

Abstract
We measure the desorption of anions stimulated by the impact of 0–20 eV electrons on highly uniform thin films of plasmid DNA-diaminopropane. The results are accurately correlated with film thickness and composition by AFM and XPS measurements, respectively. Resonant structures in the H−, O−, and OH− yield functions are attributed to the decay of transient anions into the dissociative electron attachment (DEA) channel. The diamine induces ammonium-phosphate bridges along the DNA backbone, which suppresses the DEA O− channel and in counter-part increases considerably the desorption of OH−. The close environment of the phosphate groups may therefore play an important role in modulating the rate and type of DNA damages induced by low energy electrons.
I. INTRODUCTION
Secondary electrons are the most abundant species generated by primary ionising radiation1 and most of them have energies below 20 eV.2 The ability of these low energy electrons (LEEs) to break chemical bonds in biological media and more particularly DNA has been intensively explored during the last decade.3, 4 Plasmid DNA5–7 short oligonuleotides,8–14 DNA sub-units (nucleobases, sugars, and phosphate)15–17 have been the most studied targets.
LEEs can induce bond cleavage in condensed molecules by dissociative ionization and excitation or by dissociative electron attachment (DEA). In the case of DNA, DEA can lead to fragmentation of several sub-units,18 single and double strand breaks, via phosphodiester C–O bond cleavage and bases release via rupture of the N-glycosidic bond. Below the ionization threshold, bond breakage results essentially from resonant electron attachment into an unfilled electron orbital, located at specific sites. The local transient anion thus formed can decay via DEA or autoionization. In the latter process, when the departing electron leaves the site in a dissociative electronically excited state, two or more reactive radicals are formed. The DEA channel usually results in the production of an anion and a radical. In condensed molecules, the dissociative channels are strongly influenced by the local environment and any chemical bonding of surrounding molecules. As a matter of fact, covalent bonding19 and the presence of strong electric fields20 can reduce the lifetime of electron resonance, which has a direct impact on the DEA channels.
As the first anion mass spectroscopy measurements performed on DNA films, Pan et al.21 analyzed anions desorbing from linear DNA (40 base pairs) and supercoiled plasmid DNA upon LEE impact. They showed that the yield functions for the electron stimulated desorption (ESD) of H−, O−, and OH− exhibit a single broad structure near 9 eV with a continuous rise at higher energy having an onset near 15 eV. The structure has been assigned to DEA, while the continuous rise corresponded to dipolar dissociation (DD). Comparing these results with those obtained on thymine, guanine, and cytosine, the authors conclude that H− produced by DEA to DNA arises principally from the bases and with, possibly, a smaller contribution from the sugar ring of the backbone. O− was found to be produced from fragmentation of the phosphate group in the backbone, whereas OH− could partly arise from reactive scattering of O− produced at the phosphate group. Later, Ptasinska et al.22 irradiated GCAT oligonucleotides tetramers with 3–15 eV electrons and analysed different anionic species (H−, O−/NH2−, OH−, CN−, and OCN−) by ESD. The yield functions were compared with those obtained in the gas phase on isolated elementary components (bases, desoxyribose, and phosphate groups). These authors mentioned that while some of the dissociating transient anions found in the gas phase are still present within the molecular solid of GCAT, others are likely suppressed; particularly at low energies, owing to the existence of chemical and/or hydrogen bonds within DNA or due to insufficient kinetic energy for the anions to leave the surface. They not only identified the sites and bonds involved in desorption but also demonstrated site selectivity.
More recently, Dumont et al.23 and Zheng and Sanche24 observed a protective effect on DNA damage by organic salts, particularly in the chemical buffer used in most of the experiments involving DNA (Tris-HCl, EDTA). Single and double strand breaks (SSB and DSB) were considerably reduced in plasmid DNA under irradiation with 1 eV to 60 keV electrons. It was argued that the interaction of the organic cations with DNA may decrease the capture cross-section and lifetime of transient anions formed by 1 and 10 eV electrons on the base and phosphate groups of DNA. Electron transfer from anions to Tris cations or proton transfer from Tris to anions could contribute to reduce the lifetime of the transient anions formed on DNA sub-units. These modifications, including inherent variation of the electric field close to DNA19, 23 could therefore reduce DNA damage.
The objective of the present work is to explore the specific features of the complex formed between plasmid DNA and 1,3-diaminopropane under LEE irradiation. The focus lies on the dissociative processes leading to the direct desorption of anionic species from DNA-diamine thin films or desorption following reactive scattering of the primary anion. We generate yield functions of anionic species desorbed from DNA-diamine thin films exposed to LEEs.
The DNA-diamine complexes may be considered as a first crude model system to mimic the DNA physiologic environment, more particularly the DNA-protein interaction; notably the highly alkaline proteins found in eukaryotic cell nuclei that pack and order the DNA into nucleosomes. DNA-diamine films have the advantage to be highly uniform in thickness and composition.25 So, they constitute a well-defined environment to study ESD of anions from DNA under conditions closer to those of the cell. By correlating Atomic Force Microscopy (AFM) images and X-ray photo-electron spectroscopy (XPS) spectra to ESD measurements, the present experiments also allow, for the first time, after more than a decade of investigations of DNA damage induced by LEEs, to describe accurately the effect of thickness in well-gauged DNA layers.
II. MATERIALS AND METHODS
A. Chemicals and materials
Plasmid DNA (pUC21, 3151 bp) at an initial concentration of 1 mg/ml in water was obtained from PlasmidFactory GmbH & Co. KG (Germany). The solution contained no chemical buffer and 95% of the DNA was in the super-coiled configuration. Without further purification, a stock solution of this pUC21 DNA was prepared by dilution in ultrapure water having a resistivity of 18.2 MΩ cm. The DNA concentration was determined by measuring the absorbance at 260 nm taking the molar extinction coefficient to be ε260 = 5.3 × 107 cm−1 M−1.26 1,3-Diaminopropane dihydrochloride (98%) was purchased from Sigma-Aldrich and kept at 4 °C. Solutions were freshly prepared before each experiment.
B. Target preparation
The DNA-diaminopropane thin films were deposited on graphite HOPG substrate using the protocol described extensively in a previous paper.25 Briefly, a solution of the plasmid of 40 ng/μl is mixed with an equal volume of a solution containing protonated 1,3-Diaminopropane molecules (Dap2+) to attain the desired ratios R of the molar concentrations of the divalent ions to that of anionic phosphate moieties of DNA in the solution, i.e., R = [Dap2+]/[PO4−] = 8, 16, and 32. The resulting solutions were mixed and incubated for 15 min at 25 °C. The DNA-Dap deposits were prepared by soft adsorption25: A droplet of 50 μl of the DNA-Dap mixture was deposited onto a freshly cleaved graphite (HOPG, ZYA grade, NT-MDT) surface and incubated during 15 min. Next, the surplus mixture solution was removed using filter paper. This protocol ensures a circular smear of constant diameter 8 ± 1 mm to be created on the freshly cleaved HOPG surface. After drying in clean air for 3 min, 12 samples were introduced into a load-lock chamber to be vacuum-dried 12 h under 10−8 Torr. Afterwards, they were transferred via a gate valve into an ultra-high-vacuum (UHV) chamber (2 × 10−10 Torr) for analysis by XPS and ESD measurements of anions. Sample analysis by AFM was performed in air with another system. The dap samples used in part III-B. Electron Stimulated Desorption and shown in Figure 3 were prepared using the very same protocol than the one presented above.
FIG. 3.

(a) and (b) Incident electron energy dependence of respectively O−/NH2− and H− yields from thin films of 1,3-diaminopropane(Dap2+), supercoiled plasmid DNA, and DNA-Dap2+ complexes deposited on HOPG. (c) Normalized yields of O−/NH2− signals for the Dap2+ and Dap2+-DNA layers. (d)–(f) AFM (3 × 2 μm) tapping mode images of the corresponding samples; they were all recorded with the same magnification (see (d) for the scale). The AFM images show (d) a Dap2+-DNA thin film of 4 nm thickness, (e) the surface of a Dap2+deposit, where the molecules are not visible on the image owing to the limited spatial (lateral) resolution of the AFM in air, and (f) individual DNA plasmids spread on the HOPG surface.
C. XPS and AFM characterization
The morphology, composition, and DNA/diamine ratio of the deposits were characterized by AFM and XPS, respectively. The XPS measurements were performed in a UHV chamber at an operating pressure of 4 × 10−10 Torr using a Perkin Elmer ESCA 4.0 XPS system equipped with a dual anode (Mg/A1) X-ray source (P = 400 W, Physical Electronics, 04-548), a hemispherical electron energy analyzer (Physical Electronics, 10-360), and a channeltron detector. The X-ray beam was incident at 70° relative to the sample surface, and the hemispherical electron energy analyzer was positioned normal to the surface. The pass energy of the electron energy analyzer was set at 2.95 eV, which correspond to an XPS spectral resolution of E = 0.76 eV. The optics of the spectrometer sees a maximum 6 mm diameter area (i.e., 28 mm2) on the analyzed sample. The AFM images were obtained with a Molecular Imaging scanning probe microscope (Agilent, USA). The topography of the surface could be recorded in air, at standard temperature and pressure, with silicon nitride tips coated with aluminium (Nanoandmore) at a resonant frequency of 300 kHz in the tapping mode.
D. ESD analysis
The ESD system and the LEE-irradiation procedure have been described in details in Ref. 27. Briefly, an electron gun produces a beam of 1.5 nA on a 4 mm2 spot at the surface of the DNA target with an energy spread of 0.5 eV FWHM. The beam makes an angle of 70° with respect to the normal to the target surface. A Quadrupole Mass Spectrometer, positioned perpendicular to the film surface probes the ESD yields of the desorbed anions. In the present experiment, the masses 1, 16, and 17 amu, were selected and their intensities were recorded as a function of incident electron energy in the range 0–20 eV. The relative yield was normalized to the current measured during the experiment at the graphite substrate. The total incident electron current transmitted through the sample was recorded with a picoammeter (Keithley). The beam energy was determined relative to the vacuum level by measuring the onset of current transmitted through the film.
III. RESULTS AND DISCUSSION
A. Plasmid DNA – 1,3 diaminopropane complexes
In an aqueous environment, the distance that separates the two opposite terminal amino groups, in the equilibrium conformation of diaminopropane (Dap), matches well the distance between two consecutive phosphate groups (O1P–O1P separation) along the DNA backbone in its natural B form (Figure 1(a)).28, 29 Under neutral pH conditions, the amino terminal groups of the Dap molecule are ionized (ammonium cations). The Dap moiety is a constitutive pattern in allbiogenic polyamines and is able to substitute water molecules in the first-hydration shell of the DNA polyanion.28 It was shown recently that the affinity of doubly charged, 1-3 Dap for DNA permits the growth on HOPG substrates, of plasmid DNA films of known and uniform thickness.25 The deposition method is based on soft-adsorption and enables very dense and well-characterized layers of a Dap2+-plasmid DNA mixture to be fabricated with reproducible thicknesses ranging from 2 nm (the cross-sectional diameter of DNA double strand) to several dozen of nanometres. Those layers have a total area on HOPG that depends on the volume of the drop of aqueous solution deposited. In our experiments, the circular area covered by the layers has a constant diameter of 8 mm. AFM snapshots of DNA films prepared with this method are presented in Figure 2 for the thicknesses prepared in this study.
FIG. 1.

(a) Schematic view of the specific sites where Dap2+ cations interact with the DNA phosphate moieties: (1) corresponds to the linking of two double-strands belonging or not to a same plasmid DNA, (2) corresponds to the electrostatic binding of a Dap2+ cation to two consecutive anionic phosphate moieties along the DNA’s backbone (the special case where Dap2+ is capable of pronounced narrowing of the minor groove through direct binding to the phosphate groups across the groove29 is not shown here for clarity), (3) illustrates the case where one Dap2+ cationic site remains free, (4) shows the Dap2+ diamine where both ammonium sites are counterbalanced with chloride anions in solution. (b) Size and structure comparison of the Dap molecule with the three basic amino acids histidine, arginine, and lysine that interact with the DNA phosphate moieties in histones in the cellular environment. (b) are HyperChem(TM) snapshots of molecular mechanics optimizations based on the Block-diagonal (Newton-Raphson) method.
FIG. 2.
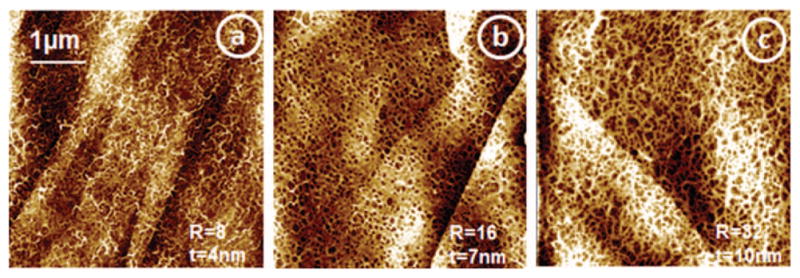
AFM tapping mode images of DNA-Dap films constructed on HOPG with molar ratios R = [Dap2+]/[PO4−]DNA = 8, 16, and 32, respectively for (a), (b), and (c), prepared with CDNA = 20 ng/μl. In such conditions, the films have respectively thicknesses of 4, 7, and 10 nm.
In those deposits, the DNA inside the layers resembles the close architecture of DNA inside nucleosomes, the base unit of histones. Histone proteins are indeed composed of basic amino acids, rich in amino groups ionized at neutral pH, that pack DNA through electrostatic interactions with the phosphate groups. Nevertheless, it should be mentioned that there are no carboxylic acid moieties present in the Dap molecule contrary to the basic amino acids: arginine, histidine, and lysine that each contains one acid group and respectively 4, 3, and 2 amino groups (See Figure 1(b)). In a first approximation, it may nevertheless be possible to compare the close environment of DNA in the Dap2+-DNA complexes to heterochromatin; the tightly packed form of DNA localized to the periphery of the cell nucleus that plays a role in the expression of genes.
B. Electron stimulated desorption
In our experiments, layers of Dap, supercoiled plasmid DNA, and the [Dap-DNA] complex were deposited onto HOPG separately and exposed to LEEs in the exactly same conditions. In Figure 3, we present the anion ESD data and corresponding AFM images of the targets. It should be noticed that it is not possible to spread uniform layers of only supercoiled DNA on HOPG using our soft-adsorption protocol; only sparsely adsorbed DNA plasmids can be imaged; as for Dap2+ alone only a very thin layer could be formed. Dap2+ and DNA plamids alone can therefore only be deposited as submonolayers using the present soft adsorption technique. The average height of the DNA monolayer was measured equal to 1.7 ± 0.4 nm; as for the Dap2+ layer, its thickness is 0.3 ± 0.2 nm. The 1,3-Dap molecules could not be imaged (in Figure 3(e)), owing to their very small size and the limited spatial resolution of the AFM in air. Dap2+ adsorption on graphite was confirmed by observing modifications in the surface properties of the HOPG substrate, notably hydrophobicity (not shown here).
The ESD signals of mass 1 and 16 amu presented in Figure 3 were recorded from DNA, Dap2+, and [DNA-Dap2+] complexes. Owing to the small amounts of material deposited on the substrate, the signal is somewhat noisy, but on average a broad peak extending from 3–5 eV to 13 eV is clearly seen in both curves. The ESD signal of mass 16 amu from the Dap2+deposit obviously can be attributed to NH2− desorbed from the amino group of this diamine, considering that no H2O was found to desorb from the surface during the irradiation in UHV. However, in the case of DNA deposits, the signal mass 16 has previously been attributed to O− produced through fragmentation of the phosphate group in the DNA backbone.37 In order to make it clear, if mass 16 u originates from NH2− and/or O−, the signals from Dap2+ and [DNA-Dap2+] layers have been normalized to the same peak intensity to allow for easier comparison and are presented on the top of the second row in Figure 3. It should be noticed that the Dap2+-DNA layer used in this part of the study had a thickness less of 4 nm, in order to keep ESD yield intensities comparable with those of the submonolayers of Dap and DNA alone. With such thin layers, the signals appear to be somewhat noisy, but the general trends observed in the right-handed upper part of Figure 3 are similar. Furthermore, when fitted mathematically (Log Normal), the two yield functions are characterized by the same onset, i.e., 4 eV within a ±0.5 eV confidence interval. With pure DNA films (not complexed) prepared by lyophilisation or as Self-Assembled Monolayers (SAM) with linkers chemically bound to the surface, or even as layers composed of only DNA bases, the ESD O−/NH2− yield function generally exhibits an onset ranging from 3.5 to 5.5 eV (see Table I). The exact origin of mass 16 is sometimes difficult to determine since both oxygenated and nitrogenised moieties exist in native DNA. Here, within our samples composed of [DNA-Dap2+] layers there exists a very rich environment of ammonium groups (R–NH3+) located in the close vicinity of the phosphate groups along the DNA backbone. Moreover, when Dap alone is exposed to LEEs, no oxygen is present. Thus, as both signals at 16 u (Figure 3, second row, top) are essentially identical when Dap or Dap–DNA layers are compared, it seems likely that both signals are mainly due to NH2−. This may appear as a surprising result, since O− is generally invoked as a source of 16 u20 when DNA is analysed by ESD. If such an assumption is taken as valid, it means that the channel by which O− is produced may be masked or drastically lowered due to the peculiar environment of DNA in our layers, particularly due to the presence of diamines near to the phosphate groups. O− cannot be considered as totally absent in our ESD experiments. But considering that a Dap submonolayer produces a highest intensity of ESD signal at 16 u as compared to Dap-DNA and DNA thin layers (Figure 3), and considering in addition, that the yields functions have very similar onsets (i.e., 4 eV), there are good reasons to think that NH2− is a major component of this signal at 16 amu. The exhaustive study of the literature, summarized in Table I, was undertaken to identify all possible sources of NH2− and O− ESD signals and to compare them to the OH− signals. Additionally, we have also studied the effect of the layers thickness on the yields of anion desorption in order to provide better insight into the mechanisms taking place during exposure to LEEs.
TABLE I.
Comparison of the ratios OH−/[O−/NH2 −] obtained from ESD analysis of different targets by different authors. The numbers in parenthesis in the fifth column represent the energy of maximum DEA signal in the structure corresponding to O−/NH2 − or OH−, respectively, whereas those in the sixth column represent the threshold energy of these signals.
| Reference | Target | Thickness (nm or ML) | Substrate | Resonance energy (eV) (O−/NH2, OH−) | Threshold energy (eV) (O−/NH2, OH−) | Yields ratio OH−/[O−/NH2−] |
|---|---|---|---|---|---|---|
| This work | Plasmid DNA-Dap2+ | 4; 7 and 10 nm | HOPG | (8.0, 7.0) | (4, 2.5) | 2.15, 1.65, 3.5 |
| Ptasińska and Sanche22 | Tetramer CGAT | 1.6 ± 0.3 nm | Tantalum | (9.1, 9.1) | (3.5, 3) | 0.7 |
| Mirsaleh-Kohan et al.30 | - SAM-DNA dosed with 16O2 (at 55 K, 0.75 ML 16O2) | … | Gold | (7.2, 7.3) | (3.3, 3) | 0.14 |
| - SAM-DNA dosed with 18O2 (at 150 K, 3.5 ML 18O2) | … | Gold | (7.0, 7.0) | (3.2, 3) | 0.2 | |
| Mirsaleh-Kohan et al.31 | - SAM-DNA (10-mer) | Gold | (7.8, 7.1) | (5.3, 2.7) | 0.9 | |
| - Lyophilized DNA (10-mer) | 5 ML | Gold | (8.3, 6.7) | (4.9, 2.65) | 0.9 | |
| Pan and Sanche32 | - SAM 5S-dsDNA | 1 ML | Gold | (8, 7) | (4, 2.5) | … |
| Mirsaleh-Kohan et al.33 | - SAM 5S-dsDNA | 1 ML | Gold | (~8, 6.5) | (4–5, 2.5) | ~2 |
| Pan et al.21 | - Linear DNA, 40 base pairs | −2.9 ± 0.3 nm | - Tantalum | (9.2, 9.0) | (4.6, 4) | 0.2 |
| - Supercoiled plasmid DNA | −3 ML | - Tantalum | (9.2, 9.0) | (5, 4) | 0.2 | |
| Abdoul-Carime et al.34 | DNA base : G | 8 ML | Platinum | (9.0, 7.8) | (5, 5) | 0.9 |
| T | 8 ML | Platinum | (11.0, 9.8) | (5.5, 5) | 1 | |
| C | 8 ML | Platinum | (8.8, 9.1) | (5, <5) | 0.8 | |
| Abdoul-Carime and Sanche35 | Amino-acid: tryptophan | 5 ML | Platinum | (7.0, 7.9) | (4.5, 4.5) | 1.6 |
| Histidine | 5 ML | Platinum | (7.3, 7.7) | (5, 5) | 0.25 | |
| Proline | 5 ML | Platinum | (7.2, 7.6) | (4.5, 4.5) | 1 | |
| Abdoul-Carime and Sanche36 | Aliphatic amino-acid: glycine | 10 ML | Platinum | (7.5, 7.5) | (5, 5) | 1 |
| Alanine | 10 ML | Platinum | (7.6, 7.6) | (3.5, 2.5) | 1 | |
| Cysteine | 10 ML | Platinum | (7.2, 7.2) | (4.5, 2.5) | 0.25 |
Figure 4 shows the OH−, O−/NH2−, and H− ESD yields of three different deposits with thicknesses equal to 4, 7, and 10 nm.
FIG. 4.

The yield functions of (a) OH−, (b) O−/NH2−, and (c) H− obtained from three DNA-Dap2+ films with 4, 7, and 10 nm thicknesses and from a clean HOPG surface. In panel (b), two arrows indicate the special behaviour of the yield function observed when comparing the 7–10 nm layer, where an inversion of the yields occurs from the resonance region (DEA) compared to the monotonically increasing region of DD. The lines through the data points serve only as a guide to the eye.
It appears from these measurements that the yields of H− and OH− near to the DEA structure are proportional to the film thickness, while those of O−/NH2− are proportional to the amount of Dap2+ present in the films. The two arrows in Fig. 3(b) point to that special case and will be discussed in more details later in Sec. III on the analysis of the composition of the films by XPS. The proportionality to the amount of Dap2+ only occurs around the DEA structure. Above the onset of dipolar dissociation (12 eV), we observe again thickness dependence. As it will be shown further in this paper, only the DEA structure is affected (inhibited) by the film composition; i.e., the proportion of Dap. The ESD yield ratios OH−/[O−/NH2−] obtained by various authors are listed in Table I.21, 22, 30–36 They include ratios obtained with DNA and some of its constituents, and also amino-acids, so as to provide a comparison with biological targets rich in both oxygenated and nitrogenised moieties. Obviously, these ratios strongly depend on target composition. A rapid comparison of the different ratios OH−/[O−/NH2−] clearly reveals that in DNA-Dap2+ layers (this work) these ratios are remarkably higher than in those related to previous experiments, except for the cases of SAM 5S-dsDNA samples, where DNA monolayers are chemisorbed by sulphur atoms on gold substrates. In these latter cases, the very same thresholds are found both for O−/NH2− and OH− ESD yields.32, 33 Such high ratios OH−/[O−/NH2−] can be considered as a signature of the presence of diaminopropane molecules in our films. Additionally, in one of the two examples shown, the OH−/[O−/NH2−] ratio appears to be as high as measured in the present experiments.33 Remarkably in the work by Pan and Sanche32 it was shown that the higher the number of phosphate moieties engaged in chemical bonds with sulphur atoms, the higher the OH− ESD yield. By looking at the yield dependence of desorbed species with particular attention to the OH−/[O−/NH2−] ratio, one can learn about possible transverse processes such as, for example, anion reactive scattering between irradiation products. This mechanism was previously identified as linking O− production to OH− desorption; it was first observed in alkane-O2 mixture films.20, 37 In this process, O− produced via DEA to O2, initiates hydrogen abstraction from the hydrocarbon moieties. In general, the ratio OH−/[O−/NH2−] should depend on the abundance of each group (–O– and −OH) in the sample and additionally on the contribution of the reactive scattering of O− to form OH−. Concerning this particular point; within DNA-Dap2+films, O− leaving could therefore react (i.e., hydrogen abstraction) with the nearby ammonium group of Dap2+ and contribute to an increase of the yield of OH− des-orbed from the sample. But, with an onset of OH− production at 2.5 eV, much lower than all other onsets observed (see Table I), and particularly the O−/NH2− onset at 4 eV, this mechanism can be discarded from energy conservation considerations.
It is important to keep in mind that plasmid DNA contains no OH moieties, which can desorb from cleavage of a single bond. Thus, a hydroxyl anion (OH−) leaving pure DNA by ESD requires that the DNA phosphate charge be neutralized30 (protonation), which in the present study requires the capture of a proton. Therefore, some chemical modification during DNA-Dap2+ irradiation should be at the origin of the present observations. Zheng et al.24 studied the effect of organic salts, namely the chemical buffer Tris-EDTA (TE), on strand break formation in DNA exposed to LEEs. In that study, a protective effect of TE on DNA was observed. It is noteworthy that within the TE buffer, the Tris molecule (2-amino-2-hydroxymethyl-1,3-propanediol) contains an amino group which is mainly ionised under neutral pH and therefore can interact with the anionic phosphate moieties along the DNA backbone. The authors attribute the observed protecting effect either to an electron transfer from the anionic sites of DNA to the Tris cation or to a proton transfer from Tris to DNA, which could contribute in reducing DNA damage. An analogy with that latter pathway (proton transfer) we provide a mechanism to explain the abundant OH− ESD yields observed in the present work. To illustrate this phenomenon, we propose the following “TNI-Proton pair decay” mechanism (Figure 5), in which the DNA phosphate group and the ammonium Dap2+ react electrostatically during the Transient Negative Ion (TNI) dissociation time. As originally shown by Barrios et al. in the case of a ground state base of DNA38 and later for an excited state of a base,39 after capture by a DNA base, a LEE is transferred to a σ* orbital of the PO4 group forming a dissociative TNI state. The phosphate group thus becomes quasi doubly charged and it is likely that a labile proton of an ammonium group in a neighbouring Dap2+ molecule may be quasi-instantaneously captured by the TNI. Let us call τTNI the average dissociation time of the TNI and τPT the average time required to transfer a proton from the Dap2+ to the phosphate moiety. When τTNI > τPT, an OH group can be formed within the DNA TNI at the P-O− site during the transient ion lifetime. In this way, OH− can be a dissociating charged product (anion) resulting from DEA. When τTNI < τPT, the charged product can be O−. Since in the present experiments the O− channel appears to be essentially suppressed, we are prone to argue that the TNI lifetime must be greater than the time required for capturing a proton.
FIG. 5.

The two possible dissociative channels of the TNI where the charged products are either OH− or O−, depending on the proton transfert mean lifetime (τPT) as compared to the TNI lifetime (τTNI).
As a result, the yield at 16 u should be attenuated; it is nevertheless not off, owing to the production of NH2− at the same mass. This may explain the high ratio OH−/[O−/NH2−] observed in DNA-Dap2+ films, despite the contribution of NH2− desorbed from Dap2+ to the yield at 16 u. The present ratio can therefore be regarded as a pertinent indicator of O− dissociative channel activity in DNA-Dap2+ complexes or in other DNA complex layers exposed to LEEs.
In fact, owing to the strong reduction of O− dissociative decay from DNA-Dap2+ films, the contribution of NH2− to the total yield at 16 u should be dominant. XPS analysis of the composition of the samples provides further insight into the role of Dap2+.
C. Composition of the films – XPS analysis
In order to ascertain that the amount of Dap2+ in the films is directly linked to the O−/NH2− yields, each sample was analyzed by XPS immediately after the ESD experiments. Our deposits were always produced using a constant volume of DNA–Dap mixture (see Sec. III B), which when deposited on HOPG lead to a circular smear (film) with a constant area of 8 ± 1 mm diameter. When analyzing a sample by means of XPS, the whole area is irradiated but the optics of the spectrometer sees a maximum 6 mm diameter area (i.e., 28 mm2). On the other hand, the LEE beam used in the ESD experiments has a cross-sectional area less than 4 mm2. In terms of irradiated surface, we have a 7-fold ratio between XPS and ESD so that when XPS is performed after an ESD experiment, most of the sample has not been damaged by ESD analysis. In addition, during the course of ESD experiments a 1.5 nA current was used so that the degradation of the sample cannot be considered as significant. On the other hand, this explains the noisy signals observed in the upper part of Figure 3. To perform the analysis of the composition of the films by XPS, nitrogen (1s) and phosphorous (2p3/2) peaks were analyzed precisely and their areas where measured after background subtraction. Figures 6(a) and 6(b) show those XPS signals and the fittings realized using the XPS Peak Fitting Program.
FIG. 6.
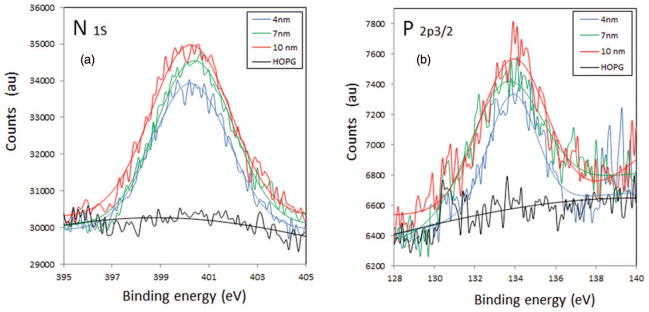
(a) N 1s and (b) P 2p3/2 XPS spectra of the three layers of DNA-Dap2+ and of the pristine HOPG substrate in the same binding energy region. The blue, green, and red lines correspond respectively to the fittings realized using the XPS peak fitting program on the 4, 7, and 10 nm thick layers.
As HOPG is freshly cleaved prior to any experiment and graphite is hydrophobic, it is not surprising to find that the amount of water and contaminants is small, as seen from the spectra presented in Figure 6. Phosphorous is found only in the composition of the DNA macromolecule whereas nitrogen is present in both DNA and Dap2+ molecules. Using the known composition of our plasmid DNA (base pairing), it is possible to compute the exact ratio N/P of the plasmid in the target films, i.e., (N/P)DNA = 3.75. From the experimentally determined ratios, the number ξ of diamines (Dap2+) per base pair is evaluated. One base pair corresponds to two phosphate groups that may each bind to one or two ammonium groups. As shown in Figure 1(a), the binding of Dap2+ following schemes 1 and 2 corresponds to ξ = 1, i.e., one diamine per base pair or per two phosphate moieties. On the other hand, scheme 3 corresponds to one Dap2+ per one phosphate group (i.e., a maximum value of ξ = 2 can be reached in that special case). For the three films bombarded 1.27 ≤ ξ ≤ 1.75. This does not mean, nevertheless, that all the phosphate groups of DNA are bound to Dap2+. Dap also can be incorporated in the layer without direct interaction with the phosphate groups. This may correspond to scheme 4 in Figure 1(a), but considering the ξ values, it is likely that most of the phosphate moieties are linked to Dap2+ cations, via schemes 1 and 3 of Figure 1(a). Manning’s theory40 of DNA condensation by multivalent counterions, where two different counterions are present, was applied by Wilson and Bloomfield41 to the case of putrescine– DNA complexes in aqueous environment. Putrescine or 1,4-diaminobutane is a diamine containing 4 methylene groups instead of 3 in 1,3-diaminopropane. These authors claim that condensation occurs for 89%–90% of the DNA negative charges neutralized. Considering that the DNA stock solution in our experiments is free of any buffer, the native DNA polyanion counterions are most probably sodium ions (Na+ or at least other biogenic cations). Thus, according to Manning’s fluctuation theory of DNA condensation by multivalent counterions, if we agree with above mentioned considerations, most of the phosphate moieties in our films are engaged in electrostatic interactions with a Dap2+ molecule and the remaining portion is neutralized by native counterions. Such an assumption is reinforced by ion competition experiments indicating that sodium ions bind to the minor groove with weak affinity, whereas ammonium ion binding is somewhat stronger.42
More interesting here is the higher ratio ξ (diamines/bp) for the 7 nm thick sample in Table II, for which we observe higher resonance amplitude than for the thicker one, i.e., 10 nm (see Figure 4(b), left arrow). A simple analysis of the ESD yields shows that there is a linear behaviour of the O−/NH2− yields versus the respective values of ξ. As for the OH− yields; a perfect linearity is observed versus film thickness (inset Figure 7). As mentioned by Tougaard,43 the composition estimated from an XPS peak area is found to be accurate within ~10%–20% depending on the particular depth distribution of the atoms. A 10%–20% is consistent with a thickness or sampling depth <3λ, where λ stands for the photoelectron Inelastic Mean Free Path (IMFP). Since anions have an IMFP much smaller than photoelectrons and LEEs, we can easily argue that due to the linearity observed in Figure 7, the condition of a sampling depth < 3λ is met; thus a ± 10% deviation appears as a reasonable estimate for the error in the results of Figure 7. As for the thickness measurements, statistical averages of various samples (at least 3 samples with each different thickness were analyzed) lead to a ± 1 nm experimental deviation.
TABLE II.
XPS experimentally determined ratios nitrogen/phosphorus (N/P) for three different DNA-Dap2+ layers obtained with R = 8, 16, and 32 and CDNA = 20 ng/μl. Nitrogen in the layers has two possible origins, from the DNA base pairs or from the Dap2+ ammonium groups. With our plasmids, (N/P)DNA = 3.75 and ξ = (N/P)exp. − (N/P)DNA, where ξ is the number of diamine per base pair in the film. AN and AP stand for the areas of the nitrogen (1s) and phosphorous (2p) peaks, respectively, after background subtraction. The DNA stock solution used in this study contained no buffer.
| R | 8 | 16 | 32 |
|---|---|---|---|
| Film thickness (nm) | 4 | 7 | 10 |
| AN | 17358.97 | 23703.78 | 26736.4 |
| AP | 3632.191 | 4529.269 | 5218.654 |
| (N/P)DNA | 3.75 | 3.75 | 3.75 |
| (N/P)exp | 5.02 | 5.50 | 5.38 |
| ξ[diamines/bp] | 1.27 | 1.75 | 1.63 |
FIG. 7.

Yields O−-NH2− versus ξ, number of Dap2+ per base pair in the films. Inset: OH− yields as a function of the thickness of the same films. The error bars represent statistical deviations (see the text for more details). The lines are linear regressions with R2 = 0.9991 and 0.9993 respectively.
From this study, we learned about the particular role of the DNA anionic phosphate groups when their charge is counter-balanced by an ammonium group. We propose that the proportionality of the O−/NH2− yields with the film composition (number of Dap2+ per base pair, ξ) may pre-dominantly be due to the desorption of NH2− from the Dap molecules as the O− desorption pathway is suppressed in the close vicinity of the phosphate moiety and finally OH− anions escape from the films due to the mechanism invoked above. The OH− yields are proportional to the film thickness and are typically 1.65–3.5-fold greater than O−/NH2− yields, when measured under the same conditions. This behavior is possible in our experiments owing to the presence of important amounts of ammonium groups in the close vicinity of DNA’s phosphate groups from the presence of Dap2+ ions. We know from different studies conducted in this area of research that under LEE irradiation, the phosphate groups of DNA play a crucial role in single and double strand break formation, either by direct attachment or by transfer from the DNA bases.16, 17, 39, 44–47 The proportionality of the OH− ESD yield with layer thickness appears nevertheless surprising, as most OH− ions would be expected to originate from the top surface layer. However, looking at the AFM images of our samples and more particularly in Figure 3(d), we can see that the layers are somewhat “porous.” Even at 10 nm thickness, when these film are exposed to LEEs in the UHV their particular fiber-like structure should contain sufficient empty volumes to allow the escape of OH− ions.
IV. SUMMARY AND CONCLUSION
In this work, we have studied the desorption of anions stimulated by the impact of 0–20 eV electrons on uniform layers of well-defined thicknesses of plasmid DNA complexed with Dap2+ diamines. The resonant structures observed by others near 9 eV have been confirmed. A comparison of the ratios OH−/[O−/NH2−] of the ESD yields resulting from this work with those gathered from earlier articles shows that the ratios determined herein are notably higher than all those measured previously. We argued that the effect of the surrounding medium favours specific reactive processes, notably the escape of OH− under LEE impact. This effect can be explained by the high labile-hydrogen concentration in the close vicinity of the DNA backbone arising from the presence of positively charged ammonium groups in the Dap molecule that are electrostatically bound to the DNA phosphate anionic moieties in the layers.
The high homogeneity and excellent flatness of the [DNA-Dap2+] layers made it possible to demonstrate that the ESD yields of OH− anions depend linearly on thickness from 4 to 10 nm, whereas the O−/NH2− yields depend linearly on the amount of Dap2+. The measured OH− yields were found to be 1.65–3.5-fold greater than the O−/NH2− yields when measured under identical conditions. Impact of the DNA close environment on the dissociative channels is therefore confirmed by the present results, particularly from the low yields of O− and the high amounts of OH− produced by ESD. Additionally, we have shown that the O−/NH2− (i.e., essentially NH2−) yields are directly proportional to the ratios ξ (diamines/bp) determined by XPS analyses of the samples; in other words, this yield depends linearly on the concentration of amino groups in the layers. The OH−/[O−/NH2−] ratio appears to be an indicator of specific dissociative channels involved in TNI decay within any DNA layer’s chemical composition.
Based on the daring hypothesis that the [DNA-Dap2+] complexes may constitute a first crude model toward LEE interactions with DNA in the cellular environment, we may anticipate that this type of environment will likely be a source of modifications in the dissociative channels of transient anions of DNA created in a real biological situation. We have thus reasons to believe that the close environment of DNA in living species may not only modify the pathways of fragments produced by DEA, but also the values of cross-sections for DNA damage production. Our group is now investigating the production of well characterized layers of plasmid DNA with electrostatic linkers containing also oxygenated groups (carboxylic acid moiety), in addition to the amino moieties, in order to get closer to the structure of basic amino acids present in the chromosomal matrix.
Acknowledgments
This research was partially financed by the Canadian Institute of Health Research (Grant No. 81356).
References
- 1.La Verne JA, Pimblott SM. Radiat Res. 1995;141:208. [PubMed] [Google Scholar]
- 2.Cobut V, Frongillo Y, Patau JP, Goulet T, Fraser MJ, Jay-Gerin JP. Radiat Phys Chem. 1998;51:229. [Google Scholar]
- 3.Baccarelli I, Bald I, Gianturco FA, Illenberger E, Kopyra J. Physics Reports. 2011;508:1. [Google Scholar]
- 4.Sanche L. Radiation Damage in Biomolecular Systems. Springer; Netherlands: 2012. p. 3. [Google Scholar]
- 5.Boudaiffa B, Cloutier P, Hunting D, Huels MA, Sanche L. Science. 2000;287:1658. doi: 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- 6.Kumar SVK, Pota T, Peri D, Dongre AD, Rao BJ. J Chem Phys. 2012;137:045101. doi: 10.1063/1.4737182. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Aleksandrov A, Orlando TM. Int J Mass Spectrom. 2008;277:314. [Google Scholar]
- 8.Huels MA, Hahndorf I, Illenberger E, Sanche L. J Chem Phys. 1998;108:1309. [Google Scholar]
- 9.Li Z, Cloutier P, Sanche L, Wagner JR. J Am Chem Soc. 2010;132:5422. doi: 10.1021/ja9099505. [DOI] [PubMed] [Google Scholar]
- 10.Solomun T, Seitz H, Sturm HJ. Phys Chem B. 2009;113:11557. doi: 10.1021/jp905263x. [DOI] [PubMed] [Google Scholar]
- 11.Orlando TM, Oh D, Chen Y, Aleksandrov AB. J Chem Phys. 2008;128:195102. doi: 10.1063/1.2907722. [DOI] [PubMed] [Google Scholar]
- 12.Solomun T, Illenberger E. Chem Phys Lett. 2004;396:448. [Google Scholar]
- 13.Ray SG, Daube SS, Naaman R. Proc Natl Acad Sci USA. 2005;102:15. doi: 10.1073/pnas.0407020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polska K, Rak J, Bass AD, Cloutier P, Sanche L. J Chem Phys. 2012;136:075101. doi: 10.1063/1.3685587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denifl S, Ptasińska S, Probst M, Hrusak J, Scheier P, Märk TD. J Phys Chem A. 2004;108:6562. doi: 10.1063/1.1649724. [DOI] [PubMed] [Google Scholar]
- 16.Bald I, Kopyra J, Illenberger E. Angew Chem. 2006;45:4851. doi: 10.1002/anie.200600303. [DOI] [PubMed] [Google Scholar]
- 17.Li Z, Zheng Y, Cloutier P, Sanche L, Wagner JR. J Am Chem Soc. 2008;130:5612. doi: 10.1021/ja077601b. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y, Cloutier P, Hunting DJ, Sanche L, Wagner JR. J Am Chem Soc. 2005;127:16592. doi: 10.1021/ja054129q. [DOI] [PubMed] [Google Scholar]
- 19.Gruyters M, Jacobi K. Surf Sci. 1995;336:314. [Google Scholar]
- 20.Sanche L, Parenteau L. J Chem Phys. 1990;93:7476. [Google Scholar]
- 21.Pan X, Cloutier P, Hunting D, Sanche L. Phys Rev Lett. 2003;90:208102. doi: 10.1103/PhysRevLett.90.208102. [DOI] [PubMed] [Google Scholar]
- 22.Ptasińska S, Sanche L. J Chem Phys. 2006;125:144713. doi: 10.1063/1.2338320. [DOI] [PubMed] [Google Scholar]
- 23.Dumont A, Zheng Y, Hunting D, Sanche L. J Chem Phys. 2010;132:045102. doi: 10.1063/1.3298895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y, Sanche L. J Chem Phys. 2010;133:155102. doi: 10.1063/1.3505046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boulanouar O, Khatyr A, Herlem G, Palmino F, Sanche L, Fromm M. J Phys Chem C. 2011;115:21291. [Google Scholar]
- 26.Manchester KL. BioTechniques. 1996;20:968. doi: 10.2144/96206bm05. [DOI] [PubMed] [Google Scholar]
- 27.du Penhoat MAH, Huels MA, Cloutier P, Jay-Gerin JP, Sanche L. J Chem Phys. 2001;114:5755. [Google Scholar]
- 28.van Dam L, Korolev N, Nordenskiöld L. Nucleic Acids Res. 2002;30:419. doi: 10.1093/nar/30.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korolev N, Lyubartsev AP, Laaksonen A, Nordenskio L. Eur Biophys J. 2004;33:671. doi: 10.1007/s00249-004-0410-7. [DOI] [PubMed] [Google Scholar]
- 30.Mirsaleh-Kohan N, Bass AD, Cloutier P, Massey S, Sanche L. J Chem Phys. 2012;136:235104. doi: 10.1063/1.4729781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirsaleh-Kohan N, Bass AD, Sanche L. J Chem Phys. 2011;134:015102. doi: 10.1063/1.3520562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan X, Sanche L. Phys Rev Lett. 2005;94:198104. doi: 10.1103/PhysRevLett.94.198104. [DOI] [PubMed] [Google Scholar]
- 33.Mirsaleh-Kohan N, Bass AD, Cloutier P, Sanche L. Proceedings of the XVI International Symposium on Electron Molecule Collisions and Swarms; Toronto. 2009; Red Hook, NY: Curran Associates, Inc; 2010. p. 29. [Google Scholar]
- 34.Abdoul-Carime H, Cloutier P, Sanche L. Rad Res. 2001;155:625. doi: 10.1667/0033-7587(2001)155[0625:leeesd]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 35.Abdoul-Carime H, Sanche L. Radiat Res. 2003;160:86. doi: 10.1667/rr3025. [DOI] [PubMed] [Google Scholar]
- 36.Abdoul-Carime H, Sanche L. J Phys Chem B. 2004;108:457. [Google Scholar]
- 37.Sanche L, Parenteau L. Phys Rev Lett. 1987;59:136. doi: 10.1103/PhysRevLett.59.136. [DOI] [PubMed] [Google Scholar]
- 38.Barrios R, Skurski P, Simons J. J Phys Chem B. 2002;106:7991. [Google Scholar]
- 39.Kumar A, Sevilla MD. Chemphyschem. 2009;10:1426. doi: 10.1002/cphc.200900025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manning GS. Q Rev Biophys. 1978;11:179. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 41.Wilson RW, Bloomfield VA. Biochemistry. 1979;18:2192. doi: 10.1021/bi00578a009. [DOI] [PubMed] [Google Scholar]
- 42.Denisov VP, Halle B. Proc Natl Acad Sci USA. 2000;97:629. doi: 10.1073/pnas.97.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tougaard S. J Vac Sci Technol A. 2013;31:3. [Google Scholar]
- 44.Simons J. Acc Chem Res. 2006;39:772. doi: 10.1021/ar0680769. [DOI] [PubMed] [Google Scholar]
- 45.Barrios R, Skurski P, Simons J. J Phys Chem B. 2002;106:7991. [Google Scholar]
- 46.Zheng Y, Wagner JR, Sanche L. Phys Rev Lett. 2006;96:208101. doi: 10.1103/PhysRevLett.96.208101. [DOI] [PubMed] [Google Scholar]
- 47.Kumar A, Sevilla MD. J Phys Chem B. 2007;111:5464. doi: 10.1021/jp070800x. [DOI] [PubMed] [Google Scholar]