

Abstract
Background
Mechanisms of glioma invasion remain to be fully elucidated. Glioma cells within glioblastoma multiforme (GBM) range from well-differentiated tumor cells to less-differentiated brain tumor-initiating cells (BTICs). The β2-subunit of Na+/K+-ATPase, called the adhesion molecule on glia (AMOG), is highly expressed in normal glia but is thought to be universally downregulated in GBM. To test our hypothesis that expression of AMOG is heterogeneous in GBM and confers a less invasive phenotype, we compared it between BTICs and differentiated cells from patient-matched GBM and then tested GBM invasion in vitro after AMOG overexpression.
Methods
Immunohistochemistry, immunoblotting, and real-time PCR were used to characterize AMOG protein and mRNA expression in tumor samples, BTICs, and differentiated cells. Matrigel invasion assay, scratch assay, and direct cell counting were used for testing in vitro invasion, migration, and proliferation, respectively.
Results
While AMOG expression is heterogeneous in astrocytomas of grades II–IV, it is lost in most GBM. BTICs express higher levels of AMOG mRNA and protein compared with patient-matched differentiated tumor cells. Overexpression of AMOG decreased GBM cell and BTIC invasion without affecting migration or proliferation. Knockdown of AMOG expression in normal human astrocytes increased invasion.
Conclusions
AMOG expression inhibits GBM invasion. Its downregulation increases invasion in glial cells and may also represent an important step in BTIC differentiation. These data provide compelling evidence implicating the role of AMOG in glioma invasion and provide impetus for further investigation.
Keywords: AMOG, Na+/K+-ATPase β2-subunit, glioblastoma, brain tumor-initiating cells, invasion
Diffusely infiltrative astrocytomas (World Health Organization [WHO] grades II–IV) are characterized by their potential to widely penetrate the normal brain parenchyma.1 Glioblastoma multiforme (GBM) is the highest-grade astrocytoma, and its patterns of dissemination have been directly linked to survival, where widespread dissemination is thought to give rise to more aggressive phenotypes.2 Increasing evidence suggests that all GBM tumors consist of heterogeneous populations of tumor cells in varied differentiation status,3 including well-differentiated tumor cells and less-differentiated stem cells. The less-differentiated stem cells from tumors are characterized by their expression of neural stem cell markers, such as CD133,4 nestin,5 and Sox2 (sex determining region Y-box 2).6 Functionally, these glioblastoma stem cells have been defined by their potent capacity for developing tumors when implanted in animals, as well as their ability to give rise to both stem cells and well-differentiated tumor cells.4,7–13 Thus, the less-differentiated tumor stem cells have also been called brain tumor-initiating cells (BTICs).4 In addition to their enhanced self-renewal and cell differentiation potential, BTICs have been shown to have increased resistance to chemo- and radiotherapy.4,7–13 Therefore, they have been identified as potentially important targets of GBM therapy.
While increasing evidence suggests that they play a role in glioma pathogenesis, very little evidence exists to date characterizing the invasive potential of BTICs. Limited evidence, based primarily on in vitro experiments, initially suggested that BTICs might be more invasive than differentiated glioma cells.14–16 However, the mechanism of the elevated invasive potential of BTICs has not been clearly delineated. Conversely, there is also evidence that cancer stem cells are not always invasive, depending on their phenotype, which further complicates our understanding of cancer stem cell biology.17
Recent research suggests that gliomas utilize ion channels, including Na+/K+-ATPase, to support their unusual growth and invasion by adjusting their shape and volume rapidly as they invade the brain parenchyma.18 The various isoforms of the β-subunit of Na+/K+-ATPase have been shown to act in regulating cellular adhesion, particularly in the context of cancer progression.19–21 The β2 isoform, also known as the adhesion molecule on glia (AMOG), is a heavily glycosylated protein that plays a role in cellular adhesion in the CNS.22,23 It is diffusely expressed in the gray matter but is expressed in only perivascular astrocytes in the white matter.24 Although AMOG is highly expressed in the normal adult CNS, evidence suggests that it may be downregulated in GBM.25 However, given the heterogeneity that exists among GBM tumors, it is not known whether this is universally true of GBM. Initial functional data characterizing AMOG suggest that it plays an active role in cellular adhesion and migration in the normal CNS.22,26 While loss of AMOG has been implicated in glioma invasion and migration, currently there are no data characterizing its function in invasion and migration specifically in human glioma cells.
To further our understanding of the role of AMOG in GBM and particularly in BTICs in the context of glioma migration and invasion, we specifically investigated (i) whether AMOG protein expression correlated with glioma grade, (ii) whether AMOG is globally downregulated in GBM, (iii) whether AMOG is differentially expressed in BTICs compared with differentiated GBM cells from patient-matched tumor tissues, (iv) whether overexpression of AMOG in low-expressing GBM cells decreases invasion and affects migration or proliferation, (v) whether AMOG knockdown increases invasion of normal glial cells, and (vi) whether downregulation of AMOG in GBM is associated with worse clinical outcome.
Methods
Preparation of Tumor Homogenates
Frozen GBM samples weighing ∼200–400 mg were thawed in 1 mL of tissue homogenization buffer and homogenized in a mechanical tissue grinder at 1300 rpm on ice (4°C). The homogenized mixture was centrifuged at 800 g for 15 min at 4°C. The supernatant was saved as raw homogenates, and the protein concentration was normalized using a standard bicinchoninic acid (BCA) assay.
Cell Lines and Cell Culture Techniques
Established cell lines (U251, G55, and U87) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 5% penicillin–streptomycin (100 units/mL penicillin and 100 μg/mL streptomycin).
Non-BTIC differentiated primary GBM cultures were established from pathologically confirmed fresh GBM tissue taken from surgical specimens (A.T.P., M.S.B.). Fresh tissue was minced, and cells were collected with Roswell Park Memorial Institute (RPMI)–1640 media supplemented with 10% heat-inactivated FBS, 5% penicillin–streptomycin, 5% nonessential amino acids, and 5% sodium pyruvate. To this mixture 1 mg/mL of collagenase IV was added and then incubated at 37°C for 1–2 h. Twenty-five milliliters of supplemented RPMI-1640 media (as noted) was added to quench collagenase activity, and the mixture was then centrifuged at 1500 rpm for 8 min. The supernatant was discarded, and the cell pellet was resuspended in 70% Percoll solution (Sigma). Subsequently, 30% Percoll solution was slowly layered on top to allow tumor cells to segregate to the top of the solution. The mixture was then spun at 1600 rpm for 20 min with no brake. The 30% Percoll solution layer was then removed and placed in supplemented RPMI-1640 media and centrifuged at 1500 rpm for 8 min. The supernatant was discarded, and the pellet was resuspended in supplemented RPMI-1640 media with 25% FBS, plated in a flask, and kept in an incubator at 37°C and 5% CO2.
BTIC cell cultures were established from pathologically confirmed fresh GBM tissue using a previously published protocol as described by Pollard et al.27 Briefly, fresh tissue was minced, enzymatically dissociated by papain, and passed through a series of cell strainers (70 μm and 40 μm). Subsequently, BTICs were cultured and expanded using Neurocult NS-A serum-free media (Stem Cell Technologies) with N2 supplement, B27 without vitamin A, epidermal growth factor (20 ng/mL), and fibroblast growth factor 2 (20 ng/mL).
Normal human astrocytes were obtained from ScienCell (#1800) and cultured in ScienCell normal human astrocyte media (#1801) in the presence of low concentration of FBS (2%) and astrocyte growth supplement (1%) (#1852).
