

Abstract
Recent studies strongly suggest an increasing role for immune responses against self-antigens (Ags) which are not encoded by the major histocompatibility complex in the immunopathogenesis of allograft rejection. Although, improved surgical techniques coupled with improved methods to detect and avoid sensitization against donor human leukocyte antigen (HLA) have improved the immediate and short term function of transplanted organs. However, acute and chronic rejection still remains a vexing problem for the long term function of the transplanted organ. Immediately following organ transplantation, several factors both immune and non immune mechanisms lead to the development of local inflammatory milieu which sets the stage for allograft rejection. Traditionally, development of antibodies (Abs) against mismatched donor HLA have been implicated in the development of Ab mediated rejection. However, recent studies from our laboratory and others have demonstrated that development of humoral and cellular immune responses against non-HLA self-Ags may contribute in the pathogenesis of allograft rejection. There are reports demonstrating that immune responses to self-Ags especially Abs to the self-Ags as well as cellular immune responses especially through IL17 has significant pro-fibrotic properties leading to chronic allograft failure. This review summarizes recent studies demonstrating the role for immune responses to self-Ags in allograft immunity leading to rejection as well as present recent evidence suggesting there is interplay between allo- and autoimmunity leading to allograft dysfunction.
Keywords: Alloimmunity, Autoimmunity, Transplant rejection, Antigen presentation, Interleukin-17
1. Introduction
Vascularized solid organ transplantation (Tx) following end-stage organ failure is a viable treatment option providing significant improvement in the quality of life. The ever increasing numbers of solid organ Tx (kidney, lung, heart, liver, pancreas etc.) demonstrates the success of this strategy. The research leading to progress in donor organ preservation, improvements in surgical techniques and management of recipients with immunosuppression following Tx have contributed to the success. However, despite advances in these key areas, acute and chronic rejection (CR) following Tx remains the most important problem for sustaining continued long-term function of the transplanted organs. Recent statistics from the Registry of Transplant Recipients 2013 shows an active waiting list of more than 75,000 patients, while there were only 30,000 transplantable organs [1]. The incidence of CR is a major hazard especially given the limitations with donor availability and re-transplantation (Table 1). This is compounded by the fact that once set-in, there are no viable treatment options to reverse CR.
Table 1.
Incidence of acute and chronic rejection in various solid organ transplants;
| Organ | Acute Rejection# | References | Chronic Rejection* | References |
|---|---|---|---|---|
| Lung | 30–60% | [80], [81] | 40–70% | [82], [83] |
| Heart | 10–25% | [84], [85] | 25–60% | [86], [87] |
| Kidney | 10–20% | [88], [89], [90] | 40–50% | [91] |
| Liver | 7–22% | [92] | 4–12% | [93] |
one year incidence,
five year incidence.
Regardless of transplanted organ, CR is characterized by enhanced inflammatory cellular infiltration around vessels and tubular structures, fibrosis of the graft parenchyma that develops anywhere from days to years after Tx. The primary targets of the recipient immune response against the allograft are the donor major histocompatibility complex (MHC) antigens (Ags). Immune recognition of mismatched donor HLA (human leukocyte antigens) results in both cellular and humoral immune activation which leads to allograft rejection. In this review we will present evidence that self-Ags play an important role in allograft rejection and often there is interplay between allo- and autoimmunity which culminates in organ failure due to rejection.
1.1. Immune responses following solid organ transplantation
The primary targets of the recipient immune response against the allograft are the donor MHC Ags present on the allogeneic tissue. Immune recognition of mismatched donor histocompatibility Ags results in both cellular and humoral immune mechanisms which leads to allograft rejection [2, 3]. Allorecognition has been proposed to occur through two unique but not mutually exclusive pathways: the direct and indirect pathways of Ag presentation. The direct pathway involves recognition of intact donor MHC molecules on the cell surface, usually by antigen presenting cells (APC). Both CD8+ and CD4+ T cells can directly recognize donor MHC molecules, MHC class I and II respectively. In contrast, the indirect pathway involves presentation of processed donor Ags by recipient APC to recipient T cells. A ‘semi-direct’ pathway has also been recently described which involves recipient APCs that acquire donor MHC through cell-to-cell contact and activate host T cell responses which may contributes to CR [4–7].
While the direct pathway is more important for acute allograft rejection, the indirect pathway plays a dominant role in CR [8, 9]. Experiments have demonstrated that inhibition of acute rejection by depleting passenger APC significantly delays but does not prevent development of CR [10]. It has been observed that the frequency of direct alloreactive T cells exceeds indirect alloreactive T cells in the early post-Tx period [11]. The frequency of direct alloreactive T cells declines with time following Tx while the continuous influx of the processed donor Ags by the recipient APC through the indirect pathways increases the number of indirect alloreactive T cells that are themselves more resistant to currently used immunosuppression [12, 13]. In addition to the above two pathways, transfer of intact MHC molecules between cells has been observed [14]. Dendritic cells (DC) have been shown to acquire intact MHC class I and II molecules from exosomes secreted by other DC and prime both naïve CD8+ and CD4+ T cells [15]. Reports from Lechler’s group proposed that this represents a third mode of allorecognition, which has been termed “semi-direct” pathway [16]. Through this pathway, DC present intact MHC molecules to directly alloreactive CD8+ T cells as well as internalized and processed donor MHC peptides to indirect alloreactive CD4+ T cells [17].
CR, the immunopathogenesis of which is not fully characterized, still remains the leading cause of long-term allograft failure in Tx recipients with no viable treatment options (Figure 1). Several risk factors have been proposed to play a role in CR, including recurrent/refractory acute rejections, cytomegalovirus (CMV) and other viral infections, HLA mismatches, organ ischemia etc. [18, 19]. Several non-specific risk factors such as donor and recipient age, graft ischemic time, and bacterial/fungal/non-CMV viral infection have also been associated with decreased long term survival of the graft [19–21]. We propose a possible unifying hypothesis that all the above mentioned inflammatory risk factors potentially lead to tissue remodeling which facilitates the induction of immune responses against self-Ags, and development of autoimmunity in CR. Further, recent evidence has demonstrated an important role of T-helper immune responses specifically Th17 responses against self-Ags along with augmentation of humoral immune responses to the self-Ags as a mechanism leading to CR. Initial studies have demonstrated prolongation of murine cardiac allograft survival following blockade of IL-17 suggesting a primary role for IL-17 in the rejection of allografts [22]. Since then, studies in the Th17 subset has been expanded considerably and current evidence demonstrate that Th17 cells are key players in developing and sustaining immune responses to self-Ags (autoimmunity) [23]. In this review, we will present evidences for cross-talk between alloimmune and autoimmune responses to self-Ags and their role as well as proposed mechanisms leading to allograft rejection..
Figure 1.
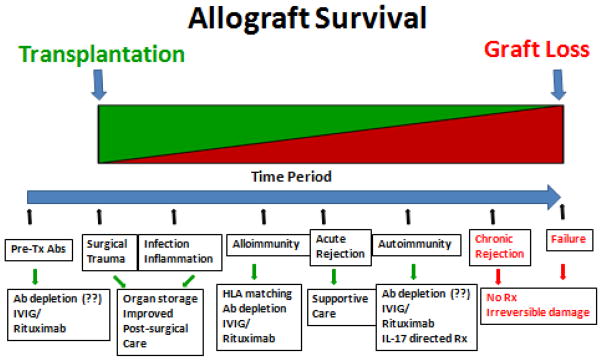
Temporal depiction of the sequence of events leading to allograft failure. The temporal sequence of initial inflammatory events following solid organ transplantation such as surgical stress, viral infections, GERD, mismatched HLA, etc leads to inflammatory injury to the allograft. These risk factors potentially play an important role in the acute rejection episode. Further, presence of pre-transplant Abs to self-antigens could also lead to higher incidence of alloimmunity and allograft rejection. This initial inflammatory injury could potentiate tissue remodeling and exposure of cryptic self-antigens and leading to autoimmunity and potential development of CR. Potential therapeutic options have been listed in the last panel. Green, indicates functioning allograft; and red, indicates non-functional allograft (color code given only for representation and not intended to indicate exact allograft survival temporal percentage).
1.2. Lung transplantation
CR after lung Tx is clinically diagnosed as bronchiolitis obliterans syndrome (BOS) and is characterized by obliteration of terminal airways within the lungs. The development of BOS following lung Tx is multifactorial and several risk factors including viral infection, primary graft dysfunction (PGD), alloimmunity and recently autoimmunity have been implicated. The development of immune responses against mismatched HLA involving both cell-mediated and humoral immunity against the mismatched donor HLA have been identified in patients diagnosed with BOS [24]. An important risk factor of interest is the transfusion of blood products. It has been well documented that transfusion-related lung injury (TRALI) can result in an acute respiratory disease syndrome (ARDS-like) picture similar to that seen with PGD following human LTx [25]. Recent multicenter studies have shown an independent association between blood product administration and increased risk for PGD, but the relationship between the two needs to be studied further [26]. The ischemia/reperfusion injury (IRI) can be an initiating event that activates inflammatory cascade leading to injury seen with TRALI [27]. Conversely, the lung injury induced by TRALI could accentuate any underlying IRI, resulting in PGD. Further, increased frequency of alloreactive CD4+ T cells against mismatched donor MHC class I and II molecules (indirect antigen presentation) has been detected in a human lung allograft recipient years after Tx and are associated with BOS [28]. Intrapulmonary lymphoid tissue has also been implicated in the pathogenesis of CR as it serves as a reservoir for effector memory T cells in high endothelial venules which can contribute to a local immune response in small airways leading to BOS [29].
Several studies have demonstrated that development of Abs to mismatched donor - HLA class I is associated with the development of CR [30–32]. Based on the reports by us [33] and others [34, 35] the presence of ‘shed’ donor HLA Ags in the bronchoalveolar lavage fluids following lung Tx, provide the substrate for Ag presentation to T helper cells and induction of alloimmunity. These T helper cells, which are engaged in indirect recognition pathways, can produce lymphokines required for the growth and maturation of alloantibody producing B cells. In addition, recent reports also demonstrated that there is strong correlation between de novo development of Abs to self-Ags in the absence of demonstrable Abs to HLA to development of BOS following human lung Tx [36–38]. Strong correlation between the development of Abs and Th17 responses to a self-protein, K-α1 tubulin (Kα1T), as well as Collagen V (ColV) with BOS have been identified in lung Tx patient diagnosed with BOS [36, 39] suggesting a pathogenic role for these Abs.
The IRI is a well known risk factor for the development of CR which is thought to be a consequence of oxidative stress injury, inflammation, and innate immune responses [40]. Studies have suggested an important role for T lymphocytes in animal models of lung IRI [41, 42] which is mediated in part by IL-17 production by the infiltrating CD4+ T cells in the lung [41, 43]. Type I invariant natural killer T (iNKT) cells have been implicated in the early innate immune response after IRI [44, 45]. Using murine models of lung IRI, studies by Sharma et al [46] have demonstrated that neutrophil infiltration following lung IRI is primarily initiated by CD41+ iNKT cells via an IL-17 dependent mechanism. However, role of IL17 and cells involved in the secretion of IL17 following lung IRI in humans are unknown at the present time.
Previous report from our laboratory have demonstrated that development of Abs to donor mismatched MHC class I precedes the development of BOS by 20 months [31]. Following development of Abs to donor HLA, these patients also developed Abs to self-Ags prior to clinical onset of BOS [47]. To determine the mechanism by which Abs to donor MHC may induce an immune response to self-Ags which lead to CR we developed a murine model of obliterative airway disease (OAD) of native lungs [39]. In this model, administration of Abs to MHC class I molecules to the native lungs resulted in cellular infiltration, epithelial hyperplasia, endothelitis, fibroproliferation, collagen deposition and luminal occlusion of the small airways- the central events seen during chronic lung allograft rejection. These animals also developed immune responses to lung associated self-Ags (Kα1T and ColV) prior to development of OAD and further more blocking of IL17 completely abrogated the immune response to self-Ags and OAD lesions supporting that immune responses to self-Ags is pathogenic for development of CR [39]. Based upon these results we instituted a preliminary observational study in which lung Tx patients who developed donor specific antibodies (DSA) but with normal lung functions were pre-emptively treated with Ab directed therapy to deplete DSA. This study demonstrated that removal of DSA following IVIG and rituximab therapy resulted in significantly better freedom from BOS in comparison to patients with persistent DSA [48]. We have reported that patients who cleared DSA as well as Abs to self-Ags following Ab directed treatment with rituximab and IVIG have greater freedom from development of BOS as compared to those that did not clear Abs to self-Ags [49]. These results demonstrated that Abs to self-Ags are induced by immune responses to donor HLA and removal of this sensitizing event results in better long term graft function indicating a role for immune responses to self-Ags in the pathogenesis of CR following human lung Tx.
Since many of the candidates for potential lung Tx are diagnosed with chronic lung diseases we determined whether they have developed immune responses to lung associated self-Ags and the presence of such an immune response affects the course following lung Tx. Towards this, we analyzed for the presence of Abs to Kα1T and Col V in the sera prior to lung Tx and its correlation to the development of PGD as well as CR following lung Tx. As shown in Table 2, these results suggest significant correlation between the presence of pre-Tx Abs to self-Ags, ColV and (Kα1T) and risk of PGD development of DSA following lung Tx and development of BOS [50]. These results strongly suggest a potential role of autoimmune responses against self-Ags prior to lung Tx in playing a critical role inducing an inflammatory cascade following Tx resulting in development of immune responses to donor mismatched MHC as well as development of CR following lung Tx.
Table 2.
Incidence of primary graft dysfunction, de novo antibodies to donor specific HLA and bronchiolitis obliterans syndrome in lung Transplantation recipients with pre-Transplant antibodies to self-antigens [50]
| Lung Tx | PGD(+) | PGD(−) | p-value |
|---|---|---|---|
| Pre-Tx Auto-Abs (+) | 35/101 (34.7%) | 6/41 (14.6%) | 0.01 |
| DSA(+) | DSA(−) | ||
|
|
|||
| Pre-Tx Auto-Abs (+) | 26/60 (43.3%) | 15/82 (18.3%) | 0.01 |
| BOS(+) | BOS(−) | ||
|
|
|||
| Pre-Tx Auto-Abs (+) | 23/59 (40%) | 15/71 (21.1%) | 0.01 |
One of the other target non-HLA Ags that have received considerable attention recently following solid organ Tx is the MHC class I related chain A (MICA). We recently analyzed the development of Abs to MICA and HLA using sera from 80 lung Tx recipients and their role in BOS after lung Tx [51]. Development of Abs either to MICA alone or MICA and HLA together significantly correlated (p< 0.01) with development of BOS. Kinetic analysis in the post-lung Tx period revealed that development of anti-HLA (7.6 ± 4.7 months) often preceded the development of anti-MICA (10.0 ± 3.5 months) and clinical diagnosis of BOS (16.3 ± 2.7 months). The development of Abs to HLA and MICA was strongly associated with the development of BOS thereby suggesting a role for MICA as an independent risk factor for development of BOS having synergistic effect with HLA Abs [51].
1.3. Kidney transplantation
Development of immune responses to lung associated self-Ags have given rise to the possibility that it is unique for the lungs at least in part, due to direct exposure of the Tx lungs to the external environment and thus prone for higher infectious and inflammatory damage leading to immune responses to self-Ags culminating in CR. However, immune responses to self-Ags have been demonstrated following other solid organ allografts succumbing to CR. Chronic allograft nephropathy (CAN) results in late graft loss following human renal Tx. The histopathologic signs of CAN include interstitial fibrosis, tubular atrophy, glomerulopathy and vasculopathy. It is thought to account for approximately 40% of graft loss at 10 years [52]. Animal models of chronic renal allograft rejection and clinical studies have implicated both cell-mediated and humoral arms of alloimmunity may contribute to its development [53]. CD4+ alloreactive T cells responding to donor derived peptides bound to recipient MHC class II have been correlated with CAN [54].
Increased levels of pre-Tx anti-HLA Abs and de novo post-Tx donor specific Abs have also been associated with development of CAN following Tx. Abs against MHC class I MICA has also been correlated with CAN following renal allografts. However, Abs developed de novo and directed at the donor HLA are not always detectable in the circulation of patients undergoing CAN, questioning the significance of Abs to HLA in the pathogenesis of CAN. Recently, refractory vascular allograft rejection in the absence of detectable anti-HLA has been associated with the presence of Abs directed at two epitopes of the second extracellular loop of a self-Ag, the angiotensin II type 1 receptor (AGTR1) [55]. It has been suggested that identification of AGTR1 receptor Abs might serve as a useful tool to identify those patients at risk for refractory allograft rejection. Other Ab targets include perlecan and Collagen types IV and VI as well as glomerular basement membrane protein, agrin [56, 57]. Reports have also suggested a role for vimentin Abs in the development of CAN [55].
Transplant glomerulopathy (TG) develops in approximately 20% patients by 5 years following renal Tx [56]. Reports suggest that TG arises as a consequence of persistent endothelial cell injury by the humoral arm of the immune system [56]. Studied by Joosten et al., using biopsy proven TG patients (n=19) has shown that anti-glomerular basement membrane Abs developed have the specificity to heparan sulphate proteoglycan agrin [58]. These results strongly suggest that immune responses to kidney associated self-Ags following renal Tx may be involved in inducing autoimmunity leading to CR following kidney Tx.
1.4. Cardiac transplantation
Ab mediated rejection (AMR) and CR in the form cardiac allograft vasculopathy (CAV) are being recognized as an important problem following cardiac Tx. Accelerated form of occlusive coronary disease, akin to CR, affecting both intramural and epicardial coronary arteries and veins occurs quite frequently and 5-year incidence of CAV has been reported to be as high as 30–40% [59]. Clinical experience has identified several risk factors including the number of acute rejection episodes, viral infections and donor HLA mismatching as independent risk factors for CAV after heart Tx [60].
Cell mediated immunity has been implicated in the pathogenesis of CAV as histologic examination of perivascular infiltrates often reveals a predominance of immune cells including T cells (CD4+, CD8+), natural killer cells, macrophages and DC. In an experimental model of CAV, depletion of recipient CD4+ but not CD8+ T cells prevented the formation of arterial lesions [61]. Alloreactive CD4+ Th1 and Th2 cells activated via the indirect pathway have been implicated in the development of CAV as well. Using a T bet−/− murine model of CAV, Th17 cells has been shown to be involved in the development of CAV in the absence of Th1 responses [62]. T cells specific to cardiac myosin has been shown to develop and persist in the absence of any alloimmune responses, indicating that response to myosin, a self-Ag, is associated with the pathogenesis of CAV [63].
Analysis of cardiac Tx recipients with AMR and CAV from our center have demonstrated significant correlation for de novo development of humoral immune responses to mismatched donor HLA and MICA to development of AMR in the early period and CAV in the late part after cardiac Tx [64]. Serial analysis also revealed that DSA (2.7 ± 1.4 months) preceded development of anti-MICA (6.5 ± 2.1 months) in recipients diagnosed with AMR at 8.3 ± 2.5 months post cardiac Tx. Similarly, development of DSA and anti-MICA (CAV+, 67–75 %, CAV−, 13%, p=0.004) was significantly associated with CAV. AMR+DSA+, and CAV+DSA+ patients demonstrated increased anti-MICA levels compared with respective DSA− patients (p=0.01). Further this study supported the induction of Ab-mediated immune responses to MICA in patients with AMR and CAV. However, molecular mechanisms by which immune responses to MICA may contribute in the pathogenesis of AMR and CAV are yet to be defined.
CR pathology have been reported following syngeneic cardiac Tx in murine models supporting that CR can be induced even in the absence of alloimmune responses [65, 66]. Following human cardiac Tx, it has been shown that Abs against vimentin, a cytoskeleton self-protein, is an independent predictor of atherosclerosis following cardiac Tx and can accelerate the course of CAV [67, 68]. Reports from our laboratory have demonstrated the presence of DSA in AMR and CAV is significantly associated with development of Abs to cardiac self-Ags, myosin and vimentin (Table 3) [69]. Furthermore, in these patients there is high frequency of CD4+ Th specific to cardiac self-Ags that predominantly secrete IL-5 and IL-17 suggesting that alloimmune responses to donor HLA may play a significant role in the induction of autoimmunity as evidenced by the development of Abs to cardiac self-Ags leading to AMR and CAV.
Table 3.
Incidence of Antibody Mediated Rejection and Coronary Artery Vasculopathy in heart Transplant recipients who develop antibodies to self-antigens following Transplantation [69].
| Heart Tx | AMR(+) n=9 |
AMR (−) n=47 |
p-value | CAV(+) n=14 |
CAV(−) n=59 |
p-value |
|---|---|---|---|---|---|---|
|
|
|
|||||
| DSA | 7/9 | 8/47 | 0.01 | 11/14 | 15/59 | 0.01 |
| Auto-Abs | 6/9 | 3/47 | 0.01 | 13/14 | 12/59 | 0.01 |
1.5. Liver transplantation
CR after liver Tx though rare can occur and is manifested as fibrous tissue replacement in the allograft, clinically mimicking cirrhosis. Fibrogenesis is a complex, dynamic process mediated by necro-inflammation and activation of hepatic stellate cells under the influence of virally induced immunomodulation. Cell-mediated and humoral immunity are both implicated in the progression of fibrosis after liver Tx [70, 71].
Studies investigating mechanisms of fibrosis following orthotopic liver Tx (OLT) with hepatitis C virus (HCV) have correlated progression of fibrosis (Table 5) with specific CD 4 T cell behavior [71]. Specifically, a lack of HCV specific Th1 type T cell immunity has been associated with the development of fibrosis and cirrhosis following recurrent HCV infection in the post-Tx period. Patients with higher degrees of fibrosis and cirrhosis have also been shown to have significantly higher levels of IL-17 production upon stimulation with HCV Ags (48). It has also been demonstrated that IL17 can lead to Induction of CXCL12 and activation of B cells [72]. CXCL12 in combination with IL-17 can induce germinal center formation and auto-Ab production to self-Ags including extracellular matrix Col I, II, and V in the liver. Our studies have demonstrated increased serum levels of IL-17, IL-6, IL-1β, IL-8 and MCP-1 following OLT who develop high grade allograft inflammation and fibrosis secondary to HCV recurrence. This was also associated with increased frequency of CD4+ T cells specific to HCV that secrete IL-17, significant decline in the frequency of HCV specific CD4+ T cells that secrete IFN-γ and increased frequencies of IL-10 secreting cells. These patients also developed Abs against Col I, II, and V [73]. All these results strongly suggest that Th17 mediated autoimmune responses and Abs to self-Ags may play a part in the development of fibrosis following re-infection of the Tx liver with HCV and the autoimmune responses may contribute to the pathogenesis of CR following OLT.
Table 5.
Evidence of Abs to specific self-antigens following solid organ Transplantation.
1.6. Interplay between allo- and autoimmunity leading to CR
The phenomenon of CR after solid organ Tx is likely the result of a multifactorial interplay of various effector arms of alloimmunity. Studies following heart, lung, liver, and kidney Tx have identified immune mechanisms against specific self-Ags (Table 5) which can result in the development of CR. An emerging and unifying theme in all of these cases is inflammation and subsequent tissue remodeling following Tx which can expose cryptic self-Ags or their determinants (Figure 2). This process along with a favorable cytokine milieu, may allow for loss of peripheral tolerance and the activation of cell-mediated immunity towards development of de novo immune responses to self-Ags. Regulatory T cells (Tregs) are known to inhibit both auto- and allo-reactive effector T cells; however, in the context of potent immunosuppression following Tx especially using calcineurin inhibitors can lead to loss of regulatory T cell proliferation leading to loss of peripheral tolerance to self-Ags [74] resulting in immune responses to self-Ags. At present, IL-17 appears to be a strong candidate for promoting such a response including the generation of auto-Abs. Further studies are needed to use this as a biomarker for risk of developing CR following solid organ Tx.
Figure 2.

An overview of the potential mechanism for the exposure of cryptic antigenic determinants leading to development of Abs to self-Ags in CR following solid organ Tx. Various risk factors such as surgical stress, viral and bacterial infections, mismatched HLA following solid organ transplantation induces tissue remodeling and exposure of cryptic self-antigenic determinants. This leads to cellular and humoral responses and upregulated expression of fibrotic signaling molecules and growth factors such as HIF1α, VEGF, FGF, etc. All these culminate to fibrosis and loss of function manifesting as CR of the allograft.
Specific binding of Abs to HLA molecules have been shown to activate endothelial and smooth muscle cells [57]. Recent studies demonstrated that surface ligation of HLA class I by specific Abs induce a intracellular signal through mammalian target of rapamycin (mTOR) promoting endothelial cell proliferation [75]. Furthermore, RNAi based knockdown of mTOR inhibited class I-mediated phosphorylation of proteins downstream of mTOR complex 1 and mTOR complex 2. These results support the role of anti-HLA in the process of Tx vasculopathy and suggest that exposure of the graft endothelium to anti-HLA may promote proliferation through the mTOR pathway [75]. We demonstrated that when airway epithelial cells (AEC) were incubated with anti-MHC class I, they underwent proliferation with the secretion of pro-fibrogenic growth factors [76]. In addition, we also demonstrated that administration of anti-MHC class I resulted in increases in growth factors and pro-apoptotic genes as well as pro-inflammatory cytokines resulting in OAD in a murine model of heterotopic tracheal Tx.[77].
In addition to HLA, at least some of the Abs to self-Ags can also induce cellular signals following ligation of their specific Ags. For example, we demonstrated that Abs to Kα1T, an epithelial cell surface gap junction protein often seen in lung Tx with BOS can upregulate pro-fibrotic growth factors [36] and lipid rafts play a critical role in the surface ligation of Abs to Kα1T to its specific receptor (Kα1T) of the AEC surface [78]. Recently we demonstrated that ligation of Kα1T on human bronchial epithelial (NHBE) cells with specific Abs caused upregulation of pro-fibrotic growth factors VEGF, HGF and TGF-β. Furthermore, incubation of NHBE with Kα1T Abs increased expression of hypoxia inducible factor (HIF-1α). The Kα1T Ab-mediated growth factor expression is dependent on HIF-1α as inhibition of HIF-1α returned fibrotic growth factor expression to basal levels. These results strongly suggest that HIF-1α-mediated upregulation of fibrogenic growth factors induced by ligation of Kα1T Abs may be involved in the development of fibrosis leading to CR following lung Tx [79].
In conclusion, alloimmune responses leading to the development of de novo autoimmunity to self-Ags appear to be an important event leading to the development of CR after solid organ Tx. Tissue inflammation and remodeling following an alloimmune response can expose self-Ags or their antigenic determinants leading to activation of both cell-mediated and humoral effector arms. This autoimmune response in part mediated by IL-17 can also facilitate the activation of fibrinogenic pathways seen in CR. Future work is warranted to identify the mechanistic role for auto-Abs in the development of rejection and their clinical utility as a biomarker for CR. Intervention of the IL-17 pathway may also represent a new strategy to expand the immunosuppressive armamentarium. Since current results strongly suggest that alloimmune responses to mismatched HLA as evidenced by circulating Abs to donor HLA precedes the development of Abs to self-Ags, it is logical to intervene as early after detection of Abs to HLA to prevent the development of Abs to self-Ags. We propose that this may prevent or delay the onset of CR. Studies in lung Tx recipients are currently underway to test this hypothesis by administering IVIG and Rituxan as early as circulating Abs to donor specific HLA were identified following lung Tx. Further, identification of de novo development of Abs to self-Ags may be valuable as a serum biomarker for identification of Tx recipients who are at risk for developing CR.
Table 4.
Incidence of autoimmunity in Hepatitis C Virus infected patients with native (non-Orthotopic Liver Transplant) and orthotopic liver Transplan (post-Orthotopic Liver Transplant) recipients with and without fibrosis [71].
| Non-OLT | p-Value | Post-OLT | p-Value | |||
|---|---|---|---|---|---|---|
|
|
|
|||||
| No Fibrosis | Fibrosis | No Fibrosis | Fibrosis | |||
|
| ||||||
| Col-I Abs+ | 2/25 (8%) | 22/25 (88%) | 0.001 | 2/10 (20%) | 9/10 (90%) | 0.001 |
| Col-II Abs+ | 3/35 (12%) | 20/25 (80%) | 0.001 | 2/10 (20%) | 8/10 (80%) | 0.001 |
| Col-IV Abs+ | 2/25 (8%) | 3/25 (12%) | 0.86 | 1/10 (10%) | 1/10 (10%) | 1 |
| Col-V Abs+ | 3/25 (12%) | 20/25 (80%) | 0.001 | 3/10 (30%) | 9/10 (90%) | 0.001 |
Acknowledgments
This work was supported by NIH HL092519 and HL056643, and the BJC Foundation (TM). The authors also thank Ms. Billie Glasscock for her help in preparing this manuscript.
Abbreviations
- Ab
antibody
- AEC
airway epithelial cells
- Ag
antigen
- AGTR1
angiotensin II type 1 receptor
- AMR
antibody mediated rejection
- APC
antigen presenting cell
- BOS
bronchiolitis obliterans syndrome
- CAN
chronic allograft nephropathy
- CAV
cardiac allograft vasculopathy
- CR
chronic rejection
- ColV
Collagen V
- CMV
cytomegalovirus
- DC
dendritic cells
- DSA
donor specific antibodies
- HCV
hepatitis C virus
- HIF-1α
hypoxia inducible factor
- HLA
human leukocyte antigen
- iNKT
invariant natural killer T cells
- IRI
schemia/reperfusion injury
- Kα1T
Kα1 Tubulin
- MHC
major histocompatibility complex
- MICA
MHC class I related chain A
- mTOR
mammalian target of rapamycin
- NHBE
human bronchial epithelial
- OAD
obliterative airway disease
- OLT
orthotopic liver transplantation
- PGD
primary graft dysfunction
- TG
transplant glomerulopathy
- TRALI
transfusion-related lung injury
- Tx
transplantation
Footnotes
Disclosures
None of the authors have any conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.2013 http://optn.transplant.hrsa.gov/
- 2.Game DS, Lechler RI. Pathways of allorecognition: implications for transplantation tolerance. Transpl Immunol. 2002;10(2–3):101. doi: 10.1016/s0966-3274(02)00055-2. [DOI] [PubMed] [Google Scholar]
- 3.Hernandez-Fuentes MP, Baker RJ, Lechler RI. The alloresponse. Rev Immunogenet. 1999;1(3):282. [PubMed] [Google Scholar]
- 4.Herrera OB, Golshayan D, Tibbott R, Salcido Ochoa F, James MJ, Marelli-Berg FM, Lechler RI. A novel pathway of alloantigen presentation by dendritic cells. J Immunol. 2004;173(8):4828. doi: 10.4049/jimmunol.173.8.4828. [DOI] [PubMed] [Google Scholar]
- 5.Pimenta-Araujo R, Mascarell L, Huesca M, Cumano A, Bandeira A. Embryonic thymic epithelium naturally devoid of APCs is acutely rejected in the absence of indirect recognition. J Immunol. 2001;167(9):5034. doi: 10.4049/jimmunol.167.9.5034. [DOI] [PubMed] [Google Scholar]
- 6.Mandelbrot DA, Kishimoto K, Auchincloss H, Jr, Sharpe AH, Sayegh MH. Rejection of mouse cardiac allografts by costimulation in trans. J Immunol. 2001;167(3):1174. doi: 10.4049/jimmunol.167.3.1174. [DOI] [PubMed] [Google Scholar]
- 7.Afzali B, Lombardi G, Lechler RI. Pathways of major histocompatibility complex allorecognition. Curr Opin Organ Transplant. 2008;13(4):438. doi: 10.1097/MOT.0b013e328309ee31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shilling RA, Wilkes DS. Immunobiology of chronic lung allograft dysfunction: new insights from the bench and beyond. Am J Transplant. 2009;9(8):1714. doi: 10.1111/j.1600-6143.2009.02690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hachem RR. Lung allograft rejection: diagnosis and management. Curr Opin Organ Transplant. 2009;14(5):477. doi: 10.1097/MOT.0b013e32832fb981. [DOI] [PubMed] [Google Scholar]
- 10.Heeger PS. T-cell allorecognition and transplant rejection: a summary and update. Am J Transplant. 2003;3(5):525. doi: 10.1034/j.1600-6143.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- 11.Benichou G, Valujskikh A, Heeger PS. Contributions of direct and indirect T cell alloreactivity during allograft rejection in mice. J Immunol. 1999;162(1):352. [PubMed] [Google Scholar]
- 12.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166(2):973. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 13.Sawyer GJ, Dalchau R, Fabre JW. Indirect T cell allorecognition: a cyclosporin A resistant pathway for T cell help for antibody production to donor MHC antigens. Transpl Immunol. 1993;1(1):77. doi: 10.1016/0966-3274(93)90063-e. [DOI] [PubMed] [Google Scholar]
- 14.Huang JF, Yang Y, Sepulveda H, Shi W, Hwang I, Peterson PA, Jackson MR, Sprent J, Cai Z. TCR-Mediated internalization of peptide-MHC complexes acquired by T cells. Science. 1999;286(5441):952. doi: 10.1126/science.286.5441.952. [DOI] [PubMed] [Google Scholar]
- 15.Andre F, Chaput N, Schartz NE, Flament C, Aubert N, Bernard J, Lemonnier F, Raposo G, Escudier B, Hsu DH, Tursz T, Amigorena S, Angevin E, Zitvogel L. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol. 2004;172(4):2126. doi: 10.4049/jimmunol.172.4.2126. [DOI] [PubMed] [Google Scholar]
- 16.Jiang S, Herrera O, Lechler RI. New spectrum of allorecognition pathways: implications for graft rejection and transplantation tolerance. Curr Opin Immunol. 2004;16(5):550. doi: 10.1016/j.coi.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol. 2002;3(12):1156. doi: 10.1038/ni854. [DOI] [PubMed] [Google Scholar]
- 18.Kroshus TJ, Kshettry VR, Savik K, John R, Hertz MI, Bolman RM., 3rd Risk factors for the development of bronchiolitis obliterans syndrome after lung transplantation. J Thorac Cardiovasc Surg. 1997;114(2):195. doi: 10.1016/S0022-5223(97)70144-2. [DOI] [PubMed] [Google Scholar]
- 19.Stanbrook MB, Kesten S. Bronchial hyperreactivity after lung transplantation predicts early bronchiolitis obliterans. Am J Respir Crit Care Med. 1999;160(6):2034. doi: 10.1164/ajrccm.160.6.9801037. [DOI] [PubMed] [Google Scholar]
- 20.Kshettry VR, Kroshus TJ, Savik K, Hertz MI, Bolman RM. Primary pulmonary hypertension as a risk factor for the development of obliterative bronchiolitis in lung allograft recipients. Chest. 1996;110(3):704. doi: 10.1378/chest.110.3.704. [DOI] [PubMed] [Google Scholar]
- 21.Tilney NL. Chronic rejection and its risk factors. Transplant Proc. 1999;31(1–2A):41S. doi: 10.1016/s0041-1345(98)02080-6. [DOI] [PubMed] [Google Scholar]
- 22.Antonysamy MA, Fanslow WC, Fu F, Li W, Qian S, Troutt AB, Thomson AW. Evidence for a role of IL-17 in organ allograft rejection: IL-17 promotes the functional differentiation of dendritic cell progenitors. J Immunol. 1999;162(1):577. [PubMed] [Google Scholar]
- 23.Heidt S, Segundo DS, Chadha R, Wood KJ. The impact of Th17 cells on transplant rejection and the induction of tolerance. Curr Opin Organ Transplant. 2010;15(4):456. doi: 10.1097/MOT.0b013e32833b9bfb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith CR, Jaramillo A, Duffy BF, Mohanakumar T. Airway epithelial cell damage mediated by antigen-specific T cells: implications in lung allograft rejection. Hum Immunol. 2000;61(10):985. doi: 10.1016/s0198-8859(00)00175-0. [DOI] [PubMed] [Google Scholar]
- 25.Webert KE, Blajchman MA. Transfusion-related acute lung injury. Transfus Med Rev. 2003;17(4):252. doi: 10.1016/s0887-7963(03)00039-7. [DOI] [PubMed] [Google Scholar]
- 26.Covarrubias M, Ware LB, Kawut SM, De Andrade J, Milstone A, Weinacker A, Orens J, Lama V, Wille K, Bellamy S, Shah C, Demissie E, Christie JD. Plasma intercellular adhesion molecule-1 and von Willebrand factor in primary graft dysfunction after lung transplantation. Am J Transplant. 2007;7(11):2573. doi: 10.1111/j.1600-6143.2007.01981.x. [DOI] [PubMed] [Google Scholar]
- 27.Rizk A, Gorson KC, Kenney L, Weinstein R. Transfusion-related acute lung injury after the infusion of IVIG. Transfusion. 2001;41(2):264. doi: 10.1046/j.1537-2995.2001.41020264.x. [DOI] [PubMed] [Google Scholar]
- 28.Bharat A, Narayanan K, Street T, Fields RC, Steward N, Aloush A, Meyers B, Schuessler R, Trulock EP, Patterson GA, Mohanakumar T. Early posttransplant inflammation promotes the development of alloimmunity and chronic human lung allograft rejection. Transplantation. 2007;83(2):150. doi: 10.1097/01.tp.0000250579.08042.b6. [DOI] [PubMed] [Google Scholar]
- 29.Sato M, Hirayama S, Hwang DM, Lara-Guerra H, Wagnetz D, Waddell TK, Liu M, Keshavjee S. The role of intrapulmonary de novo lymphoid tissue in obliterative bronchiolitis after lung transplantation. J Immunol. 2009;182(11):7307. doi: 10.4049/jimmunol.0803606. [DOI] [PubMed] [Google Scholar]
- 30.Jaramillo A, Smith MA, Phelan D, Sundaresan S, Trulock E, Lynch J, Cooper J, Patterson GA, Mohanakumar T. Temporal relationship between the development of anti-HLA antibodies and the development of bronchiolitis obliterans syndrome after lung transplantation. Transplant Proc. 1999;31(1–2):185. doi: 10.1016/s0041-1345(98)01495-x. [DOI] [PubMed] [Google Scholar]
- 31.Jaramillo A, Smith MA, Phelan D, Sundaresan S, Trulock EP, Lynch JP, Cooper JD, Patterson GA, Mohanakumar T. Development of ELISA-detected anti-HLA antibodies precedes the development of bronchiolitis obliterans syndrome and correlates with progressive decline in pulmonary function after lung transplantation. Transplantation. 1999;67(8):1155. doi: 10.1097/00007890-199904270-00012. [DOI] [PubMed] [Google Scholar]
- 32.Sundaresan S, Mohanakumar T, Smith MA, Trulock EP, Lynch J, Phelan D, Cooper JD, Patterson GA. HLA-A locus mismatches and development of antibodies to HLA after lung transplantation correlate with the development of bronchiolitis obliterans syndrome. Transplantation. 1998;65(5):648. doi: 10.1097/00007890-199803150-00008. [DOI] [PubMed] [Google Scholar]
- 33.Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: a possible mechanism for bronchiolitis obliterans syndrome. Hum Immunol. 2003;64(5):521. doi: 10.1016/s0198-8859(03)00038-7. [DOI] [PubMed] [Google Scholar]
- 34.Chalermskulrat W, Neuringer IP, Schmitz JL, Catellier DJ, Gurka MJ, Randell SH, Aris RM. Human leukocyte antigen mismatches predispose to the severity of bronchiolitis obliterans syndrome after lung transplantation. Chest. 2003;123(6):1825. doi: 10.1378/chest.123.6.1825. [DOI] [PubMed] [Google Scholar]
- 35.van den Berg JW, Hepkema BG, Geertsma A, Koeter GH, Postma DS, de Boer WJ, Lems SP, van der Bij W. Long-term outcome of lung transplantation is predicted by the number of HLA-DR mismatches. Transplantation. 2001;71(3):368. doi: 10.1097/00007890-200102150-00005. [DOI] [PubMed] [Google Scholar]
- 36.Goers TA, Ramachandran S, Aloush A, Trulock E, Patterson GA, Mohanakumar T. De novo production of K-alpha1 tubulin-specific antibodies: role in chronic lung allograft rejection. J Immunol. 2008;180(7):4487. doi: 10.4049/jimmunol.180.7.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, Meyer KC, Hayney MS, Braun RK, Greenspan DS, Gopalakrishnan B, Cai J, Brand DD, Yoshida S, Cummings OW, Wilkes DS. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117(11):3498. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magro CM, Klinger DM, Adams PW, Orosz CG, Pope-Harman AL, Waldman WJ, Knight D, Ross P., Jr Evidence that humoral allograft rejection in lung transplant patients is not histocompatibility antigen-related. Am J Transplant. 2003;3(10):1264. doi: 10.1046/j.1600-6143.2003.00229.x. [DOI] [PubMed] [Google Scholar]
- 39.Fukami N, Ramachandran S, Saini D, Walter M, Chapman W, Patterson GA, Mohanakumar T. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182(1):309. doi: 10.4049/jimmunol.182.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Land WG. The role of postischemic reperfusion injury and other nonantigen-dependent inflammatory pathways in transplantation. Transplantation. 2005;79(5):505. doi: 10.1097/01.tp.0000153160.82975.86. [DOI] [PubMed] [Google Scholar]
- 41.Yang Z, Sharma AK, Linden J, Kron IL, Laubach VE. CD4+ T lymphocytes mediate acute pulmonary ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2009;137(3):695. doi: 10.1016/j.jtcvs.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Perrot M, Young K, Imai Y, Liu M, Waddell TK, Fischer S, Zhang L, Keshavjee S. Recipient T cells mediate reperfusion injury after lung transplantation in the rat. J Immunol. 2003;171(10):4995. doi: 10.4049/jimmunol.171.10.4995. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z, Sharma AK, Marshall M, Kron IL, Laubach VE. NADPH oxidase in bone marrow-derived cells mediates pulmonary ischemia-reperfusion injury. Am J Respir Cell Mol Biol. 2009;40(3):375. doi: 10.1165/rcmb.2008-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, Engelhard VH, Okusa MD. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178(9):5899. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 45.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203(12):2639. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma AK, LaPar DJ, Zhao Y, Li L, Lau CL, Kron IL, Iwakura Y, Okusa MD, Laubach VE. Natural killer T cell-derived IL-17 mediates lung ischemia-reperfusion injury. Am J Respir Crit Care Med. 2011;183(11):1539. doi: 10.1164/rccm.201007-1173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, Steward N, Aloush A, Hachem R, Trulock E, Meyers B, Patterson GA, Mohanakumar T. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant. 2011;30(6):624. doi: 10.1016/j.healun.2011.01.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hachem RR, Yusen RD, Meyers BF, Aloush AA, Mohanakumar T, Patterson GA, Trulock EP. Anti-human leukocyte antigen antibodies and preemptive antibody-directed therapy after lung transplantation. J Heart Lung Transplant. 2010;29(9):973. doi: 10.1016/j.healun.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hachem RR, Tiriveedhi V, Patterson GA, Aloush A, Trulock EP, Mohanakumar T. Antibodies to K-alpha 1 Tubulin and Collagen V Are Associated With Chronic Rejection After Lung Transplantation. Am J Transplant. 2012;12(8):2164. doi: 10.1111/j.1600-6143.2012.04079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bharat A, Saini D, Steward N, Hachem R, Trulock EP, Patterson GA, Meyers BF, Mohanakumar T. Antibodies to self-antigens predispose to primary lung allograft dysfunction and chronic rejection. Ann Thorac Surg. 2010;90(4):1094. doi: 10.1016/j.athoracsur.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Angaswamy N, Saini D, Ramachandran S, Nath DS, Phelan D, Hachem R, Trulock E, Patterson GA, Mohanakumar T. Development of antibodies to human leukocyte antigen precedes development of antibodies to major histocompatibility class I-related chain A and are significantly associated with development of chronic rejection after human lung transplantation. Hum Immunol. 2010;71(6):560. doi: 10.1016/j.humimm.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hertig A, Anglicheau D, Verine J, Pallet N, Touzot M, Ancel PY, Mesnard L, Brousse N, Baugey E, Glotz D, Legendre C, Rondeau E, Xu-Dubois YC. Early epithelial phenotypic changes predict graft fibrosis. J Am Soc Nephrol. 2008;19(8):1584. doi: 10.1681/ASN.2007101160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Birnbaum LM, Lipman M, Paraskevas S, Chaudhury P, Tchervenkov J, Baran D, Herrera-Gayol A, Cantarovich M. Management of chronic allograft nephropathy: a systematic review. Clin J Am Soc Nephrol. 2009;4(4):860. doi: 10.2215/CJN.05271008. [DOI] [PubMed] [Google Scholar]
- 54.Murphy B, Yu J, Jiao Q, Lin M, Chitnis T, Sayegh MH. A novel mechanism for the immunomodulatory functions of class II MHC-derived peptides. J Am Soc Nephrol. 2003;14(4):1053. doi: 10.1097/01.asn.0000057541.69641.f8. [DOI] [PubMed] [Google Scholar]
- 55.Li C, Yang CW. The pathogenesis and treatment of chronic allograft nephropathy. Nat Rev Nephrol. 2009;5(9):513. doi: 10.1038/nrneph.2009.113. [DOI] [PubMed] [Google Scholar]
- 56.Fotheringham J, Angel CA, McKane W. Transplant glomerulopathy: morphology, associations and mechanism. Nephron Clin Pract. 2009;113(1):c1. doi: 10.1159/000228069. [DOI] [PubMed] [Google Scholar]
- 57.Li F, Atz ME, Reed EF. Human leukocyte antigen antibodies in chronic transplant vasculopathy-mechanisms and pathways. Curr Opin Immunol. 2009;21(5):557. doi: 10.1016/j.coi.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joosten SA, Sijpkens YW, van Ham V, Trouw LA, van der Vlag J, van den Heuvel B, van Kooten C, Paul LC. Antibody response against the glomerular basement membrane protein agrin in patients with transplant glomerulopathy. Am J Transplant. 2005;5(2):383. doi: 10.1111/j.1600-6143.2005.00690.x. [DOI] [PubMed] [Google Scholar]
- 59.Weis M, von Scheidt W. Cardiac allograft vasculopathy: a review. Circulation. 1997;96(6):2069. doi: 10.1161/01.cir.96.6.2069. [DOI] [PubMed] [Google Scholar]
- 60.Valantine H. Cardiac allograft vasculopathy after heart transplantation: risk factors and management. J Heart Lung Transplant. 2004;23(5 Suppl):S187. doi: 10.1016/j.healun.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 61.Brennan TV, Hoang V, Garrod KR, Liu FC, Hayden T, Kim J, Kang SM. A new T-cell receptor transgenic model of the CD4+ direct pathway: level of priming determines acute versus chronic rejection. Transplantation. 2008;85(2):247. doi: 10.1097/TP.0b013e31815e883e. [DOI] [PubMed] [Google Scholar]
- 62.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M, Habicht A, Clarkson MR, Iacomini J, Glimcher LH, Sayegh MH, Ansari MJ. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205(13):3133. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rolls HK, Kishimoto K, Dong VM, Illigens BM, Sho M, Sayegh MH, Benichou G, Fedoseyeva EV. T-cell response to cardiac myosin persists in the absence of an alloimmune response in recipients with chronic cardiac allograft rejection. Transplantation. 2002;74(7):1053. doi: 10.1097/00007890-200210150-00028. [DOI] [PubMed] [Google Scholar]
- 64.Nath DS, Angaswamy N, Basha HI, Phelan D, Moazami N, Ewald GA, Mohanakumar T. Donor-specific antibodies to human leukocyte antigens are associated with and precede antibodies to major histocompatibility complex class I-related chain A in antibody-mediated rejection and cardiac allograft vasculopathy after human cardiac transplantation. Hum Immunol. 2010;71(12):1191. doi: 10.1016/j.humimm.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jindra PT, Hsueh A, Hong L, Gjertson D, Shen XD, Gao F, Dang J, Mischel PS, Baldwin WM, 3rd, Fishbein MC, Kupiec-Weglinski JW, Reed EF. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol. 2008;180(4):2214. doi: 10.4049/jimmunol.180.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Atz ME, Reed EF. Role of anti-MHC class I antibody in facilitating transplant accommodation. Crit Rev Immunol. 2008;28(6):485. doi: 10.1615/critrevimmunol.v28.i6.20. [DOI] [PubMed] [Google Scholar]
- 67.Leong HS, Mahesh BM, Day JR, Smith JD, McCormack AD, Ghimire G, Podor TJ, Rose ML. Vimentin autoantibodies induce platelet activation and formation of platelet-leukocyte conjugates via platelet-activating factor. J Leukoc Biol. 2008;83(2):263. doi: 10.1189/jlb.0607339. [DOI] [PubMed] [Google Scholar]
- 68.Rose ML, Smith JD. Clinical relevance of complement-fixing antibodies in cardiac transplantation. Hum Immunol. 2009;70(8):605. doi: 10.1016/j.humimm.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 69.Nath DS, Ilias Basha H, Tiriveedhi V, Alur C, Phelan D, Ewald GA, Moazami N, Mohanakumar T. Characterization of immune responses to cardiac self-antigens myosin and vimentin in human cardiac allograft recipients with antibody-mediated rejection and cardiac allograft vasculopathy. J Heart Lung Transplant. 2010;29(11):1277. doi: 10.1016/j.healun.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schirren CA, Jung MC, Gerlach JT, Worzfeld T, Baretton G, Mamin M, Hubert Gruener N, Houghton M, Pape GR. Liver-derived hepatitis C virus (HCV)-specific CD4(+) T cells recognize multiple HCV epitopes and produce interferon gamma. Hepatology. 2000;32(3):597. doi: 10.1053/jhep.2000.9635. [DOI] [PubMed] [Google Scholar]
- 71.Bharat A, Barros F, Narayanan K, Borg B, Lisker-Melman M, Shenoy S, Lowell J, Crippin J, Chapman W, Mohanakumar T. Characterization of virus-specific T-cell immunity in liver allograft recipients with HCV-induced cirrhosis. Am J Transplant. 2008;8(6):1214. doi: 10.1111/j.1600-6143.2008.02248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le TV, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9(2):166. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 73.Borg BB, Seetharam A, Subramanian V, Basha HI, Lisker-Melman M, Korenblat K, Anderson CD, Shenoy S, Chapman WC, Crippin JS, Mohanakumar T. Immune response to extracellular matrix collagen in chronic hepatitis C-induced liver fibrosis. Liver Transpl. 2011;17(7):814. doi: 10.1002/lt.22303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bharat A, Fields RC, Mohanakumar T. Regulatory T cell-mediated transplantation tolerance. Immunol Res. 2005;33(3):195. doi: 10.1385/IR:33:3:195. [DOI] [PubMed] [Google Scholar]
- 75.Jindra PT, Jin YP, Rozengurt E, Reed EF. HLA class I antibody-mediated endothelial cell proliferation via the mTOR pathway. J Immunol. 2008;180(4):2357. doi: 10.4049/jimmunol.180.4.2357. [DOI] [PubMed] [Google Scholar]
- 76.Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: a possible mechanism for bronchiolitis obliterans syndrome. Human immunology. 2003;64(5):521. doi: 10.1016/s0198-8859(03)00038-7. [DOI] [PubMed] [Google Scholar]
- 77.Maruyama T, Jaramillo A, Narayanan K, Higuchi T, Mohanakumar T. Induction of obliterative airway disease by anti-HLA class I antibodies. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005;5(9):2126. doi: 10.1111/j.1600-6143.2005.00999.x. [DOI] [PubMed] [Google Scholar]
- 78.Tiriveedhi V, Angaswamy N, Weber J, Mohanakumar T. Lipid raft facilitated ligation of K-alpha1-tubulin by specific antibodies on epithelial cells: Role in pathogenesis of chronic rejection following human lung transplantation. Biochem Biophys Res Commun. 2010;399(2):251. doi: 10.1016/j.bbrc.2010.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tiriveedhi V, Gelman AE, Mohanakumar T. HIF-1alpha signaling by airway epithelial cell K-alpha1-tubulin: Role in fibrosis and chronic rejection of human lung allografts. Cell Immunol. 2012;273(1):59. doi: 10.1016/j.cellimm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martinu T, Chen DF, Palmer SM. Acute rejection and humoral sensitization in lung transplant recipients. Proc Am Thorac Soc. 2009;6(1):54. doi: 10.1513/pats.200808-080GO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Christie JD, Edwards LB, Aurora P, Dobbels F, Kirk R, Rahmel AO, Taylor DO, Kucheryavaya AY, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-fifth official adult lung and heart/lung transplantation report--2008. J Heart Lung Transplant. 2008;27(9):957. doi: 10.1016/j.healun.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 82.Christie JD, Edwards LB, Kucheryavaya AY, Aurora P, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI. The Registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult lung and heart-lung transplant report--2010. J Heart Lung Transplant. 2010;29(10):1104. doi: 10.1016/j.healun.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 83.Hachem RR, Trulock EP. Bronchiolitis obliterans syndrome: pathogenesis and management. Semin Thorac Cardiovasc Surg. 2004;16(4):350. doi: 10.1053/j.semtcvs.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 84.Michaels PJ, Espejo ML, Kobashigawa J, Alejos JC, Burch C, Takemoto S, Reed EF, Fishbein MC. Humoral rejection in cardiac transplantation: risk factors, hemodynamic consequences and relationship to transplant coronary artery disease. J Heart Lung Transplant. 2003;22(1):58. doi: 10.1016/s1053-2498(02)00472-2. [DOI] [PubMed] [Google Scholar]
- 85.Wu GW, Kobashigawa JA, Fishbein MC, Patel JK, Kittleson MM, Reed EF, Kiyosaki KK, Ardehali A. Asymptomatic antibody-mediated rejection after heart transplantation predicts poor outcomes. J Heart Lung Transplant. 2009;28(5):417. doi: 10.1016/j.healun.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor DO, Edwards LB, Aurora P, Christie JD, Dobbels F, Kirk R, Rahmel AO, Kucheryavaya AY, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-fifth official adult heart transplant report--2008. J Heart Lung Transplant. 2008;27(9):943. doi: 10.1016/j.healun.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 87.Ramzy D, Rao V, Brahm J, Miriuka S, Delgado D, Ross HJ. Cardiac allograft vasculopathy: a review. Can J Surg. 2005;48(4):319. [PMC free article] [PubMed] [Google Scholar]
- 88.Terasaki P, Mizutani K. Antibody mediated rejection: update 2006. Clin J Am Soc Nephrol. 2006;1(3):400. doi: 10.2215/CJN.02311205. [DOI] [PubMed] [Google Scholar]
- 89.Mauiyyedi S, Colvin RB. Humoral rejection in kidney transplantation: new concepts in diagnosis and treatment. Curr Opin Nephrol Hypertens. 2002;11(6):609. doi: 10.1097/00041552-200211000-00007. [DOI] [PubMed] [Google Scholar]
- 90.Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol. 2005;5(10):807. doi: 10.1038/nri1702. [DOI] [PubMed] [Google Scholar]
- 91.Chapman JR, O’Connell PJ, Nankivell BJ. Chronic renal allograft dysfunction. J Am Soc Nephrol. 2005;16(10):3015. doi: 10.1681/ASN.2005050463. [DOI] [PubMed] [Google Scholar]
- 92.Yilmaz F, Aydin U, Nart D, Zeytunlu M, Karasu Z, Kaya T, Ozer I, Yuce G, Aydogdu S, Kilic M. The incidence and management of acute and chronic rejection after living donor liver transplantation. Transplant Proc. 2006;38(5):1435. doi: 10.1016/j.transproceed.2006.02.108. [DOI] [PubMed] [Google Scholar]
- 93.Demetris A, Adams D, Bellamy C, Blakolmer K, Clouston A, Dhillon AP, Fung J, Gouw A, Gustafsson B, Haga H, Harrison D, Hart J, Hubscher S, Jaffe R, Khettry U, Lassman C, Lewin K, Martinez O, Nakazawa Y, Neil D, Pappo O, Parizhskaya M, Randhawa P, Rasoul-Rockenschaub S, Reinholt F, Reynes M, Robert M, Tsamandas A, Wanless I, Wiesner R, Wernerson A, Wrba F, Wyatt J, Yamabe H. Update of the International Banff Schema for Liver Allograft Rejection: working recommendations for the histopathologic staging and reporting of chronic rejection. An International Panel. Hepatology. 2000;31(3):792. doi: 10.1002/hep.510310337. [DOI] [PubMed] [Google Scholar]
- 94.Bharat A, Fields RC, Steward N, Trulock EP, Patterson GA, Mohanakumar T. CD4+25+ regulatory T cells limit Th1-autoimmunity by inducing IL-10 producing T cells following human lung transplantation. Am J Transplant. 2006;6(8):1799. doi: 10.1111/j.1600-6143.2006.01383.x. [DOI] [PubMed] [Google Scholar]
- 95.Dunn MJ, Rose ML, Latif N, Bradd S, Lovegrove C, Seymour C, Pomerance A, Yacoub MH. Demonstration by western blotting of antiheart antibodies before and after cardiac transplantation. Transplantation. 1991;51(4):806. doi: 10.1097/00007890-199104000-00014. [DOI] [PubMed] [Google Scholar]
- 96.Jurcevic S, Ainsworth ME, Pomerance A, Smith JD, Robinson DR, Dunn MJ, Yacoub MH, Rose ML. Antivimentin antibodies are an independent predictor of transplant-associated coronary artery disease after cardiac transplantation. Transplantation. 2001;71(7):886. doi: 10.1097/00007890-200104150-00011. [DOI] [PubMed] [Google Scholar]
- 97.Nath DS, Tiriveedhi V, Basha HI, Phelan D, Moazami N, Ewald GA, Mohanakumar T. A role for antibodies to human leukocyte antigens, collagen-V, and K-alpha1-Tubulin in antibody-mediated rejection and cardiac allograft vasculopathy. Transplantation. 2011;91(9):1036. doi: 10.1097/TP.0b013e318211d2f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paul LC. Chronic renal transplant loss. Kidney Int. 1995;47(6):1491. doi: 10.1038/ki.1995.211. [DOI] [PubMed] [Google Scholar]
- 99.Dehan P, Van den Heuvel LP, Smeets HJ, Tryggvason K, Foidart JM. Identification of post-transplant anti-alpha 5 (IV) collagen alloantibodies in X-linked Alport syndrome. Nephrol Dial Transplant. 1996;11(10):1983. doi: 10.1093/oxfordjournals.ndt.a027085. [DOI] [PubMed] [Google Scholar]
- 100.Carter V, Shenton BK, Jaques B, Turner D, Talbot D, Gupta A, Chapman CE, Matthews CJ, Cavanagh G. Vimentin antibodies: a non-HLA antibody as a potential risk factor in renal transplantation. Transplant Proc. 2005;37(2):654. doi: 10.1016/j.transproceed.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 101.Eisenbarth GS, Morris MA, Scearce RM. Cytotoxic antibodies to cloned rat islet cells in serum of patients with diabetes mellitus. J Clin Invest. 1981;67(2):403. doi: 10.1172/JCI110048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maruyama T, Takei I, Matsuba I, Tsuruoka A, Taniyama M, Ikeda Y, Kataoka K, Abe M, Matsuki S. Cell-mediated cytotoxic islet cell surface antibodies to human pancreatic beta cells. Diabetologia. 1984;26(1):30. doi: 10.1007/BF00252259. [DOI] [PubMed] [Google Scholar]
- 103.Bosi E, Braghi S, Maffi P, Scirpoli M, Bertuzzi F, Pozza G, Secchi A, Bonifacio E. Autoantibody response to islet transplantation in type 1 diabetes. Diabetes. 2001;50(11):2464. doi: 10.2337/diabetes.50.11.2464. [DOI] [PubMed] [Google Scholar]
- 104.Verge CF, Stenger D, Bonifacio E, Colman PG, Pilcher C, Bingley PJ, Eisenbarth GS. Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes: Combinatorial Islet Autoantibody Workshop. Diabetes. 1998;47(12):1857. doi: 10.2337/diabetes.47.12.1857. [DOI] [PubMed] [Google Scholar]
- 105.Jaeger C, Hering BJ, Hatziagelaki E, Federlin K, Bretzel RG. Glutamic acid decarboxylase antibodies are more frequent than islet cell antibodies in islet transplanted IDDM patients and persist or occur despite immunosuppression. J Mol Med (Berl) 1999;77(1):45. doi: 10.1007/s001090050299. [DOI] [PubMed] [Google Scholar]