

Abstract
The photocycle of the retinal protein from Exiguobacterium sibiricum, which differs from bacteriorhodopsin in both its primary donor and acceptor, is characterized by visible and infrared spectroscopy. At pH above pKa ~6.5 we find a bacteriorhodopsin-like photocycle, which originates from excitation of the all-trans retinal chromophore with K-, L-, M- and N-like intermediates. At pH below pKa ~6.5 the M-state, which reflects Schiff base deprotonation during proton pumping, is not accumulated. However, using the infrared band at ~1760 cm−1 as a marker for transient protonation of the primary acceptor, we find that Schiff base deprotonation must have occurred at pH not only above but also below the pKa ~6.5. Thus, the M state is formed but not accumulated for kinetic reasons. Further, chromophore reisomerization from the 13-cis to the all-trans conformation occurs very late in the photocycle. The strongly red-shifted states that dominate the second half of the cycle are produced before the reisomerization step, and, by this criterion, they are not O-like but rather N-like states. The assignment of photocycle intermediates enables reevaluation of the photocycle; its specific features are discussed in relation to the general mechanism of proton transport in retinal proteins.
Keywords: retinal protein, FTIR, proton transport, intermediates, pH-dependence, pKa
Introduction
The bacterial retinal proteins discovered some forty years ago1,2 constitute a functional group that spans all biological kingdoms3,4. They include light-induced proton5,6 and chloride7 pumps, as well as light sensors3. Absorption of light by the chromophore triggers a cyclic sequence of molecular events, which includes isomerization of the retinal from all-trans to 13-cis, movement of ions, protein conformational changes and thermal reisomerization (for reviews of the ion pumps see5,6). All of these events are accompanied by changes in the absorption of the retinal chromophore, both in the visible and in the IR, and the sequence of transient intermediates formed after photoisomerization is conventionally described with the K-L-M-N-O notation, initially introduced for bacteriorhodopsin (BR)8. In the ion pumps the photocycle leads to the build-up of a transmembrane electrical and chemical potential, which can be utilized for energy-requiring reactions such as ADP phosphorylation2,9.
Absorption of a light quantum by the retinal contributes approx. 200 kJ/mol of energy to the chromophore, a portion of which is retained in the retinal in the early K-like state(s)10,11, i.e. at a stage where the initial photochemistry is finished and the biologically significant steps of the photocycle have not begun. In the light-induced proton pumps this energy is sufficient to destabilize the proton of the protonated retinal Schiff base linkage6. After Schiff base deprotonation that produces the M state, further proton translocation across the protein is achieved by formation and breaking of transient proton conducting chain(s), and the movement of proton(s) during the sequential states of the photocycle (for reviews see6,12). In these proton pumps two proton-conducting pathways (half-channels) are formed transiently, which alternatively connect the Schiff base to the extracellular and cytoplasmic surfaces of the membrane.
To achieve this, proton-conducting groups in the half-channels should be able to serve in turn as acceptors or as donors, transiently reversing their function in the course of the photocycle (see12 for review). One of the main lessons learned from the retinal proteins is that although in principle many amino acids side chains might serve as acceptors/donors for protons – tyrosines, histidines, arginines, or even lysines, only those with carboxylic acid side chains, i.e., the aspartic and glutamic acids, had been confirmed to perform this function12–19. In the retinal proteins their effective pKa’s range from as low as 2.620–23 to over 1224–26.
While the central part of the pumping mechanism of a retinal protein is the ability of the initially protonated Schiff base to deprotonate after photoisomerization of the retinal, the pumping mechanism is optimized by two functionally important groups: (i) the primary proton acceptor, Asp-85 in BR13,15,16 and its homologues in other retinal protein proton pumps17–19,27, and (ii) the primary proton donor, Asp-96 in BR14,28,29, and its homologues17–19,27. A third residue, of Asp-212 in BR, which is super-conserved in all retinal proteins, seems to be essential for proton pumping as well4,30.
The deprotonation of the Schiff base (and thus the protonation of the initially deprotonated primary acceptor) is the first functionally important proton transfer, found so far to be obligatory in the photocycle of any retinal based proton pump. It leads to formation of the M state8,15,16,31. Reprotonation of the Schiff base by the primary proton donor14,28,29 later in the photocycle leads to decay of M and formation of N8,31–33. Reprotonation of the primary donor appears to be a prerequisite for reisomerization of the chromophore28, and the resulting O intermediate is the only transient state in the BR photocycle with an all-trans retinal34.
The presence of an initially unprotonated primary acceptor was found to be necessary for deprotonation of the Schiff base and for proton pumping. Its inactivation either by mutation13,35 or by protonation at low pH abolishes transport in both BR36–38 and proteorhodopsin (PR)39. Unlike the primary acceptor, the primary donor does not seem to be obligatory, and the corresponding mutants (e.g., D96N, D96G and D96A in BR28,29,40,41) are able to pump although with decreased turn-over rates28. Furthermore, water molecules buried deep inside protein were shown to play important roles42–45, facilitating and optimizing the functional dynamics.
The non-archaeal retinal proteins studied, e.g., PR46, Gloeobacterrhodopsin (GR)18, fungal rhodopsin from Leptosphaeria maculans (LR)19 xanthorhodopsin (XR)27,47, and ESR48, differ from BR in their proton acceptor and/or donor, raising questions whether the best-studied BR is a good paradigm for retinal-based proton pumps. They all have a histidine residue in the immediate vicinity of their primary acceptor 49,50. In the His-62/Asp-96 complex of xanthorhodopsin His-62 forms a strong hydrogen bond with the carboxylate group of Asp-9647, and this gave grounds to hypotheses for a role of histidine in proton pumping. However, in PR further attempts to explore the specific role of histidine in the His-Asp pair produced conflicting results both in favor49 and against50 the active involvement of histidine in the proton transporting mechanism.
The recent discovery of ESR, a retinal protein from the eubacterium Exiguobacterium sibiricum51, demonstrates in further detail how the non-archaeal rhodopsins may extend the BR paradigm. In ESR, similarly to PR49 and XR47, there is an Asp-85/His-57 pair at the Schiff base, which forms the primary proton acceptor48, instead of the corresponding Asp-85/water pair in BR52. Additionally, a lysine, Lys-96, occupies the place of the primary donor instead of the carboxylic acid previously found in all other known retinal protein proton pumps4. Recently available gene sequences indicate53 that ESR is not the only retinal protein with the lysine in the Asp-96 position.
As with other retinal proteins, the absorption band of ESR in the visible is pH-sensitive48, revealing a pKa of ~6.5 for the Schiff base counterion (in liposomes). Unlike in BR, this pKa is not the pKa for full deprotonation of Asp-85 but rather the higher of the two pKa’s of the complex counterion in ESR, the Asp-85/His-57 pair. In the initial state most of Asp-85 is deprotonated above the lower pKa (~2.3); the higher one accounts for deprotonation of only the last ~10%48. On the other hand, accumulation of M in the photocycle is associated with the higher pKa, a finding that was suggested to relate to the protonation state of His-57 rather than Asp-85, which receives the Schiff base proton48. This pKa, which we will further refer to as the “the apparent pKa of the accumulation of M”, defines two pH regions, in which the photocycle differs significantly48. In the pH region above this pKa, both M-like and post-M states are readily observed48,51, resembling the phenotypes observed earlier in PR17 and XR27,54. However, unlike in PR, where proton transport is fully correlated with M accumulation in the photocycle39, in ESR below this pKa no transient M accumulation occurs, indicating a strongly perturbed photocycle, which nevertheless produces proton transport48.
This paper reports on studies of the photocycles both above and below this pKa, in the visible and in the IR. Specifically, we are interested in how far the similarity between the photocycles of ESR and BR/PR goes, and how to correlate the observed transient spectral changes in ESR with molecular processes in protein. Among the questions is whether M-like state is not produced at all, or produced but not accumulated, below this apparent pKa. Earlier, we had proposed48 the latter rather than the former. Using IR spectroscopy, we show that this is indeed the case. We also find that the red-shifted state(s) observed at the end of the photocycle are not an O-like (i.e., with all-trans retinal) but rather N-like (i.e., with 13-cis retinal). We will argue that, despite the differences in the amino acid sequences and the apparent photocycle kinetics, all major mechanisms that define the proton-translocating phenotype of BR are present in ESR as well. Further, this conclusion is not restricted to the pH region above the apparent pKa of M accumulation but below it as well.
Experimental Methods
ESR, Exiguobacterium sibiricum rhodopsin,51, was expressed, harvested, and purified as described earlier51. Wild-type and K96A mutant were used.
To restore a membrane-like environment, ESR was incorporated in liposomes (L-α-phosphatidylcholine, Avanti #840051C) at 1:100 protein-to-lipid molar ratio. For this the DDM-solubilized (DDM, n-dodecyl-ß-D-maltoside) protein collected from a Ni-NTA Agarose column (Qiagen, Germany) was subjected to dialysis (12–14k MWCO tubing from Spectrum Laboratories, Inc. California) to remove imidazole, salts and buffers. DDM (n-dodecyl-ß-D-maltoside) concentration is not affected by this procedure, which was repeated twice for 2 hours at 4°C against the x200 volume of a 10 mM buffer at pH 8.0. The protein at 1 mg/ml concentration in 0.2% DDM was mixed with DDM-solubilized lipid (3 mg/ml in 0.5% DDM), and liposome formation was initiated by drop-by-drop dilution with a 3.5 mM buffer containing 2 mM EDTA at pH 8.0 to reach a DDM concentration three times below its critical micelle concentration level of 0.0087%. The detergent was then washed out by two centrifugations of the liposomes at 250,000g for 2 hours at 4°C. The first spin was in the presence of 0.1 M NaCl; the second without. The desired pH was obtained by further washing with a 3.5 mM buffer and sedimenting the liposomes at 19,000g for 60 minutes.