After cells were grown to near confluence while media were replaced every 3–5 days, cultures were split 1:3 to 1:5. After 3–4 passages, cells were tested for the presence of stem cell markers, nestin, and Sox2 by immunoblotting.
Immunohistochemistry of Tissue Array
All tissue samples were obtained as de-identified specimens through the Brain Tumor Research Center in accordance with the ethical guidelines of the University of California, San Francisco (UCSF), and with appropriate approvals from the committee on human research. Core tissue samples from each tumor were fixed on tissue array slides containing 30 samples and stained immunohistochemically with an AMOG antibody (1:1000; Santa Cruz Biotechnology) as described previously.24 As a control, some sections on the same slide were incubated with nonimmune rabbit immunoglobulin (Ig) G at identical concentrations. Normal human cortex was used as a positive AMOG control. The scoring of staining was performed by a neuropathologist (J.J.P.) independently. Each sample was assigned an AMOG positivity score of +, ++, or +++, corresponding to 0%–25%, 26%–75%, or 76%–100%, respectively, of tumor cells staining positive. Mutation status of isocitrate dehydrogenase 1 (IDH1R132H) was assessed using IDH1 (R132H) immunohistochemistry (H09, Dianova).
Immunoblotting
Cells were lysed in lysis buffer (20 mmol/L Tris-HCl [pH 7.6], 150 mmol/L NaCl, 1 mmol/L Na2EDTA, 1 mmol/L ethylene glycol tetraacetic acid, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L beta-glycerophosphate, 1 mmol/L Na3VO4 [sodium orthovanadate], 1 mg/mL leupeptin, PhosSTOP phosphatase inhibitor cocktail [Roche], Complete protease inhibitor cocktail [Roche], and 0.1 mmol/L phenylmethylsulfonyl fluoride). Insoluble material was removed by centrifugation at 14 000 rpm for 20 min. Loading concentrations were then normalized to bovine serum albumin (BSA) standards using the BCA protein assay (Thermo Scientific). Five milligrams of protein samples were heated to 90°C in 5X sodium dodecyl sulfate (SDS) sample buffer (10% SDS, 1% b-mercaptoethanol, 20% glycerol, 0.2 mol/L Tris [pH 6.8], and 0.05% bromophenol blue) for 10 min and electrophoresed through 4%–20% Tris-glycine gels (Invitrogen), and resolved proteins were transferred to Immun-Blot polyvinylidene difluoride membranes (BioRad). Membranes were blocked with 5% milk and 3% BSA dissolved in Tris-buffered saline (TBS) containing 0.05% Tween-20 and then incubated with mouse anti-AMOG primary antibody (Santa Cruz Biotechnology) diluted 1:500 in 5% milk and 3% BSA dissolved in TBS containing 0.05% Tween-20 overnight at 4°C. After washing, membranes were incubated for 1 h at room temperature with horseradish peroxidase-conjugated goat anti-mouse antibody (Santa Cruz Biotechnology). Blots were developed with Amersham ECL (electrochemiluminescence) Immunoblotting Detection reagent (GE Healthcare).
Immunocytochemistry
Cells were plated on 20 × 20-mm glass coverslips at 2.5 × 105 cells per coverslip. When cells reached 75%–80% confluence, they were fixed in 4% paraformaldehyde for 15 min then washed 3 times in 10% FBS in phosphate buffered saline (PBS). Cells stained for intracellular markers (α-tubulin monoclonal from Cell Signaling and nestin monoclonal from Abcam) were permeabilized in 0.5% Triton X-100 (Sigma Aldrich) in 10% FBS-PBS for 10 min at 4°C then washed 3 times. Cells were blocked for 1 h in 10% FBS-PBS ± 0.1% Triton for intracellular staining. Extracellular staining with anti-AMOG primary antibody (Santa Cruz Biotechnology) was performed for 1 h at room temperature at a concentration of 1:50 in 10% FBS-PBS without Triton. Cells were washed 3 times for 10 min each then stained with secondary antibody conjugated to Alexa Fluor 488 at a concentration of 1:50. Cells were washed to remove unbound secondary antibody then mounted with 4′,6′-diamidino-2-phenylindole (DAPI; Vectashield HardSet Mounting Medium, Vector Labs). Slides were imaged using a Zeiss Axiovert 100 confocal microscope.
Quantification of mRNA Transcript
RNA was extracted using the RNeasy Mini Kit (Qiagen) and reverse transcribed using a Superscript III kit (Invitrogen) according to the manufacturer's protocol. Quantification via quantitative PCR was performed using Power SYBR Green Master Mix (Applied Biosystems) according to the manufacturer's protocols. The following primers were used: AMOG (5′-GTGGTT-GAGGAG-TGGAAG-GA/GGAGAC-AGTCTG-CAGCAT-CA-3′) and hypoxanthine-guanine phosphoribosyltransferase (HPRT) (5′-ATGCTG-AGGATT-TGGAAA-G/CTCCCA-TCTCCT-TCATCA-CA-3′). Using the standard practice, we reported the measured mRNA quantity as fold change over each respective cell line's mRNA levels of HPRT, a housekeeping gene used as a control to normalize mRNA amount used across different samples.
In vitro AMOG Overexpression in GBM Cultures
An established GBM cell line (U87) and a BTIC culture, identified to have minimal AMOG expression, were cultured under normal conditions in 6-well plates. When the cells were 70%–80% confluent, either the pcDNA3.myc.AMOG (overexpression plasmid) or the pcDNA3.myc vector (control plasmid) was transfected into the cells using FuGENE 6 Transfection Reagent (9 μL/mL). A FuGENE 6 Transfection Reagent:DNA ratio of 3:1 was used according to the manufacturer's protocol. The pcDNA3.myc.AMOG and the pcDNA3.myc vectors were courtesy of Dr David Gutmann (Washington University).
In vitro AMOG Knockdown With Short Hairpin RNA in Normal Human Astrocytes
Low passage normal human astrocytes were cultured in 6-well plates. When the cells were 70%–80% confluent, lentiviral short hairpin (sh)RNA constructs (pLKO.1 shRNA constructs for AMOG [ATP1B2] from Open BioSystems and packaged by UCSF ViraCore) were added to cells in the presence of 8 μg/mL polybrene (Millipore) for 24 h. Cells were split into 10-cm petri dishes and selected with 0.3 μg/mL puromycin 48 h after infection. Invasion assay and immunoblotting confirmation of AMOG knockdown were confirmed after 14 days of puromycin selection.
In vitro Matrigel Invasion Assay
GBM cells cultured under normal conditions were resuspended in DMEM and loaded into a transwell Matrigel invasion chamber (BD Biosciences) 48 h after transfection. Normal human astrocytes with AMOG knockdown using shRNA were assayed 16 days after infection and 14 days of antibiotic selection. Cells were allowed to migrate toward 10% FBS in normal media. This was performed in triplicate for each condition. After 24 h the chambers were washed with PBS, and the nonmigratory cells were scraped off using a cotton swab. Matrigel containing the invading cells was fixed using 4% paraformaldehyde for 15 min, washed with PBS, stained with 0.05% crystal violet for 15 min, and washed again with PBS before being allowed to dry. Invading cells were visualized under light microscopy at 10×, and at least 3 representative fields per Matrigel were assessed for cell count in order to determine the invasive index for the 2 conditions. AMOG overexpression was verified using immunoblotting of the same cells grown separately and collected for lysates at the time corresponding to the end of the 24-h invasion assay. Invasion index was calculated by normalizing the number of cells that invaded through the Matrigel in each condition to that of the control of each cell line (in paired comparisons), so that the baseline invasion of each cell line had an invasion index of 1. In multiple comparisons, the invasion index was normalized to that of the least invasive cell line.
