

Abstract
We measured the low energy electron stimulated desorption (ESD) of anions from thin films of native (TXT) and bromine monosubstituted (TBrXT) oligonucleotide trimers deposited on a gold surface (T = thymidine, X = T, deoxycytidine (C), deoxyadenosine (A) or deoxyguanosine (G), Br = bromine). The desorption of H−, CH3−/NH−, O−/NH2−, OH−, CN−, and Br− was induced by 0 to 20 eV electrons. Dissociative electron attachment, below 12 eV, and dipolar dissociation, above 12 eV, are responsible for the formation of these anions. The comparison of the results obtained for the native and brominated trimers suggests that the main pathways of TBrXT degradation correspond to the release of the hydride and bromide anions. Significantly, the presence of bromine in oligonucleotide trimers blocks the electron-induced degradation of nuclobases as evidenced by a dramatic decrease in CN− desorption. An increase in the yields of OH− is also observed. The debromination yield of particular oligonucleotides diminishes in the following order: BrdU > BrdA > BrdG > BrdC. Based on these results, 5-bromo-2′-deoxyuridine appears to be the best radiosensitizer among the studied bromonucleosides.
I. INTRODUCTION
Interaction between water – the main constituent of the cellular medium – and ionizing radiation leads to the production of a large quantity of hydrogen and hydroxyl radicals as well as low energy electrons (LEEs; energies <30 eV).1 Thus, the biological effects related to the exposure of living organisms to high-energy radiation are not chiefly induced by the primary interaction on biomolecules, but rather by the secondary species mentioned above, which are generated along radiation tracks.2 Until a decade ago, it was believed that single (SSBs) and double strand breaks, one of the most geno-toxic and mutagenic DNA lesions, are triggered by the attack of ions and OH/H radicals on cellular DNA.3 Since then, theoretical4 and experimental5 studies have demonstrated unequivocally that interactions between LEEs and DNA induce frank strand breaks in the biopolymer. LEEs have an energy distribution peaking around 9–10 eV and they are produced in large quantities by radiolysis.6 Considering that their cross sections to damage DNA are of the order of 10−18 cm2/nucleotide,7 they are expected to be responsible for a large fraction of DNA damage and consequently cell death induced, for instance, by radiotherapy.
A solid tumor – the usual target of radiotherapy – consists of a heterogeneous mass of cells with areas of necrosis surrounded by cells that have very low oxygenation level.8 Such hypoxic cells are 2.5–3 times less radiosensitive than oxygenated cells.9 This therapeutically unfavorable situation related to the hypoxic environment of tumor cells, can be ameliorated with radiosensitizers, i.e., endogenous substances or xenobiotic chemicals that interact with radiation so as to enhance inactivation of tumor cells.10 The halogenated pyrimidines, 5-halouracils and 5-halocytidines, have long been recognized as radiosensitizing agents for potential clinical applications,11–13 and are still the subject of experiments with cancer cells14–16 and biomolecular models of cellular DNA.17–20 Several clinical trials have additionally reported treatment with halogenated pyrimidines. The halogenated nucleosides of uridine and cytidine were employed in the therapy of malignant brain tumors,21 malignant glioma, and anaplastic astrocytoma.22, 23 To the best of our knowledge there are no reports on the trials involving the analogues of halogenated purine bases in anticancer radiotherapies.
The fast reaction of 5-halodeoxyuridine with electrons generated in aqueous solutions has recently been studied by time-resolved femtosecond laser spectroscopy.24 The primary anions formed due to electron attachment to halouridine undergo halide anion elimination yielding the highly reactive uridine-yl radical. The latter species, if formed in DNA, may abstract a hydrogen atom from its own or adjacent deoxribose residue, promoting further damage. Indeed, it was demonstrated that abstraction of a hydrogen atom from the sugar moiety in DNA leads to single strand break.25
The gas phase studies on interactions between LEEs and 5-halouracils or 5-halodeoxyuridines,26–31 lead to conclusions similar to those reached on the basis of experiments carried out in aqueous solution, i.e., halide anions and the uracil-yl radicals were observed as the main reaction products. Unlike water solutions, the attachment of LEEs to the gas-phase haloderivatives produces also some amount of the halogen radical and the uracil-5-yl− anion. For 5-BrU and 5-ClU, computational results confirm that the former dissociation path (i.e., that producing X− and uracil-5-yl•) is energetically more favorable.26, 29
The sensitivity towards electrons of other brominated nucleosides, i.e., 5-bromo-2′-cytidine, 8-bromo-2′-deoxyadenosine, and 8-bromo-2′-deoxyguanosine, both isolated and incorporated into DNA, have been studied in the past. For instance, Sevilla et al.32 demonstrated, using low temperature EPR, that 5-BrdC incorporated in DNA is ~3 fold more efficient electron scavenger than cytidine. Similarly, the electron affinities of 8-bromopurines exceed significantly those of their non-halogenated counterparts.33–37 Reactions between hydrated electrons and 8-bromo-2′-deoxyadenosine33–35 or 8-bromo-2′-deoxyguanosine36, 37 were addressed within radiolytic studies by the group of Chatgilialoglu. They demonstrated that electron attachment to 8-bromo-2′-deoxyadenosine leads to a rapid bromine ion release followed by a fast hydrogen atom abstraction form the C5′ position of the 2-deoxyribose moiety. The latter species undergoes cyclization to afford the 5′,8-cyclopurine derivative. Moreover, the rate constant of the formation of the 8-bromo-2′deoxyadenosine anion increases by a factor of two in comparison to that of 2′-deoxyadenosine.33 It was also shown that 8-bromo-2′-deoxyguanosine captures hydrated electrons with a quantitative formation of 2′-deoxyguanosine in the presence of hydrogen donors. Therefore, this compound was employed as an effective detection system for excess-electron transfer in a variety of 8-BrdG-labeled single- or double-stranded oligonucleotides,38, 39 and G-quadruplexes.40