The photocycle was monitored both in the visible, and in the IR. In the visible, the photochromic changes following laser excitation (7 ns, 532 nm, ~2 mJ/cm2) were recorded at selected wavelengths (indicative of specific intermediates) on a single-wavelength time-resolved spectrometer, as described previously17,56. A monochromatic beam of 6.5 nm Gaussian spectral width was used for monitoring, and was obtained with two monochromators (before and after the sample) from a power-stabilized (Radiometric power supply Model 68831 with photo-feedback, Model 68850; both by Oriel Instruments, Stratford, CT) 250 W halogen lamp. The intensity of the measuring beam on the sample was ≤0.3 mW/cm2, which corresponds to a time constant of ~100 sec for potential photo-induced processes in ESR, in the 530–570 nm region of measurements where the probability of unwanted excitation by the measuring beam is the highest. To keep the measuring beam from producing measurable photoexcitation, x10 or x100 neutral filters were used to further reduce its intensity when measuring the photocycles with components slower than 10 sec. Still slower kinetics was recorded on a spectrophotometer (UV-1601, Shimadzu, Japan), using a millisecond photoflash from a camera (PowerShot A85, Canon, Japan) for excitation.
To obtain time-resolved spectra of intermediates, single-wavelength kinetic traces were collected separately at 18 wavelengths covering the 380–720 nm region with steps of 20 nm, and subjected to analysis using the FitExp and Schemefit programs56. At first global (multi-exponential) fitting was performed with FitExp program, and the minimal sufficient number of exponential components was evaluated by F-test statistics and analysis of residuals, see56 for details. This number is equal to the number of distinct intermediates detected experimentally56. The corresponding exponentials are the apparent processes, and the difference spectra of the mixtures formed between them were calculated by SchemeFit program56. These spectra are conceptually similar to that of the so-called kinetic states57, but include additional corrections obligatory for the real-case kinetics, in which the neighboring time constants, as is the case in ESR, are of the same order of magnitude. In the oversimplified case of a unidirectional photocycle, these spectra would have corresponded to the pure spectra of the intermediates. In the more realistic case of a sequence with back reactions, A↔B↔C↔D↔E↔, as is the case for the ESR photocycle (see details in the Results section), the apparent rates do not correspond to simple transitions such as A-to-B and B-to-C but rather to transitions from an A state into a mixture of A and B, and later on into a more complex mixture of A and B and C states coexisting temporarily due to transient equilibria. The spectra calculated in this manner17,56,58 are weighted sums of the pure spectra of the individual intermediates in the mixtures formed in these transient equilibria.
IR measurements were performed at 2 cm−1 resolution on a FTIR spectrometer (IFS-66/s, Bruker, Germany) equipped with a cryostat (Optistat DM, Oxford Instruments, Abingdon, U.K.). Specific intermediates and their mixtures were selectively populated at low temperatures under illumination with a ~30 mW/cm2 light of 500±20 nm produced by combination of a 150 W fiber optic illuminator (Model 1700, Dolan-Jenner, Boxborough, MA) and an interference filter. The resulting photoexcitation flux allows detection of states with decay times slower than one second. Further details on IR sample preparation, measuring protocols and data treatment were described previously59–61.
The photocycle measurements were performed on both liposome suspensions (in the visible), and films (in the visible and in the IR) prepared from the liposomes. As in other retinal proteins, humidity affects kinetics, and to minimize these effects we used highly humidified films with 800–2500 waters per each protein molecule. Under these conditions the differences in the time constants measured in films vs. those measured in suspension were within a factor of three. For direct comparison with the FTIR results, some of the measurements in the visible were repeated on IR films. The data that required direct quantitative comparison at different pH values were measured on liposomes suspensions in a mixture of six buffer: citric acid, MES, and MOPS, BICINE, CHES, and CAPS (10 mM each, all from Sigma), the use of which allowed to smoothly vary pH between 3 and 11.
The pH-dependence of proton-transporting cycle reactions was modeled by a linear system of differential rate equations (including reversible reactions), with the aim to reproduce the pH-dependencies of the observable rates and amplitudes. The intrinsic reactions were assumed to be of one of two simple types: either (i) pH-independent (first order) transitions in the photocycle and dissociation reactions, or (ii) pH-dependent (pseudo-first order) association reactions in protonation equilibria with the bulk. The model took into account a possibility of an internal donor to the Schiff base, which might be either initially protonated or deprotonated. Its protonation state in each intermediate (e.g. M-like) was accounted by explicitly including two separate sub-states: one with the donor being protonated, M(+), another not, M(−), and the corresponding balances are governed by pKa’s, which vary from one intermediate to another. The pKa in the non-excited state determines the initial conditions. The model includes also an additional pathway bypassing the donor that would enable alternative passive transport of protons from the bulk to the Schiff base during the M-to-N reaction. A program for numeric solution of these differential equations was developed in Matlab employing the procedures for solving the eigen-problem. Its output consists of the eigen-values, the apparent time constants, and the eigen-vectors, the corresponding apparent amplitudes of the concentrations of intermediates, as functions of the intrinsic rates and the assumed pKa’s, which were the adjustable parameters. Varying the pH for any set of parameters produced the pH-dependencies, and the adjustable parameters were varied to mimic the observations with the aim to estimate the region of their variability compatible with the observations.
CLUSTAL W62, OPUS (Bruker, Germany), Matlab (The Mathworks, Inc., Natick, MA), KaleidaGraph (Synergy Software, Reading, PA), CurvExpert (http://www.curveexpert.net), FitExp56, and SchemeFit56 software was used for data analysis and presentation.
Results
Structural differences between ESR and BR
The 3D structure of ESR is unavailable. Projecting the retinal binding pocket of BR (i.e. within 5 Å from retinal in BR52) on the amino acid sequence for ESR (B1YFV8_EXIS2 in www.uniprot.org) aligned51 to that of BR63 reveals (Figure 1) that of the 24 residues that form the retinal binding pocket, only 6 are different from their corresponding homologues in BR.
Figure 1.
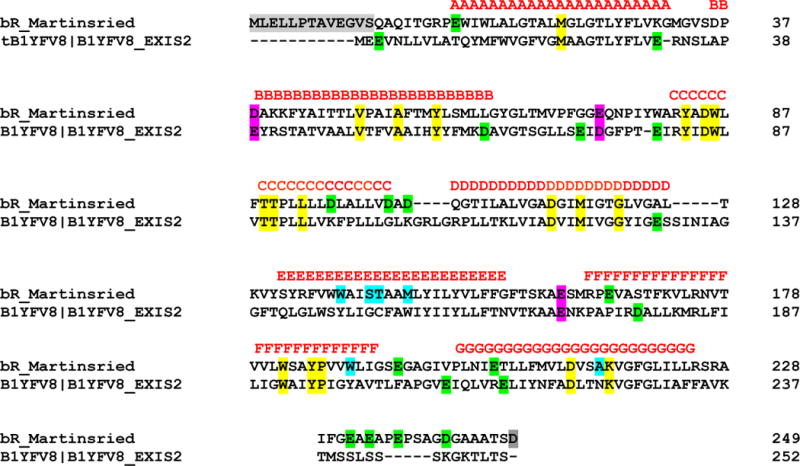
Primary structure of ESR aligned in CLUSTAL W62 with default settings. The 24 residues highlighted in yellow and cyan form the retinal-binding pocket in BR52; out of those 18 (in yellow) are conserved, 6 (in cyan) are not. In the helixes only three aspartates are conserved, Asp-85, -119 and -221, and yet another one, Asp-38 in BR, is substituted by Glu-39 (in magenta). Unconserved Asp/Glu residues are in green. Out of those, in ESR two aspartic and three glutamic acids are in helixes but only Asp-178 (displaced by a the full turn of the helix from the position of the Glu-166 in BR) and Glu-204 (immediate neighbor of the Glu-204 in BR) are sufficiently buried. Semi-conserved Asp and Glu residues are highlighted in magenta. In grey are the residues on C-and N terminus63, which are not counted in the primary sequence of BR.
In BR those are Trp-138, Ser-141, Thr-142, Met-145, Trp-189, Ala-215; they correspond to Tyr-147, Gly-150, Cys-151, Trp-154, Tyr-198, Asn-224 in ESR. Further, of the buried residues with carboxylic acid side chains, which might serve as reversible acceptor-donors12 for proton transport, both ESR and BR sequences contain 9 (Figure 1): 5 aspartic and 4 glutamic acids in ESR vs. 6 aspartic and 3 glutamic acids in BR. Asp-85, the homologue of the primary proton acceptor, Asp-85, in BR13,15,16, and Asp-119 and Asp-221, the corresponding homologues of two other functionally important aspartic acids in BR, Asp-115 and Asp-2124,64, are conserved. In ESR, the position homologous to that of the primary donor in BR, Asp-9614,28,29, is occupied by Lys-9651. Other carboxylic amino acids seem to be so far from retinal that they are unlikely to have direct acceptor or donor functions: two on the cytoplasmic, and three on the extracellular side of the membrane surface, while an additional glutamic acid, Glu-214 that does not have a corresponding homologue in BR is buried inside the protein on helix G. Unlike in BR, but similar to XR47 and GR18, the ESR sequence does not have homologues for Glu-194 and Glue-204, which play prominent role in optimizing the proton-releasing mechanism in BR44,65–69.
