PRi was calculated using the delta method (23).
Methods for using carriage data to predict changes in IPD incidence
If we assume that the invasiveness index (ratio of IPD incidence to carrier prevalence) for a given serotype (i) is an intrinsic property and does not change after vaccination (11, 24), then a change in carriage prevalence for a serotype will result in a proportional change in IPD incidence of the same serotype (e.g., a 2-fold change in carriage prevalence would result in a 2-fold change in IPD incidence). The expected post-PCV IPD incidence in a given population, then, is defined as
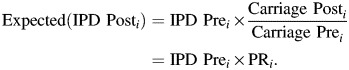 |
(1) |
The estimated change in overall IPD incidence following PCV (the incidence rate ratio (IRR)) is defined as Expected(IPD_Posti) summed across all serotypes divided by the observed pre-PCV incidence summed across all serotypes:
 |
(2) |
For this calculation, we need data on carriage prevalence before and after vaccination and a prevaccination estimate of IPD caused by each serotype (either incidence or proportion).
Estimating stabilized carriage prevalence ratios
In an ideal situation, we would perfectly measure the PRi for each serotype and plug these values into equation 1 to estimate the post-PCV7 IPD incidence for each serotype and overall. However, because of statistical uncertainty due to small numbers and the detection of short-term changes in individual serotypes, we had to use summarized or stabilized estimates in the equations. We considered 3 models that would minimize the impact of these uncertainties. In each model, we weighted log(PRi) for each serotype i by the inverse of its variance, so serotypes with few isolates (and higher variance) contributed less to the estimates.
Model 1 simply calculates the weighted average log(PRi) for broad groups of serotypes (VT/NVT or VT/high-incidence NVT/low incidence NVT). This model calculates a single estimate of log(PR) for each category of serotypes (k). This was accomplished by fitting a weighted regression model where the outcome was log(PRi), the predictor was a categorical variable (groupk) for groups of serotypes, and the weight was the inverse of the variance of each raw log(PRi). We tested 2 separate versions of this model where groupk was either VT versus NVT or VT versus NVT (high incidence pre-PCV) versus NVT (low incidence pre-PCV). An NVT was considered “high-incidence” if it represented more than 5% of NVT IPD cases during the pre-PCV7 period. This stratification allowed the more common NVTs to have a different estimate of expected(log(PRi)) than the rare NVTs (Web Appendix).
Model 2 allows the estimate of log(PRi) to vary between serotypes (using random intercepts) but pulls the expected values towards the average of the broad serotype categories used in model 1 within an individual study (25), effectively reducing the impact of statistical uncertainty due to small numbers for any one serotype (see methods in Web Appendix).
Model 3 is similar to model 2 but combines data between studies and pulls the expected value for a given serotype towards the average for broader serotype categories across studies. This equation effectively estimates log(PRi) for each serotype in each study, and the model borrows information from other studies and other serotypes in the same category, stabilizing the estimates from any one study (see Web Appendix methods). Because the impact on carriage and IPD in a population will vary with vaccine coverage, we dichotomized studies on the basis of whether the vaccine program in the population was mature or not (above or below 90% uptake).
For all 3 models, serotypes without any carriage data in either the prevaccine period or the postvaccine period did not contribute to the pooled prevalence ratio estimates. We assumed that the predicted log(PRi) for these serotypes without carriage data was the same as the average log(PRi) of other serotypes in the same category. For example, if serotype 1 did not have any carriage data in model 1, we assumed that the log(PRi) for serotype 1 was the same as that for the other nonvaccine serotypes.
All models were fitted using PROC MIXED in SAS, version 9.2 (SAS Institute Inc., Cary, North Carolina).
Estimating the IRR and errors
As demonstrated in equation 2, the overall IRR for IPD can be estimated from the expected(PRi) (calculated using one of the models described above) and the observed pre-PCV7 IPD incidence for each serotype or category of serotypes. We used Monte Carlo resampling of the model parameters and of the observed pre-PCV incidence rate (1,000 replicates) to obtain predictive intervals for the estimates (see Web Appendix for details). To demonstrate a plausible range of IRRs that would be expected given an observed change in carriage, we substituted values for percentage of pre-PCV7 disease caused by PCV7 serotypes of 30%–100% in equation 2 along with the estimated prevalence ratios.