In vitro Scratch Assay for GBM Migration
Adherent cell cultures were grown to near confluence and scratched using a stretched glass pipette, creating a uniform gap roughly 0.5 mm in width. The scratch technique has been described previously.28 After the scratch, fresh media were added, and cells were returned to incubation with time-lapse imaging every 12 min for 24 h. The amount of time for the cells to close the gap was noted, and the corresponding gap width was measured. Finally, velocity of migration was calculated. This was performed in triplicate for both cells transfected with control and AMOG constructs.
In vitro Proliferation Assay
Equal numbers of control and AMOG-overexpressing cells (∼200 000 cells) were plated in 6-well plates 48 h after transfection. At every 24 h after plating, cells were trypsinized, resuspended in 1 mL media, diluted 1:10 in trypan blue, and counted with a hemocytometer. The actual cell number was then calculated based on the hemocytometer count.
Survival Analysis Using The Cancer Genome Atlas Data
Kaplan–Meier analysis of overall survival based on differential AMOG expression of The Cancer Genome Atlas (TCGA) database was performed through the data portal and analytic tools available on the TCGA website. The group with intermediate AMOG expression (from 2-fold upregulated to 6-fold downregulated) was compared with a group with 6-fold or more downregulated AMOG expression. The intermediate group contained 175 patients, and the downregulated group contained 5 patients. Bonferroni correction for multiple hypothesis testing (5×) was applied, and thus, statistical significance was defined as P = .05/5 = .01. Analysis was performed in February 2012.
Results
The Loss of AMOG Expression Is Heterogeneous in Different Grades of Glioma
Given previous evidence suggesting that AMOG mRNA expression inversely correlated with glioma grade, we characterized AMOG protein expression in different grades of human glioma to assess whether such a relationship also existed at the protein level. Immunoblotting data showed that while AMOG was generally downregulated in GBM compared with anaplastic astrocytomas, grade II astrocytomas, and normal human brain cortex (NHB), one GBM sample had high expression of AMOG comparable to that of NHB (Fig. 1A).
Fig. 1.

AMOG expression is variably downregulated in gliomas. (A) Immunoblotting evaluation of AMOG expression in 4 GBM samples, 2 WHO grade III anaplastic astrocytomas, 2 WHO grade II gliomas, and normal human brain cortex (NHB). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Immunohistochemical evaluation of AMOG expression in different grades of gliomas (astrocytomas) in tissue arrays containing 59 GBM samples, 9 grade III anaplastic astrocytomas, and 8 grade II astrocytomas. NHB was used as a positive control, and white matter was used as a negative control (shown in right column). AMOG positivity is defined by categorizing the percentage of tumor cells that stain positive for AMOG into 3 categories (representative pictures in the left column): + for 0%–25%, ++ for 26%–75%, and +++ for 76%–100%. Scale bar: 100 μm. (C) Quantification of AMOG positivity in glioma tissue array samples in part B. The bars represent the number of tumor samples that fall into each staining category for each tumor grade. (D) Quantification of IDH1 wild types and mutants in the glioma tissue array. As expected, more IDH1 mutants were found in grades II and III, while more IDH1 wild types were in grade IV.
To further test whether AMOG protein expression correlated with glioma grade in tumor tissues and whether AMOG was globally downregulated in GBM, we immunohistochemically stained paraffin-embedded glioma tissue arrays consisting of 59 GBM tumors, 9 grade III anaplastic astrocytomas, and 8 grade II astrocytomas (Fig. 1B and C and Table 1). Using NHB as a positive control and white matter as negative control, we quantified AMOG expression based on the number of glioma cells that stained positive for AMOG (Fig. 1C). There was a trend toward decreased AMOG expression in GBM compared with lower-grade glioma specimens, but this did not reach significance (P = .257). Overall, our immunohistochemistry data showed that even though the majority of GBM samples had significant loss of AMOG expression, the expression of AMOG in GBM was heterogeneous among different tumors as well as among individual tumor cells within each tumor (Fig. 1C).
Table 1.
AMOG is heterogeneously expressed in gliomas
| AMOG Positivity |
|||
|---|---|---|---|
| Tumor grade | + | ++ | +++ |
| Grade II | 4 | 2 | 2 |
| Grade III | 3 | 3 | 3 |
| Grade IV | 35 | 16 | 8 |
Immunohistochemical evaluation of AMOG expression in different grades of gliomas (astrocytomas) in tissue arrays containing 59 GBM, 9 grade III anaplastic astrocytomas, and 8 grade II astrocytomas. The normal human brain cortex was used as a positive control, and the white matter was used as a negative control. AMOG positivity is defined by categorizing the percentage of tumor cells that stain positive for AMOG into 3 categories: + for 0%–25%, ++ for 26%–75%, and +++ for 76%–100%. The numbers of samples corresponding to 1 of the 3 categories of staining are recorded in this table for each grade of glioma.
Because previous studies showed that AMOG mRNA was differentially expressed in primary and secondary GBM, we tested whether AMOG protein expression differed based on IDH1 mutation status. Immunohistochemical staining of the GBM tumor arrays for the IDH1 R132H variant showed that while the majority of grade II and III gliomas were mutants, most GBM tumors were IDH1 wild type, both as expected (Fig. 1D). We found that overall there was no difference in AMOG positivity between IDH1 wild-type and mutant samples in each glioma grade (Table 2). Among GBM samples, AMOG positivity was low in the majority of both IDH1 wild types and mutants (Table 2). Overall, our data suggest that IDH1 mutation status and AMOG expression are not significantly associated.
Table 2.
AMOG expression does not differ by IDH1 mutation status
| AMOG Positivity | IDH1 Mutation Status (total n) |
Glioma Grade P | |
|---|---|---|---|
| Wild type (2) | Mutant (5) | Grade II | |
| + | 50% | 60% | P = .333* |
| ++ | 0% | 40% | |
| +++ | 50% | 0% | |
| Wild type (2) | Mutant (7) | Grade III | |
| + | 0% | 43% | P = .25* |
| ++ | 0% | 43% | |
| +++ | 100% | 14% | |
| Wild type (43) | Mutant (15) | Grade IV | |
| + | 53% | 73% | P = .356* |
| ++ | 33% | 13% | |
| +++ | 14% | 13% | |
*Fisher's exact test.
AMOG positivity of tumor samples from each grade was compared between IDH1 wild types and mutants. The percentages of samples that fall into each category of staining are shown. No differences were found in grades II–IV.
BTICs Overexpress AMOG Compared With Differentiated GBM Cells
Given our immunohistochemistry data showing that AMOG was variably expressed in different GBM tumors as well as in individual tumor cells within each tumor sample, we hypothesized that AMOG expression may be different in BTICs compared with more differentiated glioma cells in GBM. We therefore investigated whether BTICs differentially expressed AMOG compared with non-BTIC GBM cultures. We first established both BTIC and differentiated primary glioma cultures from surgically resected GBM tumors as described in the Methods section. All of our BTIC monolayer cultures were capable of forming neurospheres when cultured in the absence of laminin-coated substrate (Fig. 2B). The tumorigenicity of these cultured BTICs was also confirmed by injecting these cells into the brains of athymic nude mice to demonstrate that they were capable of forming brain tumors (data not shown). Additionally, our BTICs highly expressed 2 well-known stem cell markers nestin and Sox2, whereas our differentiated cultures did not (Fig. 2A and D).
Fig. 2.