FTIR characterization of the photocycle intermediates above the apparent pKa of M accumulation
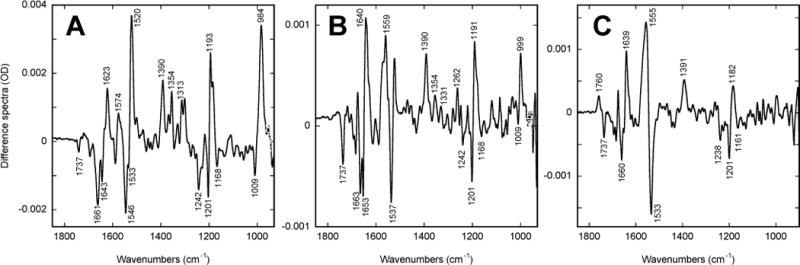
Previously, three main intermediates of the ESR cycle: the red-shifted early K-like state, the blue-shifted M-like one, and a later post-M red-shifted state, were characterized in this pH region by time-resolved spectroscopy in the visible48,51. We employed static FTIR measurements at low-temperature with the specific aims of (i) to characterize these three intermediates in more detail, and (ii) to look for additional transient states. The IR spectra in the four panels of Figure 2 reflect the presence of at least four intermediates that correspond to the K-, L-, M-, and N states of BR.
Figure 2.
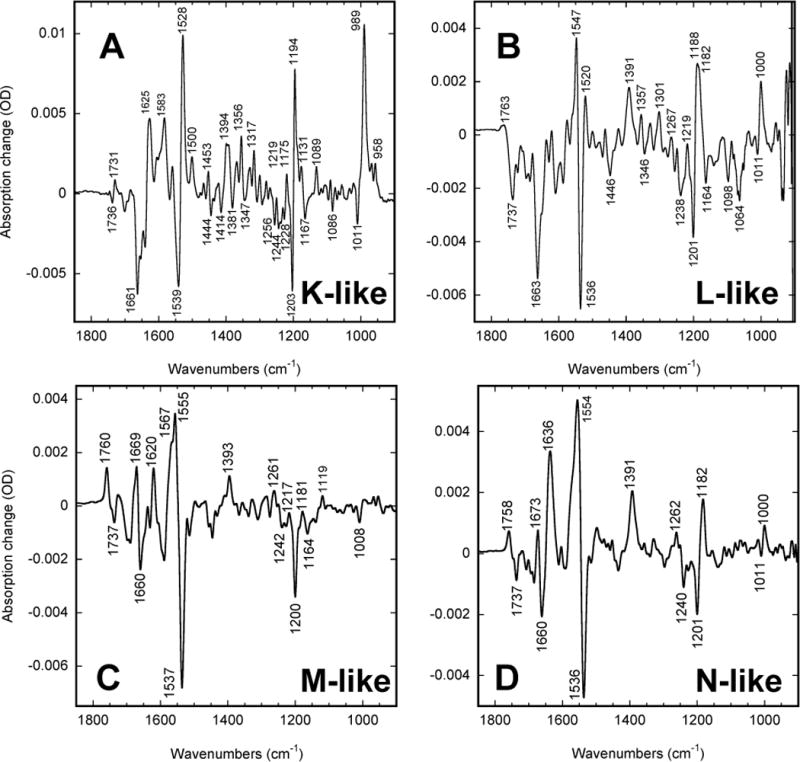
FTIR difference spectra at high pH (9.2–9.6). Mixtures of photoproducts created at low temperature under strong constant light illumination of 500 ± 20 nm in wild-type ESR: (A) a K-like state at 80 K; (B) a predominantly L-like state at 230 K; (C) a predominantly M-like state at 275 K; (D) a predominantly N-like state at 265 K. Note that the spectrum of M in panel C was obtained at a humidity level different from that used in panels A, B, and D (see text).
As evident from the combination of the negative (depletion of the initial state) and positive (formation of intermediates) bands in both the ethylenic (1500–1570 cm−1) and the finger print (1150–1250 cm−1) regions, the retinal in all four intermediates is in the 13-cis form31,70, and all of them arise from a cycle that originates from excitation of the all-trans retinal. No contribution from an alternative cycle arising from photoexcitation of the 13-cis form of the retinal is noticeable in the spectra in Figure 2. In BR the light-adaptation leads to redistribution of the isomeric ratio in favor of all-trans, traceable by a 10 nm shift in the visible1,37 and/or a distinct signature in the IR71. An attempt to detect a similar change in ESR by IR monitoring upon prolonged light-adaptation failed, revealing no redistribution in the isomer ratio. Together with the lack of any depletion bands from photo excitation of the 13-cis form of the chromophore in the spectra of intermediates (Figure 2) this implies that either non-excited ESR contain predominantly the all-trans retinal or excitation of non all-trans chromophore(s) produce intermediates sufficiently short-lived to escape our detection.
At ~80 K, similar to other retinal proteins, the cycle is blocked at an early stage59,61,72, and the trapped state is a red-shifted (see the up-shifted 1528 cm−1 positive band in the ethylenic region, indicative of a strongly red-shifted λmax and compare to the 1517 cm−1 band in low-temperature K in BR70) K-like intermediate (Figure 2A), with unique and prominent combination of the positive bands in the HOOP region (900–1000 cm−1), familiar from studies on bacteriorhodopsin61,70,71,73–76. In BR, under cryogenic trapping the contributions from the K-like intermediates are fully reversible by red light71,73,77. In ESR, unlike in BR71,73,77, but similar to GR78, some of the photo-induced changes at cryogenic temperatures are not light-reversible (data not shown). This non-reversible contribution decreases with increasing temperature, reflecting, most probably, an ability to trap a pre-K state. Further, even the component reversible with red-light is not homogeneous, and in ESR, similar to BR and XR61, there being at least two separate red-light reversible K-like states, the relative contributions from which vary with temperature. Details on these multiple early bathoproducts of ESR will be published elsewhere.
Raising the temperature allows the cycle to proceed further, and the spectrum at 230 K for panel B in Figure 2, corresponds to events that happen after the K-like state (partially) decays, but still early in the photocycle. The IR spectrum of the mixture in Figure 2B indicates the substantial presence of an L-like state. The differences from the K-like state (Figure 2A) indicate that it is a post-K state, while those vs. Figure 2C at a still higher temperature (275 K), indicate that it is a pre-M state. This assignment is based on the close similarity of the IR difference spectra in ESR with those in BR70,79–81, which identify the spectrum in Figure 2B as (i) one belonging to a 13-cis state (see the ethylenic stretch and the finger print regions, at 1490–1540 cm−1 and 1150–1250 cm−1, respectively), (ii) one with a protonated Schiff base (see the positive bands in the fingerprint and 1200–1500 cm−1 regions and compare them to the spectra of intermediates in BR in70,79–81), and (iii) one with an unprotonated Asp-85 (see the lack of a positive band at 1758–1760 cm−1 and compare to its presence in Figures 2C and 2D).
At temperatures above ~245 K (Figure 2D), as in BR, the dynamic relaxation mode is switched from distributed to conventional kinetics60, and the cycle is fully completed. Under constant illumination the resulting photostationary mixture is dominated by the slowest decaying intermediate(s) of the photocycle. Based on similarity with the IR spectra of the intermediates of BR70,79–81, the dominant intermediate in this mixture is assigned to an N-like state. It is a state with (i) a 13-cis retinal configuration, i.e. an intermediate before re-isomerization (see the ethylenic and finger print regions), which, unlike the earlier L-like state described above, has (ii) a prominent positive band at 1758 cm−1 due to transient protonation of Asp-85, (iii) a strong positive peak at 1182 cm−1 indicative of the 13-cis configuration of retinal with a reprotonated Schiff base, and (iv) a prominent positive peak at 1390 cm−1 characteristic of the N-like state in BR70,79–81. All these features indicate that, unlike the earlier L-like state described above, it is a post M state, which contains a 13-cis retinal i.e. prior to the cis-to-trans reisomerization characteristic for the N-to-O transition, and therefore we consider it N-like. Note that in ESR it is a red-shifted state (see Figure 3B and 3C), unlike the N state in BR8,82 but similar to the corresponding N-like state in PR17. The cumulative amplitude of the band at ~1760 cm−1 indicative of both M-like and post-M states increases with temperature, and follows a sigmoidal curve (not shown) similar to that described earlier for BR59,60. The midpoint of this curve is at ~250 K for pH 9.2.
Figure 3.
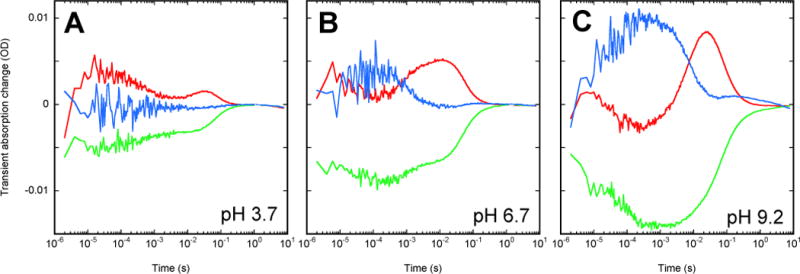
Single-wavelength traces at 420 (blue), 510 (green), and 590 (red) nm, illustrating the difference in the wild-type ESR photocycle at three different pH values. The data was measured on ESR-liposome suspensions at 22°C. The six buffer mixture at 5 mM each (see Experimental Methods) was used with no salt added.