For all models, the estimated percent change in IPD incidence was defined as (IRR – 1) × 100%. Thus, an IRR of 1.5 is equal to a change of +50%, while an IRR of 0.5 is equal to a change of −50%.
RESULTS
Predicting postvaccine IPD incidence using carriage data (model 1)
We first estimated the overall change in IPD incidence using model 1, which estimated the change in carriage and IPD for VTs and NVTs. We considered the estimate accurate if the observed value was within the 95% predictive interval of the estimate. This model accurately estimated the change in post-PCV overall IPD incidence for all of the studies that we evaluated except for England and Wales (Table 1). If we estimated the change in carriage prevalence separately for high- and low-incidence NVTs, the predicted change was modestly closer to the observed change (Table 1).
Table 1.
Observed and Predicted Changes in Overall Incidence of Invasive Pneumococcal Disease Following Administration of 7-Valent Pneumococcal Conjugate Vaccine in 6 Study Populationsa
| Observed % Change |
95% CI | Predicted % Changeb |
95% PI | Predicted % Changec |
95% PI | |
|---|---|---|---|---|---|---|
| Model 1 | ||||||
| Alaska (rural) | −36.8 | −51.8, −17.1 | −38.2 | −58.4, −10.8 | −36.5 | −62.6, 5.1 |
| Alaska (urban) | −63.0 | −72.3, −50.7 | −58.9 | −69.6, −46.1 | −60.3 | −70.3, −48.4 |
| England | −50.7 | −55.6, −45.3 | −6.6 | −35.5, 31.7 | −14.8 | −41.0, 22.7 |
| Massachusettsd | −2.4 | −27.6, 31.7 | 14.7 | −12.0, 50.2 | 7.2 | −18.6, 43.8 |
| Navajo/WMA | −49.3 | −59.2, −37.1 | −38.0 | −50.0, −22.7 | −47.0 | −58.6, −31.7 |
| The Netherlands | −54.3 | −71.6, −26.7 | −50.1 | −64.8, −28.2 | −48.7 | −64.7, −23.4 |
| Model 2 | ||||||
| Alaska (rural) | −36.8 | −51.8, −17.1 | 112.7 | −10.9, 438.3 | 172.4 | −19.5, 952.9 |
| Alaska (urban) | −63.0 | −72.3, −50.7 | −52.3 | −68.3, −23.8 | −52.1 | −69.6, −22.5 |
| England | −50.7 | −55.6, −45.3 | 65.6 | −2.8, 272.4 | 70.1 | −21.2, 361.9 |
| Massachusettsd | −2.4 | −27.6, 31.7 | 39.8 | −9.3, 115.1 | 32.9 | −19.6, 123.7 |
| Navajo/WMA | −49.3 | −59.2, −37.1 | −25.5 | −51.1, 23.0 | −26.0 | −57.6, 56.6 |
| The Netherlands | −54.3 | −71.6, −26.7 | −28.8 | −55.5, 25.3 | −19.0 | −58.1, 64.2 |
| Model 3 | ||||||
| Alaska (rural) | −36.8 | −51.8, −17.1 | 10.5 | −42.3, 112.6 | 16.0 | −40.1, 151.2 |
| Alaska (urban) | −63.0 | −72.3, −50.7 | −48.9 | −70.5, −12.9 | −48.5 | −69.2, −4.3 |
| England | −50.7 | −55.6, −45.3 | 2.1 | −37.0, 65.7 | −6.6 | −41.6, 64.0 |
| Navajo/WMA | −49.3 | −59.2, −37.1 | −22.1 | −51.4, 31.7 | −15.1 | −49.3, 57.8 |
| The Netherlands | −54.3 | −71.6, −26.7 | −16.9 | −56.8, 56.8 | −7.3 | −51.8, 100.6 |
Abbreviations: CI, confidence interval; PCV, pneumococcal conjugate vaccine; PCV7, 7-valent pneumococcal conjugate vaccine; PI, predictive interval; WMA, White Mountain Apache.
a The table shows estimates from the most recent postvaccine period for which data were available from each study site. The carriage samples shown were from Alaska (2004 and 2008), England (2008/2009), Massachusetts (2009), the Navajo/WMA (2006–2008), and the Netherlands (2010), and the invasive pneumococcal disease data were from the corresponding periods indicated in Web Table 1. Years post-PCV7 during which the data were collected for each state/country: Alaska, 4–8 years after vaccination; Navajo/WMA, 6–8 years; the Netherlands, 3–4 years; England and Wales, 3 years; Massachusetts, 8 years (see Web Table 1).
b Data in the model were stratified by vaccine serotype (vaccine type/nonvaccine type).
c Data in the model were stratified by vaccine serotype (vaccine type/high-incidence nonvaccine type/low-incidence nonvaccine type).
d The reference sample for Massachusetts was collected 1 year after vaccine introduction, not before PCV7 introduction. Massachusetts was not included in model 3 because there was no true pre-PCV sample.
In all of the studies, model 1 underestimated the decline in VTs, but the degree of underestimation varied considerably between studies (observed and expected values were −97.1% vs. −91.8% in Massachusetts and −94.8% vs. −77.3% in England and Wales (Table 2)). Model 1 accurately estimated the change in the incidence of NVTs for the Navajo, Netherlands, and Massachusetts populations but underestimated the increase in NVTs among Alaskan children and overestimated the increase in NVTs in England and Wales (Table 2). Model 1 (with stratified NVTs) accurately tracked the changes in NVTs over time (Web Figure 1) but underestimated the decline in VTs in the Navajo/WMA and Netherlands studies.
Table 2.
Observed and Predicted Changes in Incidence of Vaccine-Type and Nonvaccine-Type Invasive Pneumococcal Disease Following Administration of 7-Valent Pneumococcal Conjugate Vaccine in 6 Study Populations (Model 1)a
| Observed % Change |
95% CI | Predicted % Changeb |
95% PI | Predicted % Changec |
95% PI | |
|---|---|---|---|---|---|---|
| Vaccine serotypes | ||||||
| Alaska (rural) | −98.8 | −99.8, −91.2 | −87.6 | −95.7, −67.4 | −87.8 | −95.5, −67.6 |
| Alaska (urban) | −98.4 | −99.6, −93.6 | −86.2 | −90.9, −77.6 | −86.2 | −91.0, −78.7 |
| England and Wales | −94.8 | −96.2, −92.9 | −77.3 | −90.7, −45.1 | −77.1 | −90.4, −48.3 |
| Massachusettsd | −97.1 | −99.6, −77.9 | −91.8 | −96.6, −80.9 | −92.9 | −97.2, −81.3 |
| Navajo/WMA | −99.2 | −99.9, −94.6 | −90.3 | −95.0, −82.6 | −90.5 | −95.0, −82.4 |
| The Netherlands | −96.9 | −99.6, −77.8 | −90.6 | −94.6, −84.1 | −90.8 | −94.4, −84.0 |
| Nonvaccine serotypes | ||||||
| Alaska (rural) | 115.4 | 51.5, 206.3 | 74.0 | 23.1, 141.9 | 78.0 | 3.4, 204.1 |
| Alaska (urban) | 110.6 | 41.8, 212.8 | 65.2 | 27.7, 112.3 | 57.7 | 19.5, 107.8 |
| England and Wales | 85.5 | 59.6, 115.6 | 204.4 | 106.1, 343.1 | 171.0 | 85.3, 300.9 |
| Massachusettsd | 22.3 | −11.8, 69.7 | 41.6 | 10.0, 79.8 | 32.4 | 2.9, 73.8 |
| Navajo/WMA | 8.2 | −16.3, 39.8 | 20.2 | −0.8, 47.1 | 2.3 | −19.5, 30.0 |
| The Netherlands | 48.1 | −16.9, 164.1 | 37.0 | 4.1, 86.0 | 42.1 | 2.4, 99.6 |
Abbreviations: CI, confidence interval; PCV7, 7-valent pneumococcal conjugate vaccine; PI, predictive interval; WMA, White Mountain Apache.
a The table shows estimates from the most recent postvaccine period for which data were available from each study site. The carriage samples shown were from Alaska (2004 and 2008), England (2008/2009), Massachusetts (2009), the Navajo/WMA (2006–2008), and the Netherlands (2010), and the invasive pneumococcal disease data were from the corresponding periods indicated in Web Table 1. Years post-PCV7 during which the data were collected for each state/country: Alaska, 4–8 years after vaccination; Navajo/WMA, 6–8 years; the Netherlands, 3–4 years; England and Wales, 3 years; Massachusetts, 8 years (see Web Table 1).
b Data in the model were stratified by vaccine serotype (vaccine type/nonvaccine type).
c Data in the model were stratified by vaccine serotype (vaccine type/high-incidence nonvaccine type/low-incidence nonvaccine type).
d The reference sample for Massachusetts was collected 1 year after vaccine introduction, not before PCV7 introduction.
Adding detail to the models (models 2 and 3)
We next included estimates of change for individual serotypes in models 2 and 3. The estimates from model 2 were highly inaccurate, particularly for the studies from rural Alaska, England, and Massachusetts (Table 1). Examining the difference between observed and expected post-PCV7 IPD incidence for individual serotypes, we found that a few serotypes in each study (i.e., 5, 7F, 12F, 19A) were responsible for the poor predictions (Web Figure 2). Some of these serotypes are known to vary strongly from year to year (26).
Model 3, which pooled serotype data across all studies, provided predictions that were superior to those of model 2 but less accurate than those of model 1 (Table 1, Web Figure 2).
When we compared all of the models using mean squared errors (Web Table 2), model 1 provided the most accurate predictions. Stratifying the NVTs by pre-PCV7 IPD incidence in model 1 further improved the estimates.
Using carriage data in the absence of IPD data
Some developing countries might have carriage data but not have accurate IPD incidence data. In order to estimate the post-PCV7 IPD incidence using carriage data, we would need, at a minimum, an estimate of the proportion of IPD caused by VTs and NVTs. Ideally, this would be obtained from population-based IPD surveillance data, but it could also come from a case series as long as the cases reflected the serotype distribution of pediatric IPD in the whole community. In the absence of any IPD data, one could evaluate a range of plausible proportions of VTs and NVTs, as determined from the literature (27). The caveat is that targeting more prevalent serotypes with the vaccine might have a larger, and more unpredictable, effect on replacement because it will allow nonvaccine serotypes to increase more.
To demonstrate this, we took the prevalence ratio estimates for the Netherlands and Navajo/WMA populations calculated from model 1 and estimated the likely decline in IPD incidence in hypothetical populations where the VTs caused 30%–100% of IPD cases during the pre-PCV period. Given the observed change in carriage prevalence in the Navajo/WMA population, we would expect the vaccine to be effective over a wide range of plausible pre-PCV serotype distributions (Web Figure 3). Given the observed changes in carriage in the Netherlands population, we would expect the vaccine to be effective if approximately 50% or more of the pre-PCV7 IPD cases were caused by VTs. For both studies, the greater the percentage of baseline IPD caused by VTs the larger the reduction in overall IPD incidence.
DISCUSSION
We evaluated whether carriage data can be used to estimate overall, VT, and NVT changes in IPD incidence following the introduction of PCV. Model 1, which simply averaged the prevalence ratios of VTs and NVTs (weighted by inverse variance), provided accurate estimates of post-PCV incidence in most of the studies that we evaluated. This model assumes an average change in carriage prevalence for all serotypes within the same serotype group (i.e., VT or NVT).
Because models 2 and 3 implicitly accounted for differences in invasiveness between individual serotypes, we would have expected them to be more accurate. However, these models are sensitive to temporal fluctuations of specific serotypes in IPD incidence or carriage prevalence that occur even in the absence of vaccination (26, 28). This is a problem because carriage and IPD surveys are often conducted on different spatial and time scales, so a fluctuation might be detected in an IPD survey but not in a carriage survey or vice versa, resulting in inaccurate estimates. This means that changes in the carriage of individual serotypes detected in models 2 and 3 might be “real” changes (not the result of statistical noise), but they might not have an important impact on IPD incidence over the long term. In future studies, investigators could evaluate the importance of the length of disease surveillance and carriage surveys in the accuracy and stability of the estimates. It is also important to determine the amount of time after vaccine introduction that needs to elapse before accurate estimates of changes in carriage or disease can be obtained. Our study (Web Figure 1) suggests that the effect of vaccination is detectable in the carriage data within a year after vaccine introduction, although the impact becomes more apparent over time.