BTICs overexpress AMOG compared with differentiated glioma cells. (A) Protein expressions of AMOG in normal human brain cortex (NHB), normal human astrocytes (NHA), 3 established GBM cell lines (U251, G55, U87), 7 primary differentiated GBM cultures, and 3 BTIC cultures were assessed by immunoblotting. Nestin expression was assessed to confirm the stemness of BTIC culture. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) GBM stem cells cultured in monolayer are capable of forming neurospheres in the absence of laminin substrate. (C) AMOG mRNA expression was quantified in 4 pairs of patient-matched BTIC and differentiated GBM cultures. AMOG mRNA levels are normalized and are expressed as fold increase over HPRT mRNA levels for each sample. The comparisons are made within each pair of cultures. (*P < .01; **P < .001). (D) AMOG protein expression was assessed by immunoblotting for 5 pairs of patient-matched BTIC and non-BTIC cultures (inclusive of the 4 pairs in Fig. 2C: patients 1–4). Nestin and Sox2 expressions were assessed to confirm the stemness of BTIC culture. GAPDH was used as a loading control. (E) Representative immunocytochemical staining of AMOG and nestin staining of patient-matched BTIC and differentiated primary GBM cultures. Positive control: α-tubulin; negative control: nonspecific IgG.
To analyze the expression levels of AMOG in BTICs compared with differentiated tumor cell cultures derived from GBM, we first prepared cell lysates from 10 differentiated cultures, which included 3 established glioma cell lines (U251, G55, and U87) and 7 primary GBM cultures. The expression levels of AMOG in these differentiated cultures were compared with the levels in 3 BTIC cultures using normal human astrocytes and NHB as positive controls (Fig. 2A). We detected strong positive AMOG expression in a BTIC culture but not in any differentiated GBM culture.
To assess the true relative AMOG expression in BTIC and differentiated glioma cells, we needed to compare BTICs and glioma cells from the same tumor in order to control for the inherent heterogeneity present in GBM samples that could give rise to high AMOG expression due to random variation. We therefore established patient-matched BTIC and differentiated cultures from primary GBM in order to determine the specificity of high AMOG expression in BTICs (Fig. 2C and D). Using real-time PCR, we first measured the AMOG mRNA transcript levels in patient-matched BTIC and differentiated cultures from 4 primary GBM tumors (patients 1–4; Fig. 2C). There was a dramatic increase in AMOG mRNA transcript in each patient-matched BTIC (P < .01), and 3 of the 4 BTICs had a >100-fold increase compared with their respective differentiated GBM cultures (P < .001). Next, we investigated whether AMOG protein expression was elevated in patient-matched BTICs. Comparing the same 4 primary GBM tumors (patients 1–4) along with 1 more GBM sample (patient 5), we observed increased AMOG expression in 3 of the 5 BTIC cultures but not in any of the matching non-BTIC cultures (Fig. 2D). All BTIC cultures expressed nestin and Sox2, while differentiated GBM cells did not (Fig. 2D). Thus, among our patient-matched GBM cell cultures, only BTICs highly expressed AMOG protein. We also performed immunocytochemistry to visually confirm the expression of AMOG on the cell surface (Fig. 2E). As expected, BTICs had strong staining of AMOG and nestin, whereas primary cultures did not. Overall, our results suggested that BTICs had higher AMOG mRNA expression even when such difference in protein expression was not detectable through immunoblotting.
Interestingly, the 2 BTICs (from patients 1 and 3) with a >1000-fold increase in mRNA transcript level were the only 2 BTICs from among patients 1–4 that exhibited increased AMOG protein expression (Fig. 2C and D). Conversely, the 2 BTICs from patients 2 and 4 had a <1000-fold increase in mRNA, and we did not detect increased protein expression on immunoblotting. Given the low baseline expression of AMOG in differentiated GBM cultures, it was possible that the smaller fold increase in AMOG in BTICs from patients 2 and 4 could explain the lack of detectable amount of AMOG protein in their corresponding BTICs. Taken together, our data suggest that the degree of AMOG mRNA overexpression may correlate with that of AMOG protein overexpression.
Overexpression of AMOG Decreases GBM Cell Invasion but Not Migration or Proliferation
To assess the functional significance of differential AMOG expression between tumor-matched BTICs and differentiated GBM cells, we investigated the correlation of AMOG expression with invasion by comparing the in vitro baseline invasion of patient-matched BTICs and differentiated cultures from patients 3 and 4 (Fig. 3A). Both BTICs exhibited reduced invasion through a Matrigel-coated insert compared with their patient-matched differentiated cultures. Moreover, the BTICs from patient 3 with higher relative AMOG expression had even further reduced invasion compared with the BTICs from patient 4 (P < .05; Figs 2D and 3A). Our data suggest that higher AMOG expression correlates with slower GBM invasion.
Fig. 3.

Overexpression of AMOG decreases GBM invasion. (A) The invasion of patient-matched BTIC and differentiated cells from patients 3 and 4 was assessed by seeding the same number of cells onto the Matrigel insert and allowing them to migrate toward 10% FBS in the bottom well. The bar graphs show each cell line's invasion index, which is calculated by normalizing the number of cells that invaded through the Matrigel in each cell line to that of the least invasive cell line. The least invasive cell line has an invasion index of 1 (*P < .05, **P < .001). (B) GBM cells were transfected with either a control vector or an AMOG overexpression vector; after 48 h, equal numbers of cells from both conditions were seeded onto the Matrigel insert and allowed to migrate toward the bottom well containing 10% FBS in media. Shown are representative pictures of control cells and AMOG-overexpressing cells from the same cell line. (C) The invasion index of the control and AMOG-overexpressing cells from the U87 cell line and a low AMOG-expressing BTIC culture. The invasion index was normalized to each cell line's control so that the baseline invasion of each cell line has an invasion index of 1 (*P < .001). GAPDH, glyceraldehyde 3-phosphate dehydrogenase. Insert: The overexpression of AMOG was confirmed by immunoblotting.
To test whether AMOG overexpression alone in a GBM cell line was sufficient to inhibit its invasion in vitro, we overexpressed AMOG in the U87 cell line, which has a low baseline AMOG expression, and compared its rate of invasion with that of U87 cells transfected with the control vector (Fig. 3B and C). After 24 h, the number of U87 cells overexpressing AMOG that completely invaded through the Matrigel was about 50% that of the control U87 cells, translating to about 50% reduction in invasion among U87 cells overexpressing AMOG (Fig. 3C). We confirmed the overexpression of AMOG through immunoblotting comparing AMOG expression between control U87 cells and U87 AMOG-expressing cells (Fig. 3C).
To test whether the effect of reduced invasion after AMOG overexpression was unique to U87 cells or could be generalized to other GBM cell types, regardless of differentiation status, we selected a BTIC line (from patient 4) with one of the lowest AMOG expressions among BTICs. Thus, it was an ideal BTIC candidate for testing the effects of AMOG overexpression. We transfected this BTIC line with an AMOG expression vector and observed that the rate of invasion was reduced also by ∼50%, similar to the effect observed in the U87 cell line (Fig. 3C). Taken together, our data suggest that AMOG overexpression is sufficient to inhibit invasion of not only differentiated GBM cultures, but also BTICs that have relatively lower AMOG expressions.
Given that tumor cell migration is thought to be a process involving different cellular mechanisms than those in tumor cell invasion, we tested our hypothesis that AMOG overexpression does not change the rate of migration of GBM cells. Using the same cell lines utilized in our invasion assays in Fig. 3C, we compared the rates of migration using a scratch wound assay (Fig. 4A). The velocity at which the cells migrated across a gap of fixed width over a period of 12–24 h was not statistically significantly different between cells overexpressing AMOG and control cells for both the U87 and the BTIC cell lines (Fig. 4B).
Fig. 4.

Overexpression of AMOG does not affect GBM cell migration. (A) GBM cells were allowed to migrate across a scratched gap of fixed width and imaged every 12 min while in incubation. The distance traveled and time required for the cells to close the gap were calculated. (B) Based on distance traveled and time required to close the gap, the velocity was calculated and compared between control and AMOG-overexpressing cells in the U87 cell line and a low AMOG-expressing BTIC culture.
Next, we tested whether AMOG overexpression could alter the rate of GBM cell proliferation using the same cell lines. Due to the known function of Na+/K+-ATPase in cellular metabolism, we measured proliferation using direct cell counting with a hemocytometer, rather than with standard cell proliferation assays that typically use cell metabolites, in order to control for potential confounders. When measured serially every 24 h over a period of 72 h, there were no statistically significant differences between AMOG-overexpressing cells and control cells in either cell line (Fig. 5).
Fig. 5.

Overexpression of AMOG does not affect GBM cell proliferation. Equal numbers of GBM cells were seeded 48 h after transfection with either a control vector or an AMOG overexpression vector, and cell numbers were determined every 24 h for 72 h by direct counting on a hemocytometer. Both U87 cells and BTIC cultures were used.
AMOG Knockdown Increases Invasion in Normal Human Astrocytes
Given that AMOG is normally highly expressed in glia but lost in most GBM, we examined whether loss of AMOG could be a potential mechanism contributing to the invasiveness of malignant gliomas. To test this, we performed stable knockdown of AMOG in normal human astrocytes using lentiviral particles carrying AMOG shRNA. The in vitro invasiveness of normal human astrocytes was significantly increased after AMOG knockdown (Fig. 6), suggesting that loss of AMOG could be a contributing factor to GBM's aggressive invasion of normal brain.
Fig. 6.

AMOG knockdown increases invasion in normal human astrocytes. Normal human astrocytes with stable AMOG knockdown using lentiviral shRNA particles exhibited significantly increased invasion. AMOG knockdown was confirmed by immunoblotting, shown in insert (*P < .001). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Downregulation of AMOG Correlates With Decreased Overall Survival
Given our in vitro data suggesting that AMOG downregulation may contribute to GBM invasion, we hypothesized that dramatic loss of AMOG would be associated with a highly invasive GBM phenotype and worse overall survival. To assess whether there was a correlation between loss of AMOG expression and clinical outcome for GBM patients, we performed a Kaplan–Meier analysis of survival data for GBM using TCGA data retrieved in February 2012. First, we noted that there was a wide range of AMOG expression among GBM cells in TCGA. Second, we found that GBM with 6-fold or more downregulation of AMOG mRNA was associated with worse overall survival compared with those with intermediate AMOG expression (P < .001; Fig. 7). This correlation remained significant after Bonferroni correction for multiple hypothesis testing. We also performed a multivariate analysis controlling for age, IDH1 mutation status, and methylation status of O6-DNA methylguanine-methyltransferase (MGMT). However, AMOG was not an independent predictor of overall survival (P = .169, hazard ratio = 2.03, 95% confidence interval = 0.740–5.562).
Fig. 7.
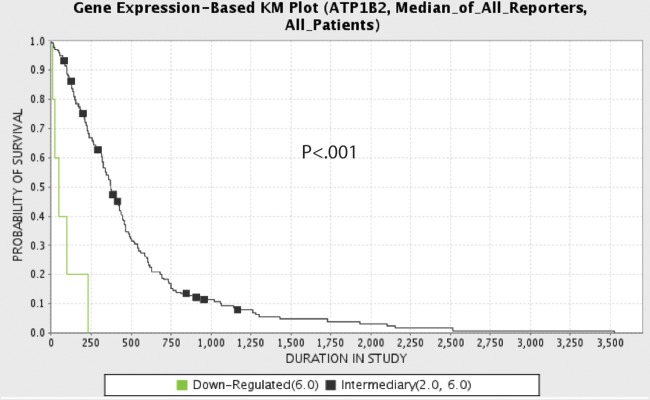
Downregulation of AMOG correlates with worse clinical outcome. Kaplan–Meier (KM) analysis comparing overall survival between patients whose GBM had intermediate (black) AMOG expression (from 2-fold upregulated to 6-fold downregulated) and patients whose GBM had downregulated (green) AMOG expression (6-fold or more downregulated) (P < .001). Expression data represent mRNA microarray expression data from TCGA database queried in February 2012.
Discussion
The aberrant expressions of the various subunits of Na+/K+-ATPase and their activity have been implicated in the general development and progression of various cancers,29,30 as well as in cancer cell proliferation, migration, and apoptosis.31,32 By using the energy from ATP to establish asymmetric distributions of ions across the cell membrane, Na+/K+-ATPase couples metabolic energy to cellular functions and to signaling events both between and within cells.33 In addition to its essential function in maintaining cell homeostasis, including ion translocation, maintenance of pH balance, and cell volume, Na+/K+-ATPase has been shown to regulate cytoskeletal remodeling, cell polarity, and lamellipodia formation.34–36 The mechanism through which Na+/K+-ATPase affects cellular migration and invasion in cancer has not been clearly delineated. However, evidence suggests that it can serve as a linker protein or a signal transducer between the extracellular environment and the proteins making up the cytoskeleton.37,38 In particular, interactions exist between cytoskeletal components and Na+/K+-ATPase through indirect interaction via ankyrin and spectrin as well as direct interaction with actin filaments.37,38 Additionally, recent evidence suggests that persistent-direction cell migration requires ion transport proteins such as Na+/K+-ATPase as directional sensors and membrane potential differences in order to maintain directedness.39
AMOG is an isoform of the beta subunit of Na+/K+-ATPase highly expressed in the CNS and thought to play a role in cellular adhesion and migration in the CNS during development. Our study is the first to show that loss of AMOG expression in gliomas is not limited to GBM, and among GBM cells its expression can be heterogeneous. While the loss of AMOG expression is heterogeneous in gliomas of WHO grades II–IV, the majority of GBM tumors have few cells that are positive for AMOG. We also found a clinical correlation between the loss of AMOG expression in GBM and a dramatic decrease in overall survival, suggesting that loss of AMOG may give rise to a more aggressive phenotype. Additionally, we found that restoring AMOG expression in GBM cells in vitro after de novo overexpression of AMOG significantly decreased invasion without affecting proliferation and migration. Knockdown of AMOG expression in normal human astrocytes produced the opposite effect of increasing their invasion. Overall, our data suggest that loss of AMOG may be one mechanism that contributes to the increased invasiveness of GBM cells.
Consistent with previously published findings,25,40 our immunoblotting and immunohistochemistry data showed that the majority of GBM cells have significantly downregulated AMOG expression. While previous reports characterized only a limited number of GBM samples and suggested that AMOG was globally downregulated in GBM, our tumor tissue arrays contained over 50 GBM tumors. With this increased sample size, our study was able to demonstrate that not all GBM cells have a dramatic loss of AMOG expression and that not all the tumor cells in GBM have the same level of AMOG expression. Given the increasing evidence showing that heterogeneity exists both across GBM tumors as well as within each GBM tumor, it is not surprising that AMOG expression varied in our samples.
One important aspect of the heterogeneity of tumor cells within GBM is the existence of glioma stem cells, that is, BTICs. BTICs have been functionally defined by self-renewal, cell differentiation potential, resistance to chemo- and radiotherapy, and increased tumor propagation and tumorigenic potential in vivo.4,7–13 To date, however, little data exist that characterize the invasive properties of BTICs. We used a known culturing technique previously described by Pollard et al27 to establish adherent BTIC cultures that express stem cell markers. The stem cell characteristics of our BTIC cultures were confirmed by expression of the stem cell markers nestin and Sox2 as well as by their ability to form neurospheres and differentiated tumors in xenotransplantation models.4,27 While this culturing technique may have its limitations, it allows us to directly compare BTICs and non-BTICs (differentiated tumor cells) from the same tumor tissue and therefore offers the potential to discover key molecular differences that underlie the ability of BTICs to recapitulate aggressively differentiated tumors in animal models. One argument against the use of adherent BTIC cultures involves the use of epidermal growth factor and fibroblast growth factor in media, which may falsely elevate protein expression. However, we have data showing that the expression levels of other known key modulators of GBM biology, such as Akt and mitogen-activated protein kinase, are not different between GBM BTIC and differentiated cultures (data not shown). Thus, we believe that overexpression of AMOG in GBM BTICs is a valid finding that warrants further investigation of downstream effectors as well as potential binding proteins of AMOG.
Recently, a few in vitro characterization studies have raised the possibility that BTICs may be potentially more invasive than non-BTICs. Given previous evidence suggesting that decreased AMOG expression may be associated with increased GBM invasion, we had expected to see a downregulation of AMOG in BTICs compared with non-BTICs. However, our data showed that AMOG expressions at both the protein and mRNA levels were increased in BTICs compared with primary differentiated GBM cells from patient-matched tumors. Therefore, our data suggest that BTICs are more likely to retain the high AMOG expression found in normal glia than are the majority of differentiated GBM cells. The loss of AMOG, therefore, may represent an important step in BTIC differentiation. This finding sheds light on the pathology of GBM, as BTICs have been thought of as progenitor cells that give rise to and sustain the growth of GBM, especially in the context of resistance to treatment. Since overexpression of AMOG could be associated with a less invasive phenotype and both BTIC lines were less invasive in our direct comparison of 2 patient-matched BTIC and differentiated cell lines, our findings suggest that they may be less invasive than previously thought, particularly when compared with differentiated GBM cells. Indeed, our own functional data comparing patient-matched BTICs and differentiated cultures showed that BTICs were less invasive in vitro. While our findings may seem to contradict existing reports showing increased invasiveness of BTICs, our methodology for testing invasion was different compared with previous studies. These previous studies used either a purely CD133-positive population or higher (>10%) concentrations of serum as a chemoattractant. Because serum differentiates stem cells and is not used during routine stem cell culture, it is possible that a very high serum concentration can act as a stronger chemoattractant for stem cells than for differentiated cells and induces greater invasion potential of stem cells in in vitro testing conditions.
If our data can be generalized, then mechanistically speaking as BTICs differentiate, they may give rise to more mobile progeny tumor cells that can then disseminate into the brain parenchyma. Consequently, glioma invasion may not be exclusively through BTICs but rather mostly through the actions of the more differentiated tumor cells. However, because the sample size of our direct comparison (Fig. 3A) was small, it is still difficult at this point to draw definitive conclusions. As more markers of GBM invasion are studied in the context of BTICs, we may find out more about the invasive properties of these cells, given their increasing importance in our understanding of GBM.
Although expression of both nestin and Sox2 appeared to mimic that of AMOG in our data, we noted high protein expression of the stem cell markers in all of our BTICs, while AMOG expression was less uniform (Fig. 2D). This suggested that the expressions of stem cell markers with AMOG are not directly correlated. Additionally, no evidence exists based on a thorough literature review and examination of the data from transcription factor chromatin immunoprecipitation sequencing of the Encyclopedia of DNA Elements (ENCODE Genome Browser) suggesting that Sox2 or nestin is directly involved in the regulation of AMOG gene expression. While a direct mechanistic relationship of Sox2 or nestin with AMOG may not necessarily exist, there is a possibility that an indirect one may be possible. Currently, the only reported transcriptional factor for AMOG is specificity protein (Sp) 1.41 A review of the literature suggests that Sp1 may play a role in the regulation of nestin and Sox2 expression in the CNS, particularly during development.42–46 This suggests that AMOG expression in BTICs may be regulated by an upstream effector common to the Sox2, nestin, and AMOG transcription mechanisms. However, in GBM, it has also been shown that AMOG downregulation is related to a hypermethylation of the promoter region,40 thus the regulation of its expression in GBM can be attributed to DNA methylation as well.
Because misexpressions of the various subunits of Na+/K+-ATPase and their activity have already been implicated in the general development and progression of various cancers, as well as in cancer cell proliferation, migration, and apoptosis, AMOG misexpression in GBM may contribute to the pathophysiology of GBM invasion. Moreover, a growing body of literature recognizes that the subunits of Na+/K+-ATPase may be important therapeutic targets for the treatment of cancer.
To that end, we tested whether restoring AMOG expression to GBM cells could reverse their invasive phenotype. While previous studies have implicated that loss of AMOG in GBM may contribute to its infiltrative and migratory capabilities, most of the experiments were based on the rat glioma cell line C6.25 To date, no data exist from functional experiments produced from a human glioma cell line specifically testing AMOG's role in glioma invasion. Our study presents functional data from 2 separate AMOG low-expressing GBM cell cultures, 1 established GBM culture, and 1 BTIC culture. After transfecting these cells with an AMOG expression vector, we observed a significant decrease in invasion for each cell line. Thus, our data demonstrate that restoring AMOG expression in these glioma cells was sufficient to slow down their invasion, regardless of the differentiation status of the original cell line. Moreover, our data showing increased invasion of normal human astrocytes after stable AMOG knockdown suggest that loss of AMOG may be associated with increased glioma invasion and may be one mechanism by which glioma cells acquire increased invasive potential. Additionally, loss of AMOG could represent an important step in BTIC differentiation.
Because AMOG was previously thought to be an adhesion molecule, it has been implicated in cellular adhesion, aggregation, and even migration.22,25,47 However, its role in glioma migration has not been tested in a human glioma cell line. Our functional data show that restoring AMOG expression in the 2 GBM lines that had decreased invasion had no effects on their rates of migration across a gap on their normal substrate in vitro. Given that AMOG had previously been implicated in normal and tumor cell migration, our data were therefore unexpected. However, because the scratch wound assay had not been used previously in migration experiments to functionally characterize AMOG, the differences in the way migration was assessed between our experiments and other studies could explain the differences in the findings. Nevertheless, our data showing that restoring AMOG expression decreases glioma invasion but not migration complicate the understanding that AMOG is simply an adhesion molecule that affects cell mobility by holding cells in place.
Our data also showed that restoring AMOG expression was not sufficient to decrease glioma proliferation in vitro. This result is consistent with previous findings based on experiments performed on both C6 rat glioma cells and U87 human glioma cells.25,47 Even though AMOG has been shown to activate the mammalian target of rapamycin/Akt pathway known to modulate glioma proliferation,47 it does not seem to affect proliferation.
Interestingly, our data showed that normal human astrocytes, which normally have high AMOG expression, became more invasive after AMOG knockdown. This suggests that loss of AMOG could be one mechanism by which malignant glial cells become invasive. One limitation of using only normal human astrocytes, however, is that there is a possibility that the same effect may not necessarily be observed in GBM cells that have high AMOG expression. While knocking down AMOG in a BTIC line with high AMOG expression would be valuable and further characterizes the role of AMOG in GBM invasion, our data based on normal human astrocytes do provide a first glimpse of the potential effects of loss of AMOG as well as the impetus for future experiments on GBM cells.