The transient equilibria of L-to-M and M-to-N are highly sensitive to the presence of water. At the intermediate level of humidification (~1000 water molecules per each ESR molecule) used for samples in Figures 2B, and 2D, these two mixtures are strongly shifted towards L- and N-like states correspondingly, i.e. away from the M-like state. Raising the humidity level above ~2000 water molecules per ESR molecule, the balance becomes more favorable to M, and the photostationary mixtures created above 250 K indicate the dominant presence of an M-like state (Figure 2C – compare it to the M spectrum in BR70,79–81). The main features allowing this assignment are (i) the positive ethylenic stretch at 1567 cm−1 (overlapping the amide perturbation band at 1555 cm−1, similar to the case of BR70) indicative of a strongly blue-shifted state (compare this to the 1565 cm−1 ethylenic band of M intermediate in BR70); (ii) the absence of any positive component in the finger print region indicative of the deprotonated Schiff base70,79–81; and (iii) the positive C=O stretch band at 1760 cm−1 indicative of Asp-85 protonation. Besides the M-like state, this spectrum (Figure 2C) seems to contain some contribution from the later N-like intermediate, which would account for the positive band at 1390 cm−1. Usually this band appears in the N-like states70,79–81, and it is indeed present with a much higher amplitude in the N-like spectrum of ESR (Figure 2D).
Besides the positive band at 1760 cm–1 indicative of Asp-85 protonation, the carboxylic region in Figure 2C and D contains a negative band at 1737 cm−1. In BR the negative bands in this region appear due to shifts in the bands of the protonated Asp-96 and Asp-11514,33. ESR lacks the homologue of Asp-96 (having a Lys-96 in the corresponding position) but Asp-119 is the homologue of Asp-115, and Asp-221 is the homologue of Asp-212 in BR. The latter is expected to be unprotonated, as in BR64, and therefore, we tentatively assign the negative band at 1737 cm−1 to the protonated Asp-119. An apparent absence of a coupled positive peak in the protonated carbonyl region (>1700 cm−1) argues against the origin of the negative band at 1737 cm−1 being due to a perturbation of Asp-119. Instead, it could be interpreted as transient deprotonation, in which case at least some of the amplitude of the positive band at 1391 cm−1 might be due to the deprotonated carbonyl stretch that arises. However, the proton-related signal of an indicator dye appears only on the time-scale of the second/slow component of the M-like state formation48, and, therefore, even if transiently deprotonated, the possible role of the Asp-119 in proton pumping is not clear. A similar problem was noted previously in BR for the Asp-96 band in L state, where the time-resolved FTIR13,83 and low-temperature trapping FTIR experiments33,84 seems to be in conflict, and there is still no consensus for its cause.
Photocycle of the wild-type ESR and its pH-dependence, from monitoring in the visible
Figure 3 presents characteristic time-resolved traces of absorption changes of ESR-liposome suspensions at three pH values, below (panel A), near (panel B), and above (panel C) of the apparent pKa in ESR.
The traces at 510 nm monitor the depletion of the initial state, while those at 420 nm are mostly from the M-like state(s). The traces at 590 nm illustrate the presence of the red-shifted product(s): the decay of the initial bathoproduct that appears faster than the 1.5 μs instrumental time-constant, and the rise and the decay of red-shifted state(s) in the second part of the photocycle. The latter, unlike in BR but similar to PR17, is not an O-like state which should have contained the all-trans retinal, but rather an N-like state because it contains 13-cis retinal (Figure 2D). The presence of yet another red-shifted intermediate, which appears upon the decay of this dominant red-shifted N-like state and decays on a time-scale of ~3-times slower than its predecessor (see more below), is revealed by the ratio of the traces at 590 and 510 nm (not shown).
The pH-dependence of the photocycle was further explored in liposome suspensions in the pH range from 3.7 to 9.2 (Figure 4A). As noted previously48, the transient accumulation of the M-like state(s) is titrated with a distinct pKa, which is ~6.5 when measured in liposomes. Figure 4B presents the comparison of the titration curve for ESR from the static absorption, the spectrum of nonexcited protein (see more in48) and the amplitude of the accumulated M-like state(s) in the photocycle (as measured at 420 nm). The two variables were fitted separately to Henderson-Hasselbalch formula, scaled to their individual magnitude of change, and overlapped in Figure 4B to illustrate that they reveal the same pH-dependence with an apparent pKa of ~6.5. Although not shown in Figure 4B, the partial decrease in the apparent accumulation of the red-shifted state(s) that follow M (see Figure 3) show the same apparent pKa (not shown).
Figure 4.
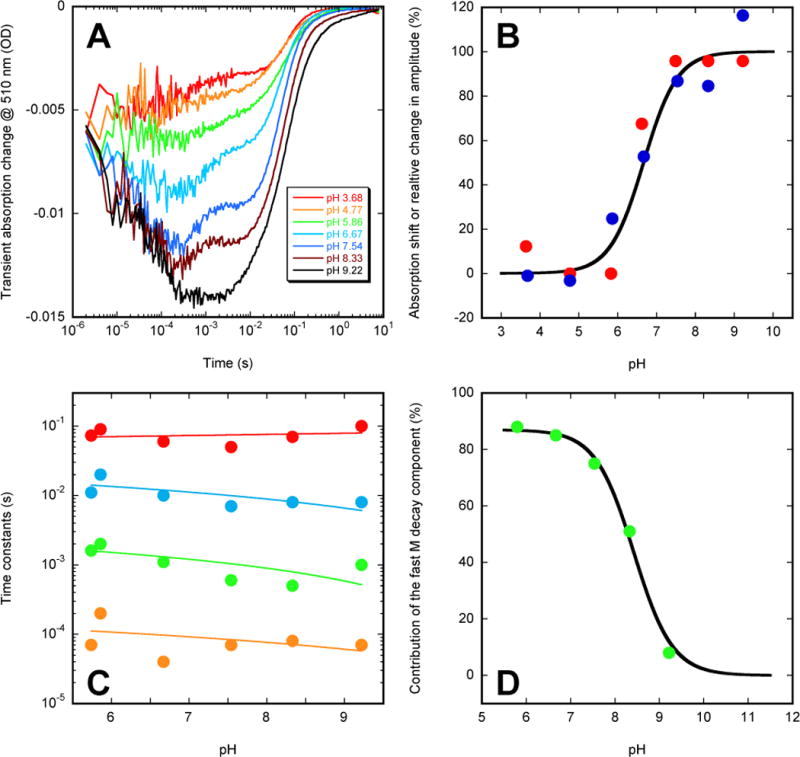
pH-dependence of the wild-type ESR photocycle. (A) kinetic traces measured at 510 nm on ESR-liposome suspension. (B) a cumulative titration curve, including (i) pH-dependence of the amplitude associated with transient accumulation the blue-shifted M-like state (blue circles), (ii) λmax shift of the non-excited state (red circles), and (iii) a Henderson-Hesselbalch curve with pKa 6.5 (solid line). The data (filled circles) are plotted after appropriate normalization. (C) time constants (filled circles) of a 4-exp fit obtained by global fitting with FitExp. The four time constants correspond to: (i) the second slower component of the M rise (in orange), (ii) the fast (in green) and (iii) the slow (in blue) components of M decay and (iv) the main component of the decay of the transient absorption due to red-shifted intermediates (in red). (D) ratio of amplitudes (filled green circles) of the two kinetic components of the M decay (corresponding to the two time constants, presented in green and blue in panel C) overlapped with a Henderson-Hesselbalch curve with pKa 8.5 (solid line).
The data measured on ESR-liposome suspensions were globally fitted with multi-exponentials, separately at each pH using the time-frame from ~10 μs to completion of the photocycle. Figure 4C illustrates the time-constants for a simple 4-exponential fit, which, although incomplete, is a sufficient approximation for the main processes discussed. The four time constants reflect (i) the second (slower) phase of the M rise (τ1 ~100 μs), (ii) and (iii) the two components (τ2 ~1 ms and τ3 ~10 ms) of the M decay and the corresponding rise of the transient absorption in the red, and (iv) the main component (τ4 ~39 ms) of the decay of the red-shifted intermediate(s). The decay of the initial K-like state(s) and the fast phase of the M rise were both faster than the chosen time-window. An additional low-amplitude process, slower than the τ4 decay, which was revealed by calculating the ratio of traces in Figure 3 (not shown), was not included in the 4-exponential fit either.
Figure 4C presents the pH-dependence of these four apparent time constants, which are nearly pH-independent in the pH range 5.7–9.2 (Figure 4C), including the biexponential decay of the blue-shifted M state, which is simultaneously the biexponential rise of the induced absorption in the red, with τ2 ~1 ms and τ3 ~10 ms time constants. On the other hand, the relative contributions of the two components of M decay are strongly pH-dependent (Figure 4D). At lower pH the faster component dominates, while the reverse is true at higher pH. If the Henderson-Hasselbalch formula is applied to the ratio of amplitudes of the two components a pKa ~8.5 is obtained (Figure 4D). This pH-dependent behavior is distinctly different from that of the titration-like changes in the overall accumulation of the M-like state, which has a pKa ~6.5 (see Figure 4B). A study on the former feature in detergent-solubilized samples is described separately85.
At pH higher than the data points shown in Figure 4D, M decay becomes pH-dependent, and above pH ~9.5 the corresponding time constant(s) increase linearly with the decreasing proton concentration in the bulk, creating a distinct inflection point in the pH-dependence of the apparent rates (see Figure 7 below), in accord with a similar effect in BR28. This is the third strongly pH-dependent feature in the photocycle kinetics, and, as we will argue below, it should reflect the titration of yet another group, different from those affected at pKa’s of ~6.5 and ~8.5.
Figure 7.