The models tended to underestimate the decline in VTs. A continuity correction (addition of 0.5 to the prevalence ratio) is required to obtain an estimate of the change when there are zero detected carriers pre- or post-PCV7. However, this limits how much of a decline in VTs can be detected. For instance, if going from 10 carriers to 0 carriers, the detected decline would be 95% rather than 100%. This is particularly a problem for smaller studies, where more of the serotypes will require continuity corrections, such as the England and Wales study (17) (192 post-PCV carriage samples compared with several hundred or several thousand swabs in other studies) (Web Table 1). Smaller sample sizes, lower prevaccine prevalence, and larger vaccine-associated declines will all be associated with less accurate estimates.
These models were fitted to data from several diverse settings, including the Navajo/WMA population, which has a high burden of pneumococcal disease and a serotype distribution that is distinct from that of the rest of the United States. In particular, serotype 12F, a highly invasive serotype, was the third-most-common cause of prevaccine disease among Navajo/WMA children. Serotype 1 was the eighth-most-common cause of prevaccine disease and was more frequent than several vaccine serotypes. However, we did not have available data from developing countries with which to test these models. In particular, it will be critical to test this approach in settings where the highly invasive serotypes 1 and 5 are dominant causes of disease and, ideally, are carried with a detectable frequency. Serotypes 1 and 5, which are included in PCV10/PCV13, are important causes of IPD in developing countries but are infrequently detected among carriage samples and produce short epidemics that might not be detected in carriage. If modeling PCV10/PCV13, serotypes 1 and 5 would be assumed to decline by the same order of magnitude as the other VTs, an assumption consistent with early effectiveness studies for serotype 1 (29).
The models presented here included several assumptions. We assumed that vaccine effectiveness against carriage is the same as vaccine effectiveness against IPD. Vaccine efficacy against IPD depends both on preventing acquisition and on protecting against progression from carriage to IPD (6). Our models, based on carriage prevalence ratios, account only for protection against acquisition in the nasopharynx (both direct and indirect effects); they do not account for direct protection against progression to IPD, and this might explain why the model underestimated the decline in VT IPD. The assumption that the vaccine protects against carriage acquisition is supported by epidemiologic studies (reviewed by Simell et al. (6)).
Second, the conceptual approach of using carriage changes to predict changes in IPD risk assumes that the invasiveness of a serotype does not change during the period following PCV introduction. This assumption is supported by evidence (11, 24), but it is possible that new clones of a particular serotype of greater or lesser virulence than the prevaccine strains could emerge.
Finally, the models assume that the underlying susceptibility of the population for IPD does not change during the sampling periods. This is a reasonable assumption in most communities over the short periods of time relevant for vaccine impact assessments (i.e., 5–10 years) but may not be true in areas with significant changes in the prevalence of risk factors (human immunodeficiency virus, influenza, malaria), and associated interventions, that strongly affect pneumococcal IPD.
The use of mixed-effects models (models 2 and 3) reduces the total variance of the prevalence ratio estimates for individual serotypes but could introduce bias. We compared the raw prevalence ratios and prevalence ratios estimated from model 2 for each serotype with the observed IRRs. The association was modestly stronger for the modeled estimates than for the observed estimates (Pearson's ρ = 0.82 vs. 0.75). This indicates that the modeled prevalence ratio estimates for the serotypes are more accurate than the raw estimates.
Because most countries that have reliable pre-PCV IPD surveillance will probably continue surveillance during the post-PCV period, the apparent utility of this method could be considered limited. However, there are several possible scenarios in which model 1 could be utilized in a real-world setting. First, if a country does not have reliable population-based pre-PCV IPD incidence data but does have representative information about the serotype distribution for IPD (i.e., from a hospital-based case series study of all pediatric IPD), these pre-PCV data could be used in model 1. Using such IPD data, it is critical that the bacterial sample accurately represent the entire population to which the model estimates will be applied. Examples where the serotype distribution of IPD may not be representative of that in the general population include sentinel hospitals or referral facilities that receive sicker patients with more underlying illnesses (e.g., human immunodeficiency virus) or a preponderance of certain syndromes (e.g., meningitis). Further studies could be performed to compare the effect of study type on the detected serotype distribution. Second, because serotype distributions are comparable within a given geographic region (27), a country could use IPD serotype information from a neighboring country with a similar demographic and socioeconomic profile. Finally, if there were no available IPD data, one could evaluate a range of plausible values for the percentage of pre-PCV IPD caused by the VTs (30%–90%) (27), as described in the Results section.
This study demonstrates that data on nasopharyngeal pneumococcal carriage, in combination with pre-PCV IPD data, can be used to evaluate the potential impact of vaccine on IPD incidence. This may provide a feasible approach for policy-makers and public health authorities who wish to assess the population-wide effect of PCV on IPD incidence. The approach has additional applicability in that it could be modified for populations without available IPD data. The estimates derived from this model could provide a sense of vaccine impact without the demands of resource-intensive IPD-based impact assessments.
Supplementary Material
ACKNOWLEDGMENTS
Author affiliations: Division of International Epidemiology and Population Studies, Fogarty International Center, National Institutes of Health, Bethesda, Maryland (Daniel M. Weinberger); Department of Epidemiology of Microbial Diseases, School of Public Health, Yale University, New Haven, Connecticut (Daniel M. Weinberger); Arctic Investigations Program, Division of Preparedness and Emerging Infections, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Anchorage, Alaska (Dana T. Bruden); Center for American Indian Health, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, Maryland (Lindsay R. Grant, Katherine L. O'Brien); Department of Epidemiology and Department of Immunology and Infectious Diseases, School of Public Health, Harvard University, Boston, Massachusetts (Marc Lipsitch); International Vaccine Access Center, Department of International Health, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, Maryland (Katherine L. O'Brien, Daniel R. Feikin); Department of Pediatrics and Section of Pediatric Infectious Diseases, School of Medicine, Boston Medical Center, Boston University, Boston, Massachusetts (Stephen I. Pelton); Department of Epidemiology, School of Public Health, Boston University, Boston, Massachusetts (Stephen I. Pelton); Department of Pediatric Immunology and Infectious Disease, Wilhelmina Children's Hospital, University Medical Center Utrecht, Utrecht, the Netherlands (Elisabeth A. M. Sanders); and Division of Preparedness and Emerging Infections, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia (Daniel R. Feikin).
D.M.W. was supported by the Division of International Epidemiology and Population Studies, Fogarty International Center, National Institutes of Health. M.L. was supported by National Institutes of Health grant 2R01AI048935. Support for the Navajo/WMA studies came from the Bill and Melinda Gates Foundation (grant 37875), The Native American Research Centers for Health (grants U26IHS300013/03 and U269400012-01), the Centers for Disease Control and Prevention National Vaccine Program Office, the Thrasher Research Fund (grant 02820-9), Wyeth Vaccines (Pfizer Inc.), the US Agency for International Development, and the World Health Organization. Support for the Alaskan postvaccine carriage study was provided, in part, by a grant from Pfizer Inc. (grant WS950636).
We thank Drs. Kari Auranen, Stefan Flasche, Elske van Gils, and Cécile Viboud for helpful discussions on these analyses. We also thank Drs. Maya Dutta-Linn, Jonathan Finkelstein, and Grace Lee for discussions about the Massachusetts carriage data.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. None of the funders contributed to the design of these analyses or the decision to publish the results.
D.M.W. has received research funding from Pfizer Inc. (New York, New York). K.O.B. and L.R.G. have received research grant support related to pneumococcal vaccines from GlaxoSmithKline (GSK; London, United Kingdom) and Pfizer. K.O.B. has received honoraria for participation in external expert advisory committees on pneumococcal vaccines convened by Merck & Company (Whitehouse Station, New Jersey), Sanofi-Pasteur (Swiftwater, Pennsylvania), and GSK. E.A.M.S. has been given research grant support related to pneumococcal vaccines from Wyeth (Overland Park, Kansas)/Pfizer and GSK and has received honoraria for participation in external expert advisory committees on pneumococcal vaccines convened by Pfizer and GSK. S.I.P. has received honoraria for participation in advisory board activities related to pneumococcal vaccine or pneumococcal disease from Pfizer, Merck Vaccines, Novartis (Basel, Switzerland), and GSKbio. M.L. has accepted honoraria or consulting fees from Pfizer and Novartis.
REFERENCES
- 1.Weinberger DM, Malley R, Lipsitch M. Serotype replacement in disease after pneumococcal vaccination. Lancet. 2011;378(9807):1962–1973. doi: 10.1016/S0140-6736(10)62225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pilishvili T, Lexau C, Farley MM, et al. Sustained reductions in invasive pneumococcal disease in the era of conjugate vaccine. J Infect Dis. 2010;201(1):32–41. doi: 10.1086/648593. [DOI] [PubMed] [Google Scholar]
- 3.Miller E, Andrews NJ, Waight PA, et al. Herd immunity and serotype replacement 4 years after seven-valent pneumococcal conjugate vaccination in England and Wales: an observational cohort study. Lancet Infect Dis. 2011;11(10):760–768. doi: 10.1016/S1473-3099(11)70090-1. [DOI] [PubMed] [Google Scholar]
- 4.Mulholland K, Satzke C. Serotype replacement after pneumococcal vaccination [letter] Lancet. 2012;379(9824):1387. doi: 10.1016/S0140-6736(12)60588-1. [DOI] [PubMed] [Google Scholar]
- 5.Weinberger DM, Malley R, Lipsitch M. Serotype replacement after pneumococcal vaccination—authors’ reply [letter] Lancet. 2012;379(9824):1388–1389. doi: 10.1016/S0140-6736(12)60590-X. [DOI] [PubMed] [Google Scholar]
- 6.Simell B, Auranen K, Käyhty H, et al. The fundamental link between pneumococcal carriage and disease. Expert Rev Vaccines. 2012;11(7):841–855. doi: 10.1586/erv.12.53. [DOI] [PubMed] [Google Scholar]
- 7.Rinta-Kokko H, Dagan R, Givon-Lavi N, et al. Estimation of vaccine efficacy against acquisition of pneumococcal carriage. Vaccine. 2009;27(29):3831–3837. doi: 10.1016/j.vaccine.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Shea KM, Weycker D, Stevenson AE, et al. Modeling the decline in pneumococcal acute otitis media following the introduction of pneumococcal conjugate vaccines in the US. Vaccine. 2011;29(45):8042–8048. doi: 10.1016/j.vaccine.2011.08.057. [DOI] [PubMed] [Google Scholar]
- 9.Weinberger D, Harboe Z, Flasche S, et al. Prediction of serotypes causing invasive pneumococcal disease in unvaccinated and vaccinated populations. Epidemiology. 2011;22(2):199–207. doi: 10.1097/EDE.0b013e3182087634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang SS, Hinrichsen VL, Stevenson AE, et al. Continued impact of pneumococcal conjugate vaccine on serotypes, antibiotic resistance, and risk factors for carriage in young children. Pediatrics. 2009;124(1):e1–e11. doi: 10.1542/peds.2008-3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott JR, Millar EV, Lipsitch M, et al. Impact of more than a decade of pneumococcal conjugate vaccine use on carriage and invasive potential in Native American communities. J Infect Dis. 2012;205(2):280–288. doi: 10.1093/infdis/jir730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Brien KL, Millar EV, Zell ER, et al. Effect of pneumococcal conjugate vaccine on nasopharyngeal colonization among immunized and unimmunized children in a community-randomized trial. J Infect Dis. 2007;196(8):1211–1220. doi: 10.1086/521833. [DOI] [PubMed] [Google Scholar]
- 13.Millar EV, Watt JP, Bronsdon MA, et al. Indirect effect of 7-valent pneumococcal conjugate vaccine on pneumococcal colonization among unvaccinated household members. Clin Infect Dis. 2008;47(8):989–996. doi: 10.1086/591966. [DOI] [PubMed] [Google Scholar]
- 14.Singleton RJ, Hennessy TW, Bulkow LR, et al. Invasive pneumococcal disease caused by nonvaccine serotypes among Alaska native children with high levels of 7-valent pneumococcal conjugate vaccine coverage. JAMA. 2007;297(16):1784–1792. doi: 10.1001/jama.297.16.1784. [DOI] [PubMed] [Google Scholar]
- 15.Park SY, Moore MR, Bruden DL, et al. Impact of conjugate vaccine on transmission of antimicrobial-resistant Streptococcus pneumoniae among Alaskan children. Pediatr Infect Dis J. 2008;27(4):335–340. doi: 10.1097/INF.0b013e318161434d. [DOI] [PubMed] [Google Scholar]
- 16.Spijkerman J, van Gils E, Veenhoven R, et al. Carriage of Streptococcus pneumoniae 3 years after start of vaccination program, the Netherlands. Emerg Infect Dis. 2011;17(4):584–591. doi: 10.3201/eid1704101115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flasche S, Van Hoek AJ, Sheasby E, et al. Effect of pneumococcal conjugate vaccination on serotype-specific carriage and invasive disease in England: a cross-sectional study. PLoS Med. 2011;8(4):e1001017. doi: 10.1371/journal.pmed.1001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yildirim I, Stevenson A, Hsu KK, et al. Evolving picture of invasive pneumococcal disease in Massachusetts children: a comparison of disease in 2007–2009 with earlier periods. Pediatr Infect Dis J. 2012;31(10):1016–1021. doi: 10.1097/INF.0b013e3182615615. [DOI] [PubMed] [Google Scholar]
- 19.Weatherholtz R, Millar EV, Moulton LH, et al. Invasive pneumococcal disease a decade after pneumococcal conjugate vaccine use in an American Indian population at high risk for disease. Clin Infect Dis. 2010;50(9):1238–1246. doi: 10.1086/651680. [DOI] [PubMed] [Google Scholar]
- 20.van Deursen AM, van Mens SP, Sanders EA, et al. Invasive pneumococcal disease and 7-valent pneumococcal conjugate vaccine, the Netherlands. Emerg Infect Dis. 2012;18(11):1729–1737. doi: 10.3201/eid1811.120329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wenger JD, Zulz T, Bruden D, et al. Invasive pneumococcal disease in Alaskan children: impact of the seven-valent pneumococcal conjugate vaccine and the role of water supply. Pediatr Infect Dis J. 2010;29(3):251–256. doi: 10.1097/INF.0b013e3181bdbed5. [DOI] [PubMed] [Google Scholar]
- 22.Park IH, Pritchard DG, Cartee R, et al. Discovery of a new capsular serotype (6C) within serogroup 6 of Streptococcus pneumoniae. J Clin Microbiol. 2007;45(4):1225–1233. doi: 10.1128/JCM.02199-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosner B. Fundamentals of Biostatistics. Belmont, CA: Thomsen Higher Education; 2006. [Google Scholar]
- 24.Yildirim I, Hanage WP, Lipsitch M, et al. Serotype specific invasive capacity and persistent reduction in invasive pneumococcal disease. Vaccine. 2010;29(2):283–288. doi: 10.1016/j.vaccine.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Efron B, Morris C. Stein's paradox in statistics. Sci Am. 1977;236(5):119–127. [Google Scholar]
- 26.Harboe ZB, Benfield T L, Valentiner-Branth P, et al. Temporal trends in invasive pneumococcal disease and pneumococcal serotypes over 7 decades. Clin Infect Dis. 2010;50(3):329–337. doi: 10.1086/649872. [DOI] [PubMed] [Google Scholar]
- 27.Hausdorff WP, Bryant J, Paradiso PR, et al. Which pneumococcal serogroups cause the most invasive disease: implications for conjugate vaccine formulation and use, part I. Clin Infect Dis. 2000;30(1):100–121. doi: 10.1086/313608. [DOI] [PubMed] [Google Scholar]
- 28.Hoti F, Erästö P, Leino T, et al. Outbreaks of Streptococcus pneumoniae carriage in day care cohorts in Finland—implications for elimination of transmission. BMC Infect Dis. 2009;9(102) doi: 10.1186/1471-2334-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller E, Andrews NJ, Waight PA, et al. Effectiveness of the new serotypes in the 13-valent pneumococcal conjugate vaccine. Vaccine. 2011;29(49):9127–9131. doi: 10.1016/j.vaccine.2011.09.112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.