Moreover, our in vitro results would be further validated by a set of parallel in vivo experiments showing that GBM cells that stably overexpress AMOG after transfection would be less invasive and result in improved survival in animals. Conversely, GBM cells with AMOG knockdown grown in animals would potentially result in tumors with a more invasive phenotype, leading to shorter survival. While this report is limited by the lack of in vivo data, the in vivo experiments we propose are the subject of ongoing and future investigations.
Finally, our Kaplan–Meier analysis of TCGA data showed a direct correlation between the downregulation of AMOG and decreased overall survival among GBM patients. One limitation of our analysis, however, was that it was only a univariate analysis. Thus, we performed a Cox regression analysis controlling for age, IDH1 mutation, and MGMT methylation status and found that AMOG expression did not independently predict overall survival. In this multivariate model, however, only age was an independent predictor, and neither IDH1 mutation nor MGMT methylation status was an independent predictor (data not shown). Overall, we were not surprised that the downregulation of the AMOG gene was not an independent predictor of survival outcome considering that survival depends on multiple factors clinically, and very few genes to date have been shown to independently predict survival by virtue of varying gene expression. While obtaining the same correlation from multiple datasets would be ideal, given that it is difficult to obtain another set of expression data of the scale comparable to TCGA's, we were not able to perform independent validation using another, equivalent set.
The mechanism by which glioma cells can lose AMOG expression has not been thoroughly delineated. Some evidence suggests that downregulation is partially contributed by the methylation of 5′ cytosine–phosphate–guanine islands of the AMOG gene.40 Additionally, because the AMOG gene (ATP1B2) maps to chromosomal band 17p3.1 adjacent to the TP53 tumor suppressor gene, it is located in a region known to have frequent allelic losses in diffuse astrocytic gliomas.48 Notably, our immunoblot data showing that overexpressed AMOG in low-expressing GBM cells has identical molecular weight to AMOG in normal brain suggest that these GBM cells are capable of fully glycosylating this Na+/K+-ATPase enzyme subunit. Thus, even though AMOG may be downregulated in these cells, their posttranslational machinery for modifying and glycosylating this protein appears to be intact.
It has been shown that the routes of glioma invasion are associated with distinct anatomic structures.49,50 Specifically, they follow myelinated axons, the basement membranes of blood vessels, and the subependymal lining.49,50 If loss of AMOG is one mechanism of glioma invasion, then our data showing that AMOG expression is downregulated in the white matter compared with the cortex are consistent with the idea that glioma cells can more easily invade through brain via white matter tracts.
In summary, we present evidence that AMOG expression in GBM inhibits its invasion, suggesting that the loss of AMOG in GBM may therefore contribute to its invasive properties. While the loss of AMOG expression is variable in different grades of glioma, the majority of GBM tumors have dramatic loss of AMOG expression. GBM cells with restored de novo AMOG expression by an overexpression vector exhibited dramatically decreased invasion but no difference in migration or proliferation compared with control GBM cells. Because BTICs overexpress AMOG at both protein and mRNA transcript levels compared with differentiated GBM cells from tumor-matched samples, it is possible that loss of AMOG may represent an important step in BTIC differentiation. In normal human astrocytes, downregulation of AMOG led to an increase in their invasiveness, suggesting that loss of AMOG may be one possible mechanism contributing to GBM invasion. Moreover, among GBM patients, the loss of AMOG is associated with worse overall survival based on TCGA data. Our study warrants further investigation characterizing AMOG's role in GBM biology and, more specifically, the role of AMOG in BTIC migration and invasion.
Funding
M.Z.S. is a Howard Hughes Medical Institute Medical Research Fellow. M.S. was supported by a grant from the Doris Duke Charitable Foundation. Dr Oh received a National Research Service Award from the National Institutes of Health (F32NS073326-01). Dr Ivan is funded by the National Research Education Foundation through the American Association of Neurological Surgeons. Dr Parsa is partially funded by the Reza and Georgianna Khatib Endowed Chair in Skull Base Tumor Surgery.
Conflict of interest statement. None declared.
Acknowledgments
We thank Dr David Gutmann (Washington University in St Louis) for providing the AMOG overexpression vectors. We also thank King Chiu and Cynthia Cowdrey for providing assistance with tissue array preparation and immunohistochemistry assistance. Finally, we thank Dr Joseph Costello for providing one of the BTIC lines.
References
- 1.Alves TR, Lima FR, Kahn SA, et al. Glioblastoma cells: a heterogeneous and fatal tumor interacting with the parenchyma. Life Sci. 2011;89(15–16):532–539. doi: 10.1016/j.lfs.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 2.Parsa AT, Wachhorst S, Lamborn KR, et al. Prognostic significance of intracranial dissemination of glioblastoma multiforme in adults. J Neurosurg. 2005;102(4):622–628. doi: 10.3171/jns.2005.102.4.0622. [DOI] [PubMed] [Google Scholar]
- 3.Venere M, Fine HA, Dirks PB, Rich JN. Cancer stem cells in gliomas: identifying and understanding the apex cell in cancer’s hierarchy. Glia. 2011;59(8):1148–1154. doi: 10.1002/glia.21185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 5.Messam CA, Hou J, Major EO. Coexpression of nestin in neural and glial cells in the developing human CNS defined by a human-specific anti-nestin antibody. Exp Neurol. 2000;161(2):585–596. doi: 10.1006/exnr.1999.7319. [DOI] [PubMed] [Google Scholar]
- 6.Ellis P, Fagan BM, Magness ST, et al. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci. 2004;26(2–4):148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- 7.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 8.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66(16):7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 9.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 11.Shi L, Zhang S, Feng K, et al. MicroRNA-125b-2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. Int J Oncol. 2012;40(1):119–129. doi: 10.3892/ijo.2011.1179. [DOI] [PubMed] [Google Scholar]
- 12.Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu SP, Yang XJ, Zhang B, et al. Enhanced invasion in vitro and the distribution patterns in vivo of CD133+ glioma stem cells. Chin Med J (Engl) 2011;124(17):2599–2604. [PubMed] [Google Scholar]
- 15.Cheng L, Wu Q, Guryanova OA, et al. Elevated invasive potential of glioblastoma stem cells. Biochem Biophys Res Comm. 2011;406(4):643–648. doi: 10.1016/j.bbrc.2011.02.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu BZD, Tao J, Tie X, Wu A, Wang Y. Human brain glioma stem cells are more invasive than their differentiated progeny cells in vitro. J Clin Neurosci. 2012;19:130–134. doi: 10.1016/j.jocn.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Biddle A, Liang X, Gammon L, et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011;71(15):5317–5326. doi: 10.1158/0008-5472.CAN-11-1059. [DOI] [PubMed] [Google Scholar]
- 18.Ransom CB, O'Neal JT, Sontheimer H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J Neurosci. 2001;21(19):7674–7683. doi: 10.1523/JNEUROSCI.21-19-07674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu K, Zhang J, Ren J-J, Wang X-J, Yang H-L, Lin P. [Interference of human Na/K-ATPase B1 subunit on proliferation and migration of gastric adenocarcinoma cell line SGC-7901] Ai zheng=Aizheng = Chin J Cancer. 2009;28(3):225–231. [PubMed] [Google Scholar]
- 20.Vagin O, Dada LA, Tokhtaeva E, Sachs G. The Na-K-ATPase alpha1beta1 heterodimer as a cell adhesion molecule in epithelia. Am J Physiol Cell Physiol. 2012;302(9):C1271–1281. doi: 10.1152/ajpcell.00456.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vagin O, Tokhtaeva E, Sachs G. The role of the beta1 subunit of the Na,K-ATPase and its glycosylation in cell-cell adhesion. J Biol Chem. 2006;281(51):39573–39587. doi: 10.1074/jbc.M606507200. [DOI] [PubMed] [Google Scholar]
- 22.Antonicek H, Persohn E, Schachner M. Biochemical and functional characterization of a novel neuron-glia adhesion molecule that is involved in neuronal migration. J Cell Biol. 1987;104(6):1587–1595. doi: 10.1083/jcb.104.6.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gloor S, Antonicek H, Sweadner KJ, et al. The adhesion molecule on glia (AMOG) is a homologue of the beta subunit of the Na,K-ATPase. J Cell Biol. 1990;110(1):165–174. doi: 10.1083/jcb.110.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boer K, Spliet WGM, van Rijen PC, Jansen FE, Aronica E. Expression patterns of AMOG in developing human cortex and malformations of cortical development. Epilepsy Res. 2010;91(1):84–93. doi: 10.1016/j.eplepsyres.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 25.Senner V, Schmidtpeter S, Braune S, et al. AMOG/beta2 and glioma invasion: does loss of AMOG make tumour cells run amok? Neuropathol Appl Neurobiol. 2003;29(4):370–377. doi: 10.1046/j.1365-2990.2003.00473.x. [DOI] [PubMed] [Google Scholar]
- 26.Müller-Husmann G, Gloor S, Schachner M. Functional characterization of beta isoforms of murine Na,K-ATPase. The adhesion molecule on glia (AMOG/beta 2), but not beta 1, promotes neurite outgrowth. J Biol Chem. 1993;268(35):26260–26267. [PubMed] [Google Scholar]
- 27.Pollard SM, Yoshikawa K, Clarke ID, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4(6):568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 29.Einbond LS, Shimizu M, Ma H, et al. Actein inhibits the Na+-K+-ATPase and enhances the growth inhibitory effect of digitoxin on human breast cancer cells. Biochem Biophys Res Comm. 2008;375(4):608–613. doi: 10.1016/j.bbrc.2008.08.054. [DOI] [PubMed] [Google Scholar]
- 30.Mijatovic T, Van Quaquebeke E, Delest B, Debeir O, Darro F, Kiss R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim Biophys Acta. 2007;1776(1):32–57. doi: 10.1016/j.bbcan.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Prassas I, Diamandis EP. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov. 2008;7(11):926–935. doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]
- 32.Prassas I, Paliouras M, Datti A, Diamandis EP. High-throughput screening identifies cardiac glycosides as potent inhibitors of human tissue kallikrein expression: implications for cancer therapies. Clin Cancer Res. 2008;14(18):5778–5784. doi: 10.1158/1078-0432.CCR-08-0706. [DOI] [PubMed] [Google Scholar]
- 33.Mobasheri A, Avila J, Cózar-Castellano I, et al. Na+, K+-ATPase isozyme diversity: comparative biochemistry and physiological implications of novel functional interactions. Biosci Rep. 2000;20(2):51–91. doi: 10.1023/a:1005580332144. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan JH. A moving new role for the sodium pump in epithelial cells and carcinomas. Sci STKE. 2005;(289):pe31. doi: 10.1126/stke.2892005pe31. [DOI] [PubMed] [Google Scholar]
- 35.Schwab A. Ion channels and transporters on the move. News Physiol Sci. 2001;16:29–33. doi: 10.1152/physiologyonline.2001.16.1.29. [DOI] [PubMed] [Google Scholar]
- 36.Schwab A. Function and spatial distribution of ion channels and transporters in cell migration. Am J Physiol Renal Physiol. 2001;280(5):F739–F747. doi: 10.1152/ajprenal.2001.280.5.F739. [DOI] [PubMed] [Google Scholar]
- 37.Alper SL, Stuart-Tilley A, Simmons CF, Brown D, Drenckhahn D. The fodrin-ankyrin cytoskeleton of choroid plexus preferentially colocalizes with apical Na+K(+)-ATPase rather than with basolateral anion exchanger AE2. J Clin Invest. 1994;93(4):1430–1438. doi: 10.1172/JCI117120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cantiello HF. Actin filaments stimulate the Na(+)-K(+)-ATPase. Am J Physiol. 1995;269(5 Pt 2):F637–F643. doi: 10.1152/ajprenal.1995.269.5.F637. [DOI] [PubMed] [Google Scholar]
- 39.Ozkucur N, Perike S, Sharma P, Funk RH. Persistent directional cell migration requires ion transport proteins as direction sensors and membrane potential differences in order to maintain directedness. BMC Cell Biol. 2011;12:4. doi: 10.1186/1471-2121-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Boom J, Wolter M, Blaschke B, Knobbe CB, Reifenberger G. Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real-time reverse transcription-polymerase chain reaction. Int J Cancer. 2006;119(10):2330–2338. doi: 10.1002/ijc.22108. [DOI] [PubMed] [Google Scholar]
- 41.Kawakami K, Watanabe Y, Araki M, Nagano K. Sp1 binds to the adhesion-molecule-on-glia regulatory element that functions as a positive transcription regulatory element in astrocytes. J Neurosci Res. 1993;35(2):138–146. doi: 10.1002/jnr.490350204. [DOI] [PubMed] [Google Scholar]
- 42.Cheng L, Jin Z, Liu L, et al. Characterization and promoter analysis of the mouse nestin gene. FEBS Lett. 2004;565(1–3):195–202. doi: 10.1016/j.febslet.2004.03.097. [DOI] [PubMed] [Google Scholar]
- 43.Huang YL, Shi GY, Lee H, et al. Thrombin induces nestin expression via the transactivation of EGFR signalings in rat vascular smooth muscle cells. Cell Signal. 2009;21(6):954–968. doi: 10.1016/j.cellsig.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Kovacevic Grujicic N, Mojsin M, Krstic A, Stevanovic M. Functional characterization of the human SOX3 promoter: identification of transcription factors implicated in basal promoter activity. Gene. 2005;344:287–297. doi: 10.1016/j.gene.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 45.Luster TA, Rizzino A. Regulation of the FGF-4 gene by a complex distal enhancer that functions in part as an enhanceosome. Gene. 2003;323:163–172. doi: 10.1016/j.gene.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 46.Mojsin M, Stevanovic M. PBX1 and MEIS1 up-regulate SOX3 gene expression by direct interaction with a consensus binding site within the basal promoter region. Biochem J. 2010;425(1):107–116. doi: 10.1042/BJ20090694. [DOI] [PubMed] [Google Scholar]
- 47.Scheidenhelm DK, Cresswell J, Haipek CA, Fleming TP, Mercer RW, Gutmann DH. Akt-dependent cell size regulation by the adhesion molecule on glia occurs independently of phosphatidylinositol 3-kinase and Rheb signaling. Molec Cell Biol. 2005;25(8):3151–3162. doi: 10.1128/MCB.25.8.3151-3162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reifenberger G, Collins VP. Pathology and molecular genetics of astrocytic gliomas. J Mol Med (Berl) 2004;82(10):656–670. doi: 10.1007/s00109-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 49.Scherer H. Structural development in gliomas. Am J Cancer. 1938;34:333–351. [Google Scholar]
- 50.Scherer H. The forms of growth in gliomas and their practical significance. Brain. 1940;63:1–35. [Google Scholar]