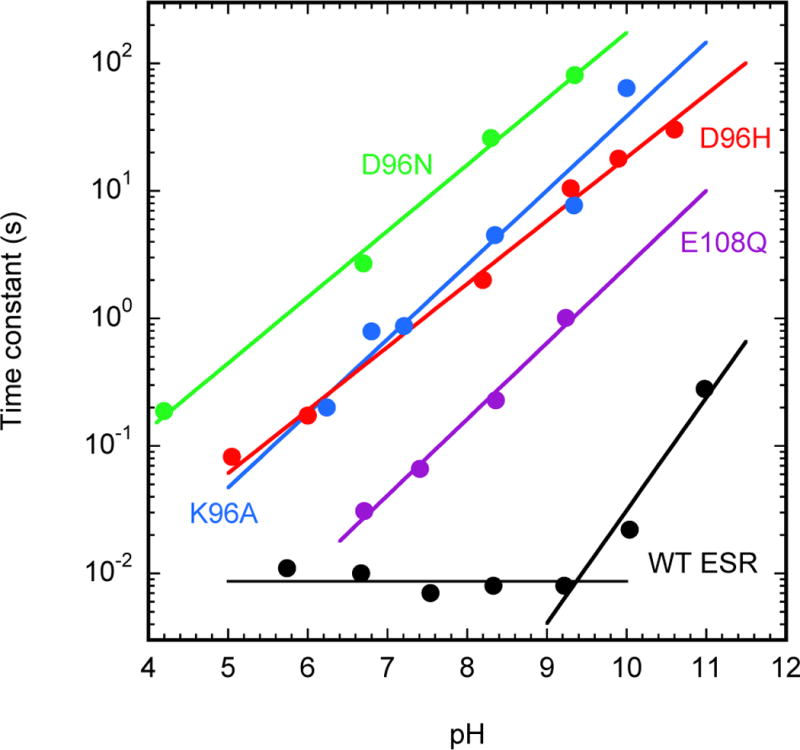
pH-dependences of M decay for: the slow component in the wild-type ESR (black), the K96A (blue) mutant in ESR, the D96N (green) and D96H (red) mutants in BR and the E108Q (magenta) mutant in PR. The K96A data were obtained on ESR-liposomes (in a 5 mM buffer, no salt); those on D96N and D96H mutants of BR, on purple membrane fragments (in a 5 mM buffer in the presence of 100 mM NaCl); the data on E108Q mutant of PR – on a crude (after French press but before solubilization/purification) membrane fraction (in a 10 mM buffer in the presence of 100 mM NaCl).
Monitoring the photocycle in the visible reveals four interesting features. First, as we noted previously, the apparent accumulation of M in the photocycle is dependent on pH, the decrease in the overall observable transient amplitude of M with decreasing pH follows a titration-like pattern, and transient accumulation of the M-like state is completely eliminated at low pH48. Second, not only the apparent accumulation of the M-like state but also the apparent contribution from the red-shifted post-M state(s) is also affected by pH. However, unlike the M-like state(s), the accumulation of the red-shifted post-M state(s) is decreased but not eliminated at low pH. Third, M decay and the resulting formation of N are clearly not monoexponential48. Both contain at least two components, and the ratio of their relative contributions is strongly pH-dependent (see more below). Fourth, unlike in BR, the build-up of the M-like state leads not only to the strong depletion in the main absorption band (monitored at 510 nm) but it is traceable also in the time-resolved absorption changes in the red (monitored at 590 nm) in the hundreds of microsecond time-range (see more in the section on the K96A mutant below). The reverse is also true, and the concomitant formation of the red-shifted intermediates (see the 590 nm trace in Figure 3) on the millisecond time-scale leads to an apparent depletion observable at 420 nm as well. This latter feature is very different from what is observed in BR, and, as a consequence, could create some strongly misleading impression on the dynamics of the ESR photocycle if one compares it to the photocycle of BR.
Detection of the post-M states in the photocycle below the apparent pKa of M accumulation
While the absence of accumulation of the M-like state at low pH is evident (Figures 3A and 4B), the question remains whether an M-like state is not produced at all or produced but is not accumulated below the apparent pKa of ~6.5 (Figure 4B). Earlier48, we proposed that the latter is the case, based on the mostly anionic state of the proton acceptor, Asp85, in this pH region, and the apparent discrepancy between the inability to detect a transient accumulation of M-like state in time-resolved experiments below the corresponding pKa and the transport activity observed48. To reconcile these observations we proposed48 that M-like state is produced but not accumulated, for kinetic reasons. To explore this option in detail, we performed additional measurements on films, which were prepared at low pH (~5.0) and their effective pH was verified by monitoring the photocycle at room temperature (data not shown). We then attempted to capture, if not an M-like state itself, then a post-M state by photostationary measurements at low-temperature with IR monitoring.
The most characteristic IR feature of an M-like state in both BR and ESR is a strong positive band at ~1760 cm−1 arising from the asymmetric stretch of the protonated carbonyl band of the primary acceptor, Asp-85. At high pH, this band is indeed absent in the pre-M intermediates (K-like and L-like in Figure 2A and 2B) but present in the post-M states, e.g. N-like state, which is produced from its M-like predecessor – see Figure 2D for ESR and compare to the corresponding spectra in BR70,79–81. We used the band at ~1760 cm−1 as a marker for the M and post-M states, which are associated with the proton pumping cycle. Similar to the high pH data (Figure 2), at low pH (~5.0) little if any positive band at ~1760 cm−1 is seen up to 250 K (Figures 5A and 5B).
Figure 5.
Infrared difference spectra at low pH (5.0). Mixtures of photoproducts trapped as in Figure 2: (A) a K-like state trapped at 160 K, (B) the predominantly L-like state in the photostationary mixture at 250 K with some residual contributions (see the HOOP region) from the K-like one, and (C) the N-like state at 260 K. The latter could be observed both at 260 K and above with reduced amplitude when the photocycle is becoming faster than the photo-induced rate provided by our illumination. The data measured at 270 K fully overlapped with that at 260 K (see text).
However, photostationary measurements at 260 K (Figure 5C) and 270 K (scaleable to the spectrum in Figure 5C but reflecting decreased population; not shown) give rise to a pronounced band at ~1760 cm−1. These spectra (see Figure 5C) are comparable to the spectrum of the N-like state at higher pH (Figure 2D), and have all the IR features characteristic for the mixture of post-M states, with a possible minor contribution from an M-like state. The IR spectrum in Figure 5C can be unambiguously assigned as containing a majority of N-like state, based on the combination of its characteristic bands: (i) the positive band in the finger print region at 1182 cm−1, (ii) the ethylenic stretch band at 1554 cm−1 and the (iii) bilobe band in the carbonyl region at 1760/1737 cm−1. All these features are very similar in the pH 5.0 spectrum at 260 K (Figure 5C) and in the N-like spectrum at pH 8.0 (Figure 2D), and very different from the corresponding spectra of the pre-M states at high pH (Figure 2A and 2B) or low pH (Figures 5A and 5B). Further, comparing the amplitudes of the protonated Asp-85 band (at ~1760 cm−1) to those of the bands in the ethylenic (1520–1570 cm−1) and fingerprint (1160–1240 cm−1) regions at low pH (Figure 5C) vs. high pH (Figure 2D) confirms that at low pH, as at high pH, Asp-85 protonation occurs in all of the cycling fraction and does not reflect a minor contribution.
Photocycle of the K96A mutant
Figure 6 presents data on the photocycle of the K96A mutant.
Figure 6.
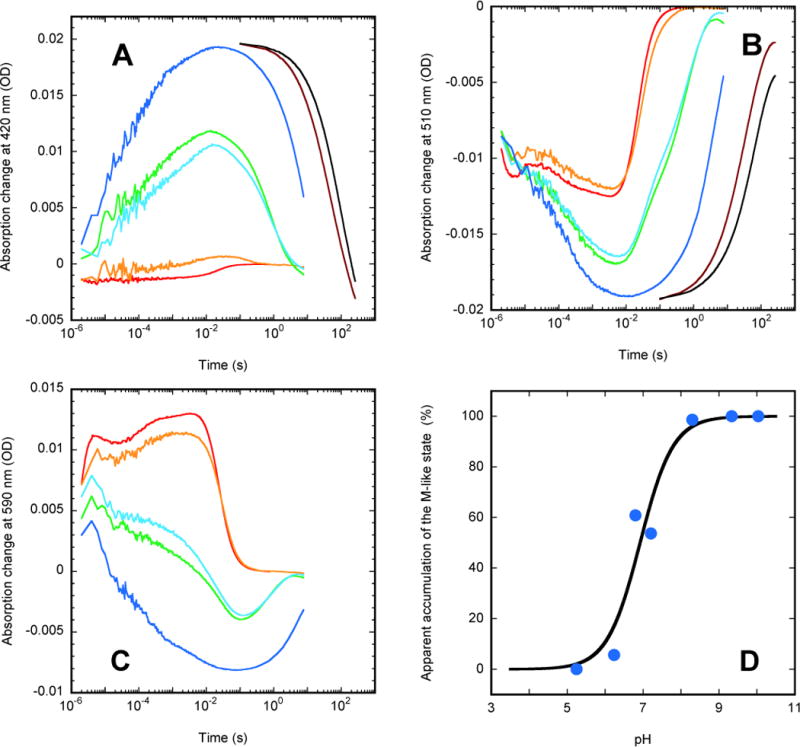
pH-dependence of single-wavelength kinetics in the K96A mutant: time-resolved traces for monitoring (A) at 420 nm, (B) at 510 nm, (C) at 590 nm, and (D) the transient accumulation of the blue-shifted M-like state in the photocycle. In panel (D) the amplitudes (solid circles) observed in time-resolved measurements are overlapped with a Henderson-Hesselbalch dependence with pKa 7.0 (solid line). The pH values for traces in panels A-C are: 5.25 (red), 6.24 (orange), 6.80 (green), 7.21 (light blue) 8.35 (dark blue), 9.34 (brown), 10.04 (black).