The observations mentioned above suggest that DNA labeled with halonucleobases becomes radiosensitive owing to the presence of the halogen, which increases the electron affinity of nucleobases. The higher electron affinity increases the probability that any thermalized excess electron originating from water radiolysis can localize on the halonucleobase. Such electron attachment may trigger the release of a halogen anion and formation of a reactive nucleobase radical ultimately leading to DNA strand breaks18, 19 as well as inter-strand41 and intra-strand42 cross-links. Taking into account these advantageous features of halonucleobases, in the present work, we study the sensitivity towards LEEs of bromonucleobases bound within a short DNA strand. Systematic measurements of the electron stimulated desorption (ESD) of anions from the thin films of oligonucleotides, whose structures are shown in Fig. 1 are reported. These oligonucleotides are composed of three thymidine (T) units (TTT) substituted in the center with 5-bromodeoxyuridine (5-BrdU), 5-bromodeoxycytidine (5-BrdC), 8-bromodeoxyadenosine (5-BrdA) or 8-bromodeoxyguanosine (5-BrdG). The desorbed yields of the anions H−, CH3−/NH−, O−/NH2−, OH−, CN−, and Br− from the brominated trimers TBrXT (X = U, C, A, G) are recorded in the 0–20 eV incident electron energy range. To better probe the effect of the Br atom on ESD from the brominated trimers, the data are compared with those from the native oligonucleotides, TXT (X = T, C, A, G, corresponding to thymidine, deoxycytidine, deoxyadenosine, and deoxyguanosine, respectively). Since the data are recorded under identical experimental conditions, the results not only provide a precise systematic comparison of the effect of halogen substitution in TTT on ESD anion yields, but also the relative efficiency of brominated bases to break DNA upon LEE impact. As a result, we find that the substitution of the bromine atom in TXT trimers, on the one hand, blocks the electron-induced degradation of the heterocyclic ring of a nucleobases and, on the other, increases the yields of the OH− desorption. Moreover, the interaction between LEEs and brominated trimers leads to the efficient release of the Br− anion, which corresponds to the second most intense signal of the measured anion yield functions. Finally, if the yield of Br− ESD at low energies reflects the radiosensitizing potential of bromonucleobases, our results suggest 5-bromo-2′-deoxyuridine to be the best radiosensitizer among the studied bromonucleosides.
FIG. 1.
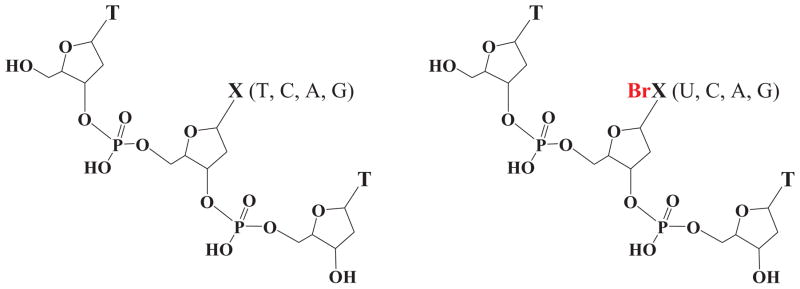
Structures of the nonbrominated and brominated trimers investigated.
II. EXPERIMENTAL
A. Preparation of samples
The ESD measurements were performed with the two types of trimers mentioned in the Introduction. The brominated trimers were purchased from Genomed (Warsaw, Poland) and the native ones from Alpha DNA (Montreal, QC). All oligonucleotides were purified by high performance liquid chromatography with an analytical YMC-Pack ODS-A column (250 × 6 mm), held at 30 °C. A linear gradient from 1% to 10% acetonitrile in a volatile buffer containing triethylamine acetate (20 mM, pH = 7) was maintained over an interval of 60 min, at the flow rate of 1.0 mL/min. Each sample was dissolved in 25 μl of sterile deionized (millipore) water. The DNA solution was prepared without adding any salt. It was deposited on the chemically clean gold substrate, frozen at liquid nitrogen temperature, lyophilized with hydrocarbon-free sorption pump at 5 mTorr and transferred into a load-lock vacuum system (~1 × 10−8 Torr). After evacuation for about 12 h, the sample was transferred to an adjacent ultrahigh vacuum chamber (~2 × 10−10 Torr) for electron bombardment. Recently, it was demonstrated that under such experimental conditions an oligonucleotide film of almost uniform distribution is formed.43
B. ESD-TOF
Anion ESD from thin films of trimers bombarded with 0–20 eV electrons was measured with an apparatus described in Refs. 44 and 45. The beam was produced by a commercial electron gun (Kimball physics ELG – 2) having a resolution of 0.5 eV full width at half maximum. The electrons collided with the sample film at an incident angle 45° with respect to the plane of the substrate and were focused into an estimated spot size of 1 mm2. The transmitted current was set at 4 nA. The electron gun was operated in a pulsed mode with 800 ns duration at the rate of 5 kHz. Anions that desorbed after the LEE pulse were measured by time-of-flight (TOF) mass analysis. The mass resolution (m/Δm, where Δm is the minimum peak separation at which two ion signals of average mass m) can be distinguished, is greater than 500.45
Owing to the short penetration depth of low-energy anions within molecular solid,46 the probing depth of the ESD signal in the present experiment is at the most two MLs for all detected anions with the exception of H−. The latter could arise from deeper probing into the oligonucleotide multilayer.46 For this reason, the thickness of our multilayers for optimum signal was chosen by measuring the H− signal as a function of film average thickness from 0.5 to 5 nm with 8.5 eV incident electrons. The maximum signal was observed at 2.5 MLs of oligonucleotides. All ESD measurements were carried out at this thickness.
III. RESULTS AND DISCUSSION
Two basic mechanisms are responsible for LEE-induced molecular fragmentation accompanied by desorption of a stable negative ion: dissociative electron attachment (DEA) and dipolar dissociation (DD).47 DEA is initiated by the resonant attachment of an electron into a usually unfilled orbital of either a molecule or a molecular subunit of a large molecule. For dissociation into a stable negative ion and a neutral radical fragment (i.e., DEA) to occur, the additional electron must be retained in that orbital for a time of the order of, or larger than a vibrational period along the dissociation coordinate. The transient anion may also stabilize or autodetach the electron. DEA is observed as a peak in the yield of fragments.48 DD, on the other hand, results from the heterolytic dissociation of an electronically excited molecule into an anion and a cation. It produces a monotonic increase in LEE-anion yield functions above a certain energy threshold.48
As indicated by our ESD results and a similar experiment on thin film of 5-bromouracil carried out several years ago in our group,49 both DEA and DD are observed in the 0–20 eV electron energy range. The yield functions of H−, CH3−/NH−, O−/NH2−, OH−, and CN− desorption from TXT and TBrXT are shown in Figs. 2, 3, 4, 5, and6, while those of Br− desorption are depicted in Fig. 7. For all desorbed anions these curves exhibit a resonance signal, characteristic of DEA, with the maximum between 6.0 and 8.5. The continuous rise of the signal above 13–14 eV is due to DD.
FIG. 2.

Incident-electron energy dependence of H− yields desorbed from the TXT (black lines) and TBrXT (blue lines) trimers deposited on a gold substrate. The structures of the trimers are shown in Fig. 1.
FIG. 3.

Incident electron energy dependence of CH3−/NH− yields desorbed from the non- and brominated trimers deposited on a gold substrate. The zero-signal base line is shifted upwards for clarity.
FIG. 4.

Incident electron energy dependence of O−/NH2− yields desorbed from the non- and brominated trimers deposited on a gold substrate. The zero-signal base line is shifted upwards for clarity.
FIG. 5.

Incident electron energy dependence of OH− yields desorbed from the non- and brominated trimers deposited on a gold substrate. The zero-signal base line is shifted upwards for clarity.
FIG. 6.

Incident electron energy dependence of CN− yields desorbed from the non- and brominated trimers deposited on a gold substrate. The zero-signal base line is shifted upwards for clarity.
FIG. 7.

Incident electron energy dependence of Br− yields desorbed from the brominated trimers deposited on a gold substrate. The zero-signal base line is shifted upwards for clarity.
All fragment anions with the exception of OH− and Br− were also observed in DEA studies of gas phase nucleobases.47, 48 The energy required to form a negative anion from a condensed molecule is generally lower than that in the gas phase due to the polarization induced by the anion on surrounding media. However, to be desorbed from the solid or its surface anions have to overcome this induced polarization potential. Moreover, post-dissociation energy transfer may prevent the anionic fragment from leaving the surface. Thus, it is not surprising that the observed maxima of desorption peaks in the yield functions are shifted significantly (e.g., by 1–2 eV) toward higher energies with respect to those observed in the gas phase.43
A. H− desorption
The H− desorption signals in Fig. 2 exhibit a broad resonance structure with a threshold near 5 eV and maximum around 9.0 eV, due to DEA. It is followed by a weak increase of the hydrogen anion signal above 13 eV corresponding to non-resonant DD. As seen from Table I, H− is the most abundant signal for all films studied. The tabulated data underestimates the intensity of H− desorption relative to other anions by about an order of magnitude due to a lower detection efficiency of this ion; only those H− ions desorbed in the final 50 ns of the electron pulse are detected.44 Nevertheless, these results are similar to those reported by Ptasińska and Sanche43 in anion ESD studies from films of the tetramer GCAT. In gas phase studies, the loss of H− was observed only for thymine50, 51 and adenine,52, 53 whereas no H− signal was detected for a sugar analog.54 On the other hand, H− desorption from thin films of a deoxyribose analog was observed,48 although much weaker than that from physisorbed thymine. Moreover, the susceptibility to release the hydride anion is significantly larger for the TTT trimer than for the other non-brominated oligonucleotides as indicated by the integrated DEA signals gathered in Table I. These facts suggest that the main source of H− in the studied TXT and TBrXT trimers is DEA to thymine. Additionally, it is also worth noticing that substitution of the middle thymine with 5-bromouridine decreases the integrated H− desorption to the level characteristic for the unsubstituted TCT, TAT, and TGT trimers. This observation might suggest that the desorbed hydride anions mainly originate from the methyl groups of thymine. Indeed, the gas phase studies on unsubstituted and methylated thymine indicate that high energy resonance is related to the H− loss from CH3.50 The small shift of the resonance maximum, from 8.5 eV in the gas phase thymine to 9.0 eV in Fig. 2 is probably due to the induced polarization potential in the solid. Other DEA experiments on small organic molecules containing the methyl group also indicate that —CH3 is probably a main source of H− in these systems.55 Obviously, our data do not exclude a contribution from the N-H bonds dissociation to the total H− signal. However, as indicated by gas phase DEA experiments on the native and N1 methylated thymine,50 the signal related to the formation of H− from the N—H bonds almost disappears due to methylation and simultaneously the yield of the CH2—H dissociation increases. Since in oligonucleotides all N1/N9 positions are blocked (i.e., are involved in N-glycosidic bond with 2′-deoxyribose) this further suggests that the H− signal measured in the current work mainly originates from methyl group dissociation.
TABLE I.
H−, CH3−/NH−, O−/NH2−, OH−, CN−, and Br− LEE-induced desorption yields (in arbitrary units) integrated over the entire energy range from thin films of the nonbrominated and brominated trimers. The average and standard deviation values result from three independent experiments.
| Anion | System | Area (a. u) | Standard deviation (%) |
|---|---|---|---|
| H− | TTT | 241 174.1 | 10.1 |
| TCT | 168 208.4 | 3.5 | |
| TAT | 176 193.0 | 0.3 | |
| TGT | 191 393.7 | 5.6 | |
| TBrUT | 178 536.8 | 13.2 | |
| TBrCT | 161 529.0 | 1.3 | |
| TBrAT | 214 892.0 | 1.7 | |
| TBrGT | 183 491.1 | 3.4 | |
| CH3−/NH− | TTT | 477.3 | 22.4 |
| TCT | 136.9 | 24.5 | |
| TAT | 140.2 | 15.5 | |
| TGT | 145.4 | 15.9 | |
| TBrUT | 217.3 | 5.5 | |
| TBrCT | 178.4 | 24.6 | |
| TBrAT | 185.2 | 7.3 | |
| TBrGT | 202.1 | 15.3 | |
| O−/NH2− | TTT | 704.4 | 3.1 |
| TCT | 959.9 | 10.8 | |
| TAT | 708.9 | 23.7 | |
| TGT | 855.6 | 1.7 | |
| TBrUT | 983.6 | 19.7 | |
| TBrCT | 814.7 | 2.9 | |
| TBrAT | 958.4 | 4.5 | |
| TBrGT | 582.0 | 13.1 | |
| OH− | TTT | 454.8 | 21.3 |
| TCT | 437.0 | 2.1 | |
| TAT | 406.2 | 21.1 | |
| TGT | 452.9 | 6.6 | |
| TBrUT | 1722.5 | 11.3 | |
| TBrCT | 989.6 | 24.9 | |
| TBrAT | 1520.0 | 1.7 | |
| TBrGT | 837.6 | 3.6 | |
| CN− | TTT | 149.2 | 6.0 |
| TCT | 797.8 | 23.2 | |
| TAT | 175.5 | 2.3 | |
| TGT | 209.1 | 17.4 | |
| TBrUT | 0.0 | · · · | |
| TBrCT | 0.0 | · · · | |
| TBrAT | 0.0 | · · · | |
| TBrGT | 0.0 | · · · | |
| Br− (79) | TBrUT | 9445.8 | 8.3 |
| TBrCT | 1729.4 | 7.5 | |
| TBrAT | 4446.0 | 16.8 | |
| TBrGT | 3298.3 | 14.0 |
B. CH3−/NH− desorption
The yield function of the negative ion with the mass of 15 amu is shown in Fig. 3. At priory, it can be attributed to the formation of CH3− or NH−. The resonant DEA peak has a maximum around 8.5 eV and an onset near 6 eV. Probably due to the weakness of the signal, DD above 14 eV is visible only for the TTT trimer. The small yields in the formation of the CH3−/NH− anion may explain why no desorption of these ions was observed in the DEA signal from the GCAT tetramer.43 Table I shows that the CH3−/NH− yields are three orders of magnitude smaller than the measured yields for H− desorption and similar to each other, except for that of the TTT trimer. In the latter case, the integrated desorption is 2–3 fold larger in comparison to other oligonucleotides. Since only the TTT trimer contains three metyl groups this DEA process should be attributed to the formation of CH3− rather than that of NH−. Moreover, releasing of NH− from TTT would require damage to the relatively stable pyrimidine ring via breakage of two chemical bonds, while the formation of CH3− implies only detachment (splitting of a single chemical bond) of the methyl substituent. Nevertheless, the 50% increase of CH3 content in TTT cannot explain the much larger desorption signal of CH3− (i.e., up to threefold) from this trimer.
This large enhancement may be related to differences in conformations acquired by the oligonucleotides in the bombarded films, which may affect the initial coherent enhancement of the captured electron wave function, and hence also the electron capture probability.56 Furthermore, coupling of the initially diffracted wave within the trimer, with local transitory electron states is expected to depend on configuration and base substitution.57 Since temporary electron localization on a specific unfilled orbital usually favors a particular dissociation pathway (i.e., desorption of a given stable anion via DEA), we expect the anion yields also to depend on the type of base content in the oligonucleotide and its morphology. Thus, the quantum mechanical aspects of the electron capture process in oligonucleotides, which has been treated theoretically by Caron and co-workers,56–60 may partly explain, not only the larger CH3− yields in TTT, but also differences in the yields of other anions upon base substitution.
C. O−/NH2− desorption
The 16-amu negative ions, whose yield functions are shown for all oligomer in Fig. 4, correspond to either O− or NH2−, since without isotopically labeled compounds or/and high resolution mass assignment, the portion of the signal arising from each anion cannot be assigned unambiguously. There are, however, reasons to believe from consideration of previous gas and condensed phase data that the major portion of the 16-amu signal arises from O−. For example, no anionic fragment was observed at 16 amu in gas-phase DEA experiments on adenine, which contains many nitrogen groups but no oxygen atoms.53 Moreover, the comparison between the shape of desorption of O− from films of NaH2PO4 and that of the 16-amu fragment from GCAT films strongly suggests that in DNA oxygens form the phosphate groups rather than the NH residues are responsible for this ESD signal.43 This conclusion is also in line with the previous investigations on DNA films, which show that O− production arises from temporary electron localization on the phosphate group.61
Thus, the resonance observed in Fig. 4 with a maximum and onset around 8.5 eV and 5.0 eV, respectively, is tentatively ascribed to O− desorption from all trimers. Above ~14 eV, nonresonant DD dominates these O− yields. Both the line shapes of the signals and their integrated magnitudes are similar for the native and brominated oligomers. In fact, variation in the magnitude of the O− signal is less than 30% of that of the most intense yields, as shown in Table I. Although three systems, TTT, TAT, and TBrGT, are characterized by significantly lower yields (see Table I), two of them are non-brominated trimers and one is a brominated oligonucleotide; this suggests no correlation with base content. Moreover, signal standard deviations for two of the three trimers are pretty high (13% (TBrGT) and 24% (TAT)). Thus, one can conclude that the 16-amu yields are indeed similar for all studied systems. This similitude further confirms that these yields originate from the phosphate oxygens rather than oxygens belonging to bases. Indeed, it is likely that, if the O− desorption derived from the bases, one should observe a clear difference between the O− yields from the TBrXT and TXT trimers, since adenine contains no oxygen and the quantum mechanical mechanisms mentioned in Sec. III B should be operative. The electron capture probability and the following DEA process are expected to be much less dependent on morphology and the nature of the middle base, if O− desorption arises from transient anion formation on the backbone, which suffers minimal modifications upon base substitution.
D. OH− desorption
The yield functions of OH− ESD from all trimers are shown in Fig. 5. A clear resonance with a maximum around 6 eV and threshold at 2.5 eV is observed only for the TBrXT oligomers. Only a weak and broad “hump” around 8 eV is observed in the yield functions of the TXT trimers. Above ~ 13 eV the ESD curves are dominated by nonresonant DD. The OH− yield from the brominated oligomers is 2–4 times larger than that from their native counterparts (see Table I). The OH− ions could arise from the phosphate group in the backbone of DNA, when the counter-ion is a proton. These ions could also arise from the OH groups of terminal sugars or the reactive scattering of O−. If the latter possibility would be the case, the shape and position of the OH− yield function should resemble that registered for the O− desorption; but there is no such correlation as indicated by comparison of Figs. 4 and 5. Moreover, if OH− came principally from the reactive scattering of O−, the intensity of the OH− desorption signal would be much smaller than that of the O−. As seen from Table I, this is not the case either, as in previously recorded data for OH− and O− desorption from the NaH2PO4 and GCAT films. These results imply that O− reactive scattering is not significant in DNA films.43 We must point out, however, that the large difference between the OH− desorption yields observed from TXT and TBrXT samples could always be partially explained by reactive scattering of Br− at the terminal OH groups of TBrXT. Such SN2 type reactions between Br− and alcohols are well known in basic organic chemistry. In this case, the line shape of both the OH− and Br− signals seen from a comparison of Figs. 5 and 7 complies with the requirements of reactive scattering. The differences in the relative magnitude of the OH− and Br− signals are not sufficiently large, however, to exclude contributions from other mechanisms.
In previous work, it has been shown that in anion ESD from films of long single DNA strand (≥20 bases), OH− arises principally from DEA to the phosphate group, whereas in films of GCAT, which present a much higher density of terminal OH of a deoxyribose sugar, ESD of OH− from the latter moiety is prominent. We therefore suggest that in ESD from films of TBrXT, which are even shorter than GCAT, a sizeable contribution to the OH− signal arises from DEA to the terminal sugar. In this case, the much larger DEA OH− signal from the brominated trimers could be related to the high electron affinity of bromine, which creates a deep potential well for incoming electrons. The added attractive potential in TBrXT trimers could increase the initial electron capture cross section and hence increase at least certain subsequent DEA reactions, including those occurring at the terminal sugar. If such a potential is involved in OH− dissociation from a core-excited anion state, the DEA maximum is expected to lie at a lower energy for the brominated trimer, as observed experimentally.
E. CN− desorption
Interestingly, resonance desorption with a maximum around 7 eV and threshold at ~4 eV is observed only for the non-brominated trimers (see Fig. 6 and Table I). The source of the cyanide anions is nucleobases and their formation must be accompanied by a substantial rearrangement of their heterocyclic rings. This pathway of damage seems to be completely blocked in the brominated trimers. It indicates that the presence of the bromine atom protects nucleobases from the electron-induced degradation of their rings (i.e., release of CN−).
This lack of CN− signal suggests involvement of an electron transfer process. Indeed, if an electron is captured by a flanking thymine in TBrXT, it could be strongly attracted by the deep potential well created by the bromine atom and transferred to the middle BrX where the C–Br bond dissociation is then triggered. In fact, very small kinetic energy barriers have been calculated for the release of Br− form the brominated nucleobases.26 All brominated bases, as opposed to the non-brominated ones, possess positive adiabatic electron affinities (AEAs) which create a favourable thermodynamic stimulus for electron transfer. Actually, the AEAs for 5-BrMeU, 5-BrMeC, 8-BrMeA, and 8-BrMeG (5/8-BrMeX stands for methylated at the 1 or 9 position bromonucleobase), calculated in the gas phase at the B3LYP/6-31++G(d,p) level, amounts to 15.9, 10.4, 16.4, and 2.5 kcal/mol, respectively.62 Thus, any electron localized on the brominated base is expected to swiftly dissociate releasing Br−, since this is the most favourable path. In other words, the lifetime of transient anions formed on any bases of the TBrXT molecule see their lifetime shorten in comparison to that of the bases of the native oligonucleotides. We can therefore conjecture that this reduced lifetime is insufficient to allow for rearrangement of the heterocyclic ring, which leads to CN− formation. Modification of interbase electron transfer could therefore explain the lack of CN− ESD signal from TBrXT films. In fact, in anion ESD from BrU films, where no such transfer is possible, the CN− signal is still observed.49 Many parameters could cause the difference in signal between CN− ESD from BrU and the present brominated trimers. First, the energy of the anion state is expected to be different in both cases. Since the lifetime is sensitive to the energy of the initial transient anion and bonding to thymine, the net result may be a larger reduction in the lifetime for the brominated trimers. Similar arguments may also be valid to explain the higher CN− signal from TCT, compared to that of the other TXT trimers, as shown in Table I.
F. Br− desorption
The yield function of the Br− ESD signal from all trimers is shown in Fig. 7. As seen from Table I, the Br− yield is the most abundant of all observed anions, after H−. Because dissociating H− has a much larger velocity than the heavy halogen, such a large Br− signal seems surprising. Most of the Br− anions are expected to stay on the surface owing to induced polarization forces. Thus, the significant Br− desorption suggests that the cross section for the formation of the bromide anion must be very high in accordance with AEAs mentioned in Sec. III E.
The yield functions in Fig. 7 have maxima around 6–6.5 eV and onsets at ~2.5 eV. The weak increase in the Br− signal above 13 eV provides evidence for the DD process. The integrated desorption yields, gathered in Table I, demonstrate that besides the release of H−, DEA leading to the C–Br bond dissociation is the main electron-induced degradation pathway in TBrXT trimers. As an outcome of this DEA process, reactive base-centered radicals are formed63 that may trigger secondary processes involving a hydrogen atom abstraction from the sugar moiety, which eventually leads to the formation of SSBs.25 Since the electron-induced dissociation of the C–Br bond is suspected to be responsible for the increased radiosensitivity of brominated DNA,18, 19, 41 one can compare the reactivity of particular trimers in terms of the yield of Br− release. Thus, the following series of decreasing sensitivity toward LEEs can be postulated on the basis of data shown in Table I: TBrUT > TBrAT > TBrGT > TBrCT. Although the yield of Br− formation is significantly larger for the TBrUT trimer than for the other brominated oligonucleotides (see Table I), LEE-induced loss of Br− is still substantial for the remaining systems, especially for the trimers containing brominated purines.
IV. SUMMARY
Electron stimulated desorption induced by electrons of 0–20 eV from native (TXT, X = base) and brominated (TBrXT) oligonucleotide films deposited on a gold substrate has been studied. Bombardment of the oligonucleotides with LEEs produces desorption of the anionic fragments, H−, Br−, O−, OH−, CH3−, and CN−, which proceeds via two distinctive mechanisms: dissociative electron attachment and nonresonant dipolar dissociation. The maxima of resonant electron attachment occur between 5 and 9 eV, while dipolar dissociation begins around 13–14 eV.
Based on the literature data and on the fact that bromination of nucleobases does not affect the ESD signal of 16 amu, we conclude that it originates from the O− anions, which are formed due to direct interaction between the incident electron and phosphates. Dissimilarity between the O− and OH− DEA signals suggests that the latter are not formed by reactive scattering of O−. However, the much larger OH− signals observed from TBrXT oligomers with respect to those found in TXT and a similarity between the Br− and OH− signals indicates that reactive scattering of Br− could be partially responsible for the OH− desorption; an increase of the electron capture probability owing to the presence of Br could also contribute to the difference in OH− ESD. The increase in DEA signal from TTT observed for the anion of 15 amu suggests that it could be ascribed to CH3−. Finally, the presence of the bromine atom protects bases from the electron-induced degradation of their rings as indicated by the complete disappearance of the CN− signal in brominated oligonucleotides.
The yields of Br− formation induced by electron impact on thin films of brominated trimers indicate that the sensitivity of particular oligonucleotides to LEEs diminishes in the following order: BrdU > BrdA > BrdG > BrdC. Consequently, our findings confirm in a simple model that substitution of a nucleobase for any of the four brominated nucleobase should increase LEE-induced damage to DNA and hence its radiosensitivity. Thus, besides brominated pyrimidines that were already tested in clinical trials, brominated purines also appear as promising radiosensitizers.
Acknowledgments
This work was supported by the Polish Ministry of Science and Higher Education (MNiSW), under the Grant Nos. N N204 023135 (J.R) and the Canadian Institutes of Health Research (L.S.).
References
- 1.Buxton GV, Greenstock CL, Helman WP, Ross AB. J Phys Chem Ref Data. 1988;17:513. and references therein; Ross AB, Mallard WG, Helman WP, Buxton GV, Huie RE, Neta P. NDRL-NIST Solution Kinetic Database, version 3. Notre Dame Radiation Laboratory; Notre Dame, IN: NIST Standard Reference Data; Gaithersburg, MD: 1998.
- 2.Von Sonntag C. The Chemical Basis for Radiation Biology. Taylor & Francis; London: 1987. [Google Scholar]
- 3.O’Neil P. Radiation Chemistry. Elsevier; Dordrecht, The Netherlands: 2001. Radiation-induced damage in DNA; pp. 585–622. [Google Scholar]
- 4.Kumar A, Sevilla M. Theoretical modeling of radiation induced DNA damage. In: Greenberg M, editor. Radicals and Radical Reactivity in Nucleic Acid Chemistry. Chap 1 Wiley; New York: 2009. [Google Scholar]
- 5.Sanche L. Reactive Intermediates in Chemistry and Biology. In: Greenberg M, editor. Radicals in Nucleic Acids. Wiley; Hoboken, NJ: 2009. [Google Scholar]
- 6.LaVerne JA, Pimblott SM. Radiat Res. 1995;141:208. [PubMed] [Google Scholar]
- 7.Panajotovic R, Martin F, Cloutier P, Hunting DJ, Sanche L. Radiat Res. 2006;165:452. doi: 10.1667/rr3521.1. [DOI] [PubMed] [Google Scholar]
- 8.Oronsky BT, Knox SJ, Scicinski J. Trans Oncol. 2011;4:189. doi: 10.1593/tlo.11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daşu A, Denekamp J. Radiother Oncol. 1998;46:269. doi: 10.1016/s0167-8140(97)00185-0. [DOI] [PubMed] [Google Scholar]
- 10.Wardman P. Clin Oncol. 2007;19:397. doi: 10.1016/j.clon.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Djordjevic B, Szybalski W. J Exp Med. 1960;112:509. doi: 10.1084/jem.112.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erickson RL, Szybalski W. Radiat Res. 1963;20:252. [PubMed] [Google Scholar]
- 13.Cramer JW, Prusoff WH, Welch AD, Sartorelli AC, Delmore IW, Von Essen CF, Chang PK. Biochem Pharmacol. 1962;11:761. doi: 10.1016/0006-2952(62)90044-8. [DOI] [PubMed] [Google Scholar]
- 14.Brust D, Feden J, Farnsworth J, Amir C, Broaddus WC, Valerie K. Cancer Gene Ther. 2000;7:778. doi: 10.1038/sj.cgt.7700168. [DOI] [PubMed] [Google Scholar]
- 15.Dabaja BS, McLaughlin P, Ha CS, Pro B, Meyers CA, Seabrooke LF, Wilder RB, Kyritsis AP, Preti HA, Yung WK, Levin V, Cabanillas F, Cox JD. Cancer. 2003;98:1021. doi: 10.1002/cncr.11627. [DOI] [PubMed] [Google Scholar]
- 16.Prados MD, Seiferheld W, Sandler HM, Buckner JC, Phillips T, Schultz C, Urtasun R, Davis R, Gutin P, Cascino TL, Greenberg HS, Curran WJ., Jr Int J Radiat Oncol, Biol, Phys. 2004;58:1147. doi: 10.1016/j.ijrobp.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 17.Cecchini S, Girouard S, Huels MA, Sanche L, Hunting DJ. Radiat Res. 2004;162:604. doi: 10.1667/rr3267. [DOI] [PubMed] [Google Scholar]
- 18.Cecchini S, Girouard S, Huels MA, Sanche L, Hunting DJ. Biochemistry. 2005;44:1932. doi: 10.1021/bi048105s. [DOI] [PubMed] [Google Scholar]
- 19.Dextraze ME, Wagner JR, Hunting DJ. Biochemistry. 2007;46:9089. doi: 10.1021/bi062114e. [DOI] [PubMed] [Google Scholar]
- 20.Dugal PC, Abdoul-Carime H, Sanche L. J Phys Chem B. 2000;104:5610. [Google Scholar]
- 21.Matsutani M, Kohno T, Nagashima T, Nagayama I, Matsuda T, Hoshino T, Sano K. Radiat Med. 1988;6:33. [PubMed] [Google Scholar]
- 22.Prados MD, Scott CB, Rotman M, Rubin P, Murray K, Sause W, Asbell S, Cpmis R, Curran W, Nelson J, Davis RL, Levin VA, Lamborn K, Phillips TL. Int J Radiat Oncol, Biol, Phys. 1998;40:653. doi: 10.1016/s0360-3016(97)00770-0. [DOI] [PubMed] [Google Scholar]
- 23.Urtasun RC, Cosmatos D, DelRowe J, Kinsella TJ, Lester S, Wasserman T, Fulton DS. Int J Radiat Oncol, Biol, Phys. 1993;27:207. doi: 10.1016/0360-3016(93)90229-o. [DOI] [PubMed] [Google Scholar]
- 24.Wang CR, Hu A, Lu QB. J Chem Phys. 2006;124:241102. doi: 10.1063/1.2217014. [DOI] [PubMed] [Google Scholar]; Wang CR, Lu QB. Angew Chem, Int Ed. 2007;46:6316. doi: 10.1002/anie.200701559. [DOI] [PubMed] [Google Scholar]; J Am Chem Soc. 2010;132:14710. doi: 10.1021/ja102883a. [DOI] [PubMed] [Google Scholar]
- 25.Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Sanche L, Sevilla MD. J Phys Chem A. 2002;106:11248. doi: 10.1021/jp021669q. [DOI] [PubMed] [Google Scholar]
- 27.Abdoul-Carime H, Huels MA, Illenberger E, Sanche L. J Am Chem Soc. 2001;123:5354. doi: 10.1021/ja003952d. [DOI] [PubMed] [Google Scholar]
- 28.Abdoul-Carime H, Huels MA, Illenberger E, Sanche L. Int J Mass Spectrom. 2003;228:703. [Google Scholar]
- 29.Li X, Sevilla MD, Sanche L. J Am Chem Soc. 2003;125:8916. doi: 10.1021/ja034286u. [DOI] [PubMed] [Google Scholar]
- 30.Radisic D, Ko YJ, Nilles JM, Stokes ST, Sevilla MD, Rak J, Bowen KH. J Chem Phys. 2011;134:015101. doi: 10.1063/1.3525623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdoul-Carime H, Limão-Vieira P, Gohlke S, Petrushko I, Mason NJ, Illenberger E. Chem Phys Lett. 2004;393:442. [Google Scholar]
- 32.Razskazovskii Y, Swarts SG, Falcone JM, Taylor C, Sevilla MD. J Phys Chem B. 1997;101:1460. [Google Scholar]
- 33.Flyunt R, Bazzanini R, Chatgilialoglu C, Mulazzani QG. J Am Chem Soc. 2000;122:4225. [Google Scholar]
- 34.Chatgilialoglu C, Guerra M, Mulazzani QG. J Am Chem Soc. 2003;125:3839. doi: 10.1021/ja029374d. [DOI] [PubMed] [Google Scholar]
- 35.Boussicault F, Kaloudis P, Caminal C, Mulazzani QG, Chatgilialoglu C. J Am Chem Soc. 2008;130:8377. doi: 10.1021/ja800763j. [DOI] [PubMed] [Google Scholar]
- 36.Chatgilialoglu C, Caminal C, Guerra M, Mulazzani QG. Angew Chem, Int Ed. 2005;44:6030. doi: 10.1002/anie.200501087. [DOI] [PubMed] [Google Scholar]
- 37.Chatgilialoglu C, Caminal C, Altieri A, Vougioukalakis GC, Mulazzani QG, Gimisis T, Guerra M. J Am Chem Soc. 2006;128:13796. doi: 10.1021/ja062636h. [DOI] [PubMed] [Google Scholar]
- 38.Kimura T, Kawai K, Tojo S, Majima T. J Org Chem. 2004;69:1169. doi: 10.1021/jo035194q. [DOI] [PubMed] [Google Scholar]
- 39.Manetto A, Breeger S, Chatgilialoglu C, Carell T. Angew Chem, Int Ed. 2006;45:318. doi: 10.1002/anie.200502551. [DOI] [PubMed] [Google Scholar]
- 40.De Champdore M, De Napoli L, Montesarchio D, Piccialli G, Caminal C, Mulazzani QG, Navacchia ML, Chatgilialoglu C. Chem Commun. 2004;10:1756. doi: 10.1039/b404473h. [DOI] [PubMed] [Google Scholar]
- 41.Dextraze ME, Cecchini S, Bergeron F, Girouard S, Turcotte K, Wagner JR, Hunting DJ. Biochemistry. 2009;48:2005. doi: 10.1021/bi801684t. [DOI] [PubMed] [Google Scholar]
- 42.Lin G, Zhang J, Zeng Y, Luo H, Wang Y. Biochemistry. 2010;49:2346. doi: 10.1021/bi901861w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ptasińska S, Sanche L. J Chem Phys. 2006;125:144713. doi: 10.1063/1.2338320. [DOI] [PubMed] [Google Scholar]
- 44.Hedhili MN, Cloutier P, Bass AD, Madey TE, Sanche L. J Chem Phys. 2006;125:094704. doi: 10.1063/1.2338030. [DOI] [PubMed] [Google Scholar]
- 45.Mirsaleh-Kohan N, Bass AD, Sanche L. J Chem Phys. 2011;134:015102. doi: 10.1063/1.3520562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akbulut M, Sack NJ, Madey TE. Surf Sci Rep. 1997;28:177. [Google Scholar]
- 47.Sanche L. Mass Spectrom Rev. 2002;21:349. doi: 10.1002/mas.10034. [DOI] [PubMed] [Google Scholar]
- 48.Sanche L. Eur Phys J D. 2005;35:367. [Google Scholar]
- 49.Herve du Penhoat M-A, Huels MA, Cloutier P, Jay-Gerin J-P, Sanche L. J Phys Chem B. 2004;108:17251. [Google Scholar]
- 50.Ptasińska S, Denifl S, Grill V, Märk TD, Illenberger E, Scheier P. Phys Rev Lett. 2005;95:093201. doi: 10.1103/PhysRevLett.95.093201. [DOI] [PubMed] [Google Scholar]
- 51.Ptasińska S, Denifl S, Mróz B, Probst M, Grill V, Ilenberger E, Scheier P, Märk TD. J Chem Phys. 2005;123:124302. doi: 10.1063/1.2035592. [DOI] [PubMed] [Google Scholar]
- 52.Denifl S, Zappa F, Mähr I, Lecointre J, Probst M, Märk TD, Scheier P. Phys Rev Lett. 2006;97:043201. doi: 10.1103/PhysRevLett.97.043201. [DOI] [PubMed] [Google Scholar]
- 53.Hubert D, Beikircher M, Denifl S, Zappa F, Matejcik S, Bacher A, Grill V, Märk TD, Scheier P. J Chem Phys. 2006;125:084304. doi: 10.1063/1.2336775. [DOI] [PubMed] [Google Scholar]
- 54.Sulzer P, Ptasińska S, Zappa F, Mielewska B, Milosavljevic AR, Scheier P, Märk TD, Bald I, Gohlke S, Huels MA, Illenberger E. J Chem Phys. 2006;125:044304. doi: 10.1063/1.2222370. [DOI] [PubMed] [Google Scholar]
- 55.Prabhudesai VS, Kelkar AH, Nandi D, Krishnakumar E. Phys Rev Lett. 2005;95:143202. doi: 10.1103/PhysRevLett.95.143202. [DOI] [PubMed] [Google Scholar]
- 56.Caron L, Sanche L. Phys Rev A. 2005;72:032726. [Google Scholar]
- 57.Caron LG, Tonzani S, Greene CH, Sanche L. Phys Rev A. 2008;78:042710. [Google Scholar]
- 58.Caron LG, Sanche L. Phys Rev A. 2004;70:032719. [Google Scholar]
- 59.Caron LG, Sanche L. Phys Rev A. 2006;73:062707. [Google Scholar]
- 60.Caron L, Sanche L, Tonzani S, Greene CH. Phys Rev A. 2009;80:012705. [Google Scholar]
- 61.Pan X, Cloutier P, Hunting D, Sanche L. Phys Rev Lett. 2003;90:208102. doi: 10.1103/PhysRevLett.90.208102. [DOI] [PubMed] [Google Scholar]
- 62.Chomicz L, Rak J, Storoniak P. Electron-Induced Elimination of the Bromide Anion from Brominated Nucleobases. A Computational Study. J Phys Chem B. doi: 10.1021/jp3008738. (submitted) [DOI] [PubMed] [Google Scholar]
- 63.Klyachko DV, Huels MA, Sanche L. Radiat Res. 1999;151:177. [PubMed] [Google Scholar]