The traces at 420 nm (Figure 6A) reflect predominantly the dynamics of the blue-shifted M-like state(s), the traces at 510 nm (Figure 6B) reflect the initial state depletion, while those at 590 nm (Figure 6C) illustrate both the dynamics of the early red-shifted K-like state(s) and those of the later red-shifted states. Note, however, that the latter is not exclusively reporting on the red-shifted states. On the contrarily, as is evident from the pH region 6.8 – 8.35 (see the blue and green traces in Figure 6) the dominating presence of the long-lived M-like state(s) (Figure 6A) has an unexpected effect creating a profound depletion in the apparent absorption changes in the red (at 590 nm in Figure 6C) from the transient bleaching of the initial state.
Similar to wild-type ESR (Figure 4B), the apparent amount of M detected during the photocycle is strongly pH-dependent (Figures 6A and 6D). Transient accumulation of the M-like state(s) disappears with an apparent pKa of ~7 (Figure 6D), presumably due to the analogous partial titration of the Asp-85/His-57 complex, as in the wild-type with pKa ~6.5 (Figure 4B).
Similar to mutants lacking the primary proton donor Asp-96 in BR28,40,86 or the corresponding Glu-108 mutants in PR17, the M decay in the K96A mutant in the accessible pH range (Figure 6A) is the rate-limiting step, and its kinetics is virtually monoexponential. Attempts to include a second exponential component resulted in fits with this second component accounting for 5–15 % of the M decay, but both the rates and its amplitudes varied inconsistently over pH. With M decay being the rate limiting step of the photocycle in ESR, little if any post-M states could be observed in the flash experiments, resulting in the lack of induced positive absorption changes in the red in the second half of the photocycle (Figure 6C).
Comparison of the pH-dependence of M decay in the wild-type ESR to those in the K96A mutant in ESR and to D96N-like mutants in BR and PR
Similar to the D96N-like mutants in other retinal proteins, in the K96A mutant the slope in the pH-dependence of the monoexponential M decay approximation (see Figure 7) is not equal to one, as should have been expected if the protons were captured directly from the bulk in a pseudo-first order reaction. This slope is close to 0.5 instead, a value found earlier in the D96N mutant of BR40,87. A somewhat steeper slope, ~0.7, was reported elsewhere for the D96A and D96N mutants28, which is still lower than expected for the simple pseudo-first order reaction. Figure 7 presents additionally the pH-dependence of the slower component of the M-state decay in wild-type ESR in an extended pH range, together with the corresponding pH-dependencies of M decay in D96-like mutants in BR and PR.
Since the slope ~0.5 is consistently obtained in different mutants, i.e., D96N28,40,41,87, D96H, E108Q, E108H (latter not shown), K96A (in Figure 7), on different preparations (cells, membranes, proteoliposomes, detergent-solubilized preparations) in three different retinal proteins (BR, PR, ESR), its deviation seems to reflect an interesting specificity in the mechanism of passive proton translocation between the bulk and the deprotonated Schiff base. Earlier, a pH-dependent change in the surface charge was proposed as the reason for this deviation40 but there is no consensus in this.
An unexpected feature observed in the wild-type ESR is a mismatch between the apparent inflection point in Figure 7 at pH of ~9.5 and the apparent pKa ~8.5 for the titration of the ratio of the M decay amplitudes in Figure 4D. Modeling of protonation reactions during the photocycle (see below) confirmed the intuitive feeling that this discrepancy signals that the inflection point in Figure 7 at pH of ~9.5 and the apparent pKa ~8.5 in Figure 4D cannot be due to titration of a single group.
Global analysis of the transient absorption changes at pH above and below the apparent pKa ~6.5 for M accumulation
An extended series (for the 380–720 nm range) of single-wavelength measurements was performed with an aim to extract spectral information from kinetic analysis. These measurements were performed on highly humidified films prepared for FTIR measurements both above (at pH 8) and below (at pH 5) the apparent pKa (~6.5) of the M accumulation. Since our specific focus here is on the later (millisecond) part of the photocycle, only the time-resolved data starting from 1 ms after excitation were used for analysis, and the time range extended up to 3.8 sec at pH 8 and to 0.8 sec at pH 5.
Global kinetic analysis revealed that at least four distinct intermediates are needed for a statistically valid description of the measured absorption changes in the second half of the photocycle, both at pH 8 and 5.
Figures 8A and 8B present the amplitude spectra of the corresponding components produced by FitExp from data at pH 5 and 8, respectively. The time constants obtained from these humidified films agree with those measured on liposomes suspension (Figures 3 and 4A) within a factor of three. The spectra of mixtures formed in-between the correspondent decay components were then calculated17,56,58 by the SchemeFit program56. This oversimplified reconstruction does not produce the pure spectra of intermediates but rather the spectra of mixtures, i.e. weighted sums of spectra of individual states, and Figures 8C and 8D presents the difference spectra for these mixtures assuming a cycle, in which each consecutive step is slower than the previous one. The corresponding absorption spectra were reconstructed assuming, a ~15% photoconversion by non-saturating excitation flashes used, as is the case in BR under similar excitation conditions58,88,89, and they are shown in Figures 8E and 8F. The shape of the spectra (Figures 8E and 8F) unambiguously indicate that they cannot be due to pure intermediates but are indeed mixtures, indicating a substantial contribution of back-reactions in the ESR photocycle. At pH 8 (Figures 8B, 8D, 8F) the contributions from the M-like state(s) are present in two fastest (in the time-range above 1 ms) kinetic components with τ1 ~4, τ2 ~14 ms (the blue and green curves in Figure 8). These two contributions per se do not indicate the presence of two different M-like states, and as in BR biphasic decay of M most probably appears due to the presence of a back reaction28,90, and might indicate a single M-like intermediate with λmax ~ 400 nm.
Figure 8.
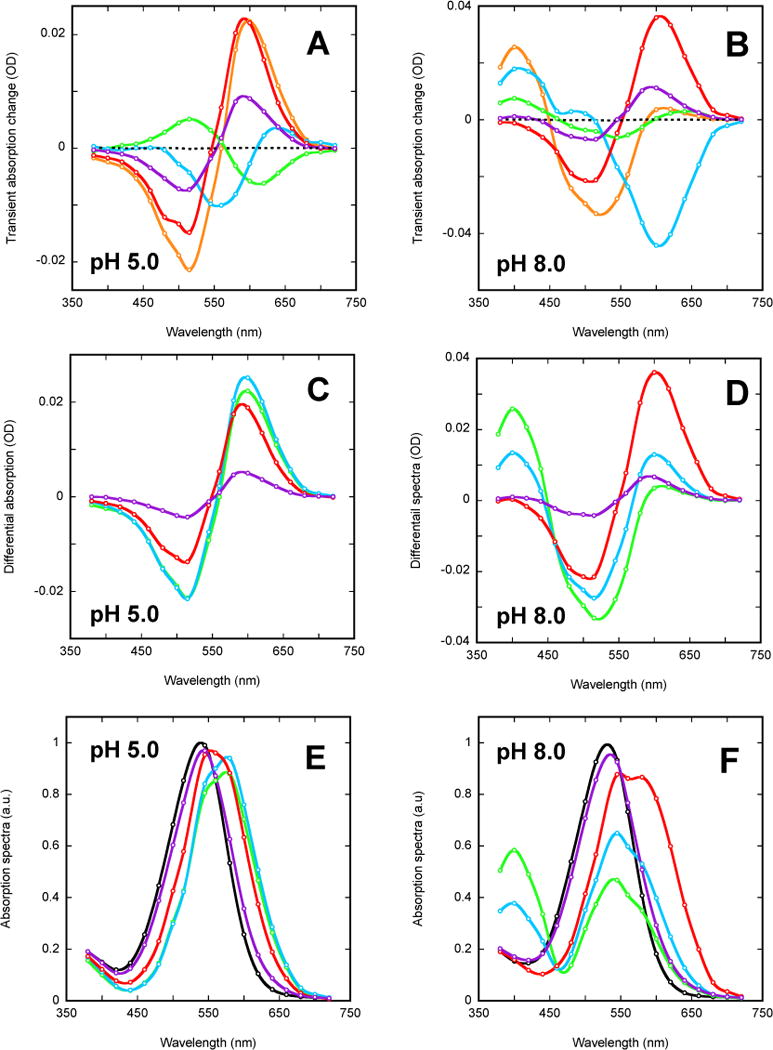
Kinetic decomposition of absorption changes in the second part of the photocycle measured on two humidified films, which were prepared from liposomes suspensions at either pH 5.0 (left column) or 8.0 (right column) In both cases the photocycle dynamics is described with four intermediates, which give rise to four kinetic components. (A and B) individual amplitude spectra of kinetic components calculated by FitExp for the four exponentials: τ1 (green), τ2 (blue), τ3 (red), τ4 (magenta) and their sum (orange), which corresponds to extrapolation for the spectrum that is observed prior to these processes. The spectra in panels C-F are color-coded in the sequence of green-blue-red-magenta for the last four apparent transitions in the photocycle. (C and D) difference spectra recalculated by SchemeFit assuming a unidirectional sequence of intermediates, when each consecutive step is slower than the preceding one. (E and F) absorption spectra reconstruction assuming a ~15% efficiency for conversion into the photocycle. The spectra of the initial states (black) were corrected for scattering. The open circles are the calculated points, and the solid curves are the cubic splines drawn through them.
The increase in absorption in the red occurs in a biphasic manner: with τ1 (minor contribution) and τ2 (major contribution) time constants. An attempt to interactively separate these two red-shifted N-like states resulted in the earlier one, N1, having a λmax(N1) of ~580 nm, while the later one, N2, being farther red-shifted with a λmax of ~605 nm. The apparent transient absorption in the red (from those N-like states) disappears in biphasic kinetics, with τ3 ~60 ms as the major component and τ4 ~240 ms the minor one. The latter component (magenta spectra) corresponds to the one revealed by the ratio of the traces in the region of ground state depletion to that in the red (Figure 3). The amplitude spectra of the τ3 (red) and the τ4 (magenta) processes at pH 8 are different, indicating that the strongly red-shifted N2, λmax(N2) ~605 nm, which decays with the τ3 time constant, is followed by an intermediate (N3), which decays with the τ4 time constant and has only a slightly red-shifted absorption band: λmax(N3) ~ 537 vs. 532 nm in the initial state, i.e. an approx. 5 nm red shift. Contributions from the N3 state (magenta in Figure 8F) might be present in the previous spectra (see the shoulder in the green, blue and red spectra in Figure 8F) as well. However, interactive subtraction of the former (spectrum in magenta) from the latter does not eliminate the corresponding shoulder, indicating that those are not due to the presence of N3 but rather reflect the contributions from the N1 and N2 states. Thus, the intermediate N3 is always present only as a minor contribution. As the result, it is not yet clear whether it has a 13-cis or an all-trans retinal, and unlike the preceding states it may or may not correspond to an all-trans O-like state. This state has a strong shoulder in the 600–650 nm region (Figure 8F), which might indicate that in reality it is a much stronger red-shifted state whose contribution to the spectrum (in magenta in Figure 8F) is minor due to its very low concentration in the mixture. Alternatively, the last spectrum might indicate the presence on the final stages of the photocycle of an intermediate similar to the “PR-prime” states reported earlier in PR at the very end of its cycle91.
The spectra reconstructed from the data at pH 5 are quite different from those at pH 8, and, unlike those for at the higher pH, they (Figure 8E) lack noticeable contributions from blue-shifted M-like state(s). The analysis reveals the presence of four transient states, and the manual decomposition of the corresponding spectra indicates that the earliest one (in blue) is moderately red-shifted: λmax(N1) ~580 nm. It is followed by a second one, N2 (in green), which is red-shifted approximately to the same extent as N1, while the last two, N3 (red) and N4 (magenta), are only slightly red-shifted: λmax ~542 nm vs. ~540 nm in the initial state.
Both the first (blue) and the second (green) spectra at pH 5.0 (and for the time range above 1 ms) in Figure 8E are mixtures of at least 2–3 states, and the apparent red shift in the corresponding transient absorption is due to a dominant presence of the N1/N2 states, both of which has λmax ~580 nm. Both of these spectra have a strong shoulder at ~545 nm and a smaller one at ~500 nm. The former indicate a substantial presence of yet another intermediate, which is much less red-shifted, and its actual λmax should be between that of the initial state, at ~540 nm, and the apparent position of the shoulder in Figure 8E, at ~545 nm. The slight shoulder at ~500 nm seems to be evidence for the residual presence in those mixtures of contribution from an L-like state, a tail of the earlier state in the photocycle due to back-reaction. Redistribution during the τ1 (~7 ms) transition leads to an increase in the absorption in the red indicating that the later N2 state might be somewhat stronger red-shifted than its predecessor, the N1 state. The τ1 transition also leads to a very small increase at ~400 nm, which might be indicative for the minor presence of a kinetically hidden M-like state (as proposed in48), the presence of which is not evident from the flash-photolysis signals (like those in Figures 3). The τ2 (~15 ms) transition is the decay of the red-shifted transient absorption. The next step (τ3~40 ms) decreases the transient absorption in the red even further, and the last of the detected intermediates has a λmax ~542 nm (Figure 8E). Currently we do not have enough data for full characterization and separation of the either the first two stronger red-shifted states (both with λmax ~580 nm) or the later less red-shifted species (both with λmax ~542 nm), and their molecular differences will be a subject of further studies.
The absence at pH 5.0 (Figure 8E) of an intermediate as strongly red-shifted as the N2 state with λmax ~605 nm at pH 8.0 (Figure 8F) results in an apparent decrease of the amplitude of the overall transient absorption in the red at low pH (Figure 3). Since unlike at high pH, the transient absorption in the red at low pH is due to much less red-shifted intermediates (λmax ~580 nm at pH 5.0 vs. λmax ~605 nm at pH 8.0), the apparently decreased amplitude of the transient absorption in the red (Figure 3A) at pH below pKa ~6.5 is not an indication for a decreased concentration of corresponding red-shifted post-M states.
Discussion
The pH-dependence of the accumulation of the M state (Figure 4B) defines an apparent pKa of ~6.5, which separates two different pH regions. There is still a third region at pH below pKa ~2.3, in which Asp-85 is fully protonated48, but limited stability of the protein, when for example 22% of the visible absorption was irreversibly lost after an attempt of a reversible transition from pH of 7.1 to 3.1 and back in 30 minutes (data not shown), precluded its detailed study. As we reported previously, at pH between 3 and 6.5, Asp-85 is mostly unprotonated but time-resolved monitoring of the photocycle in the visible does not reveal any transient accumulation of M-like state(s)48. At pH above 6.5, both M-like and post-M states are readily observed48,51, and the photocycle resembles those observed earlier for PR17 and XR27. This is observed both in liposomes (this study) and in detergent-solubilized samples48,85, and once the difference in pKa’s for the two systems is taken into account, their behavior is nearly indistinguishable, indicating that the pH dependent changes in the photocycle arise from the protein rather than its environment.
Intermediates of the ESR photocycle in the pH range above the apparent pKa of the M accumulation
The photocycle of wild-type ESR revealed a phenotype similar to that of BR, and contains K-, L-, M-, and N-like intermediates. The presence of at least three spectrally distinct intermediates, an early red-shifted K-like, a blue-shifted M-like, and a late red-shifted state were described earlier in48,51. Initially51 the late red-shifted state was assumed to be an O-like type. IR spectra of intermediates at low temperature reveals that the red-shifted state(s) in the second half of the photocycle contain a retinal chromophore still in the 13-cis configuration (see Figure 2D). Therefore, these are states before re-isomerization of the retinal, which in BR marks the N-to-O transition34,92. In spite of their red-shifted absorption, they are actually N-like. This finding is in accordance with such red-shifted N states observed previously in PR17.
As revealed by kinetic analysis (Figure 8), the transient absorption in the red in the second half of the photocycle cannot be accounted for by a single red-shifted intermediate. Detailed kinetic analysis of multi-wavelength time-resolved data revealed the existence of at least three or four transient states after M above and below the pKa ~6.5 of the apparent M accumulation, respectively. The absorption maxima of these states vary from ~537 to ~605 nm but all of them are red-shifted, and all of them except probably the last one, have a 13-cis chromophore, indicating that they are N-like states. The latest state, however, might be a genuine O-like state with an all-trans-retinal. Unfortunately, it is always present only as a minor component.
The presence of the L-like state is not immediately evident from the data in the visible (Figure 3) but is revealed by FTIR spectroscopy at low temperature (Figure 2B). L-like states are observed both above (Figure 2B) and below (Figure 5B) the apparent pKa ~6.5 of the accumulation of M. Further, the dependence of the amount of a particular intermediate trapped at particular temperature follows a sigmoidal curve59,60. In ESR the mid-point in this dependence for the K-like state(s), as monitored by the intensity of the HOOP band at 988 cm−1, is at ~200 K, while that for the M-like and post-M states, as traced by the intensity of the protonated Asp-85 band at ~1760 cm−1, is at ~250 K (not shown). Therefore, the temperature range between these two midpoints, 200 and 250 K, is where the L-like state is the most prominent. In BR the corresponding mid-points characterizing the regions of stability of intermediates are at ~143 K for the K state59, and at 220 and 260 for M and N states60 respectively.
Summarizing our data (see also48,85), the photocycle of ESR is represented by the following tentative sequence of intermediates:
where the subscripts refer to the absorption maxima in the visible. They were calculated assuming that each consecutive step in the photocycle is slower than the previous one.
The photocycle below the apparent pKa for the accumulation of M
When the pH is lowered to below the pKa ~6.5 (Figures 3A and 4B) the BR-like photocycle disappears. A spectral transition with the same pKa is observed for the non-excited protein (Figure 4B), which in the wild-type corresponds to a bathochromic shift of the absorption maximum by ~8 nm. This magnitude is much smaller than the corresponding 37 nm shift in BR with pKa 2.5–2.91,37, and, revealingly, nearly 5-times smaller than the corresponding shift (with pKa ~4.5) in the H57M mutant (see more in48). We attribute the observable pKa ~6.5 to partial titration of the counterion, which in the wild-type ESR is more complex than in BR and involves His-57/Asp-85 interactions48. Full protonation of Asp-85 occurs with a pKa ~2.3, and between pH 3 and 6.5 Asp-85 is about ~90% unprotonated48.
At pH below pKa ~6.5 in the wild-type ESR, a strongly altered photocycle is observed, which is, however, not analogous to the acid photocycle of BR observed when its counterion is fully protonated. In BR, the acid cycle contains K-like and L-like states but no post-L states that correspond to either M-like or N-like intermediates93,94. This is in contrast to the ESR photocycle below pKa ~6.5, which contain those post-L states. Although M-like state(s) are not transiently accumulated at pH below the pKa ~6.5 (Figures 3A and 4B), Asp-85 does become transiently protonated in the course of the photocycle not only above but below this pKa as well (Figure 5C). We interpret this as the evidence for the existence of post-M states in the main path of the photocycle, in accord with our earlier proposal48 that the lack of observed accumulation of M during the photocycle in this pH range is for kinetic reasons only. The detection of these post-M states at pH below the apparent pKa ~6.5 for M accumulation is also in accord with our previous results on proton transport measurements in liposomes: although the proton transport is decreased by approx. half with a similar pKa (Fig. 10B in48), it does not disappear concomitant with disappearance of the transient accumulation of the M-like state(s). Consistent with this, the large positive C=O stretch difference band from the protonated Asp-85 (Figure 5C) could arise only if this residue were (largely) unprotonated in the initial state. This confirms our earlier conclusion48, from different evidence, that Asp-85 is mostly unprotonated in the initial state in this pH region.
Alternatively, one could imagine that at low pH Asp-85 becomes transiently protonated (Figure 5C) by a proton that does not originate from the Schiff base. Since this occurs in the pH region where the proton transport is not abolished48, this would require a mechanism that bypasses Schiff base and, apparently, does not even include destabilization of its proton by light either. In conceptual terms, this means a mechanism that is completely different from that in any known proton pump in retinal proteins. Currently, we consider this option as too extreme.
pH-dependence of M decay in the wild-type ESR photocycle
The most unexpected result is the virtual pH-independence of the time constants in a wide range from pH 5.7 to 9.2 (Figure 4C). The transient accumulation of M disappears (Figure 4B) and concomitant absorption in the red is decreased by approximately a factor of two (Figure 3) with a pKa of ~6.5, but this happens with little if any effect on the apparent time constants (Figure 4C). The same is true for the pH-dependence of the ratio for the two components of the M decay, which are found to be strongly affected by pH, with a pKa of ~8.5 (Figure 4D). However, similarly to the pH-dependence of the overall amplitude of the apparent M accumulation (pKa ~6.5 in Figure 4B), this occurs without any noticeable effect on the apparent rates (Figure 4C). Thus, in the pH range from pH 5.7 to 9.2 we observe two separate strong titration-like pH-dependences of the vital photocycle features involving the key M intermediates. Counterintuitively, both of these features are expected to be correlated with kinetics, yet neither of them is reflected in the apparent time constants.
The two-component M decay with its strongly pH-dependent ratio of contributions (Figure 4D) could originate from at least three possible causes. First, it might reflect a superposition of kinetics from two separate photocycles (i.e., inhomogenity in the non-excited state), with pH-dependent relative contributions. Second, it might reflect interplay of pH-dependent and pH-independent reactions in shaping of the M↔N↔ transient equilibrium8,28,57,90,92. Third, it might reflect the presence of two distinct consecutive M-like intermediates in a single photocycle. The seemingly paradoxical combination of (nearly) pH-independent time constants (Figure 4C) and strongly pH-dependent amplitudes of the two components of M decay (Figure 4D) seems to favor the former two over the third option. The first option – the initial pH-dependent inhomogeneity resulting in two parallel photocycles – is always possible. However, such a strong assumption should be probably the last to be considered.
We believe that the second option, a complex interaction of pH-dependent and pH-independent steps in the M↔N→ segment of the photocycle, is the most promising one (see more in the next section). For the oversimplified M↔N→ sequence of intermediates, the relationship between the intrinsic and the apparent kinetics is described by a quadratic equation41,95, which is readily solved analytically. In this case the three measurable quantity – the two apparent rates of the M decay (Figure 4C) and the ratio of their corresponding amplitudes (Figure 4D) – define in full the three intrinsic rates involved, and the true intrinsic (rather than the apparent) rates for the (i) M→N, (ii) N→M and (iii) N→ transitions and, therefore, their pH-dependences could be explicitly recalculated41,95. Previous analysis of similar cases in HR95 and in the D96N mutant of BR41 resulted in a clear separation of the pH-dependent and pH-independent intrinsic contributions, thus confirming the initial hypothesis. In the case of ESR, however, the situation was found to be less straightforward: our attempt to recalculate the three intrinsic rates using the approach of41,95 failed to reveal the expected pH-independent step but instead resulted in intrinsic rates all three of which are pH-dependent, in violation of the hypothesis.
Insights from modeling of the photocycle kinetics and its pH-dependence
To explore the role of the proton donor for Schiff base reprotonation in the ESR photocycle, two sub-states, with and without protonated donor, X(+) and X(−), respectively, were included for each intermediate “X”. Excluding the steps up to formation of the L state, this results in the following two-dimensional (2D) extension of the photocycle scheme:
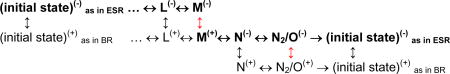 |
The vertical transitions are governed by pH, and the corresponding pKa’s at each step are the adjustable parameters. As we will argue below, the actual photocycle pathway in ESR (in bold) is different from that in BR with respect to the reactions that require proton equilibration with the bulk (marked in red). In ESR such a reaction affecting the donor occurs during the life-time of M, but in BR it takes place in the post-M states. Thus, for ESR we propose a sequence: ESR(−) … ↔ L(−)↔ M(−) ↔ M(+) ↔ N(−) ↔ N2(−) ↔ ESR(−), while for BR the corresponding (oversimplified) sequence will be: BR(+) … ↔ L(+) ↔ M(+) ↔ N(−) O(+) ↔ BR(+)12.
To reproduce the pH-dependence of the amplitudes of M decay in ESR (Figure 4D), proton uptake from the bulk has to occur either (i) during the life-time of M-like state, in the M(−) ↔ M(+) transition, (i.e., necessarily with an active participation of a donor), or (ii) during formation of N, in the M(−) ↔ N(−) transition (possibly bypassing a donor, in direct re-equilibration with the bulk). In the former case, the pKa of ~8.5 in Figure 4D is the pKa of the donor in the M-state; in the latter it corresponds to the pKa of the Schiff base during the M-to-N transition. The observed virtual pH-independence of the apparent time constants (Figure 4C) enforces further limitations. It could be reproduced in both cases if the overall rate of Schiff base reprotonation (in the pH range of the data in Figure 4C) is limited by a pH-independent component. This could be either an internal proton transfer from a donor or a rate-limiting step in the transport of the proton through an aqueous chain in the cytoplasmic half-channel. The latter, however, also implies an explicit proton donor, e.g., a water molecule or a network of water molecules, on which the proton transiently resides. Thus, a proton donor seems to be obligatory.
Importantly, the observed pH-dependence of the M decay (Figure 4D) could be reproduced only if the donor is unprotonated in the initial state but becomes transiently protonated during the cycle, and this causes the observed pH-dependence. Conversely, attempts to set the donor pKa such that the donor is initially protonated inevitably led to disappearance of the pH-dependence, reproducing the phenotype for M decay in BR where the donor is initially protonated14. Thus, the titration-like dependence of amplitudes (Figure 4D) is incompatible with a donor that is protonated already in the non-excited state, like Asp-96 in BR14, and indeed conditions were found where proton uptake, which protonates an internal donor in ESR, precedes reprotonation of the Schiff base85.
The mid-point of the pH-dependence of amplitudes (Figure 4D) and the inflection point in the pH-dependence of the time constants (for the wild-type ESR in Figure 7) differ by ~1 pH unit. If both were to originate from the same group, the two should coincide not only for the single elementary transition but for the photocycle as well. Therefore, the two are likely to reflect titrations of two different groups. The former, at ~8.5 (Figure 4D), most probably belongs to the donor(see also 86), while the latter, at ~9.5 (Figure 7), originates from the Schiff base. The observed difference between these two pKa values is explicitly the ΔpKa between that of the donor and the Schiff base in the M- and N-like state. The latter sets the upper limit to the ratio of the two components of the M decay, and is in accord with the observed ~85% level at low pH in Figure 4D.
The K96A phenotype
(Figure 6) and its difference from that of the wild-type ESR (Figure 7) are very similar to that observed in BR and its D96N-like mutants. Therefore, we tentatively interpret the switch with an apparent pKa of ~9.5 (Figure 7) from pH-independent to pH-dependent rate of M decay as a transition that reveals the pKa in M of the Schiff base itself. Previously, in BR evidence analogous to that in Figure 7 allowed proposing that Asp-96 is the primary donor for the Schiff base reprotonation28. FTIR spectroscopy confirmed this role for the Asp-96 in BR14 as well as a similar role for Glu-108 in PR17. In ESR the homologue of Asp-96 (in BR) is Lys-96, and unlike the well-separated strong band of the protonated carbonyl stretch of the Asp-96 in BR14, in ESR the bands of interest for the transiently protonated ε-amino group of Lys-96 are hidden under those of the abundant amino groups in protein. In the absence of direct evidence from IR, the difference between the phenotypes of the wild-type and K96A mutant could be interpreted in at least two ways. The first option is to assume that Lys-96 is the donor itself. In this case, the ε-amino group donates its exchangeable proton to the Schiff base during the M-to-N transition. The expected pKa’s for an initially buried lysine, and a lysine with increased exposure to water in the M state, as Asp-96 in BR96, would be consistent with protonation of Lys-96 from the bulk and transfer of its proton to the Schiff base85.
The second option is that the proton that reprotonates the Schiff base originates from a water molecule in the half-channel, which connects the deprotonated Schiff base to the bulk. Such half-channel is expected to contain water molecules in any case, and their existence seems to be a required mechanism for the Schiff base reprotonation in D96N-like mutants in BR, E108Q-like mutants in PR, and in the K96A mutant in ESR, and this is supported by molecular simulation studies45,97. If this is the case, the role of Lys-96 would be limited to maintaining this proton-conducting half-channel transiently, during the M-to-N transition, but the actual donor is a water molecule inside this half-channel.
Replacing Lys-96 (as in the K96A mutant) results in substantial deceleration of the photocycle (see Figure 7 and85) but this per se is not an evidence for the actual participation of an exchangeable proton of the ε-amino group of Lys-96 in the Schiff base reprotonation, as the function of the primary donor would have required. Whether the donor is Lys-96 or an aqueous network associated with Lys-96 cannot be decided from the available evidence at this time.
Comparison of the ESR vs. BR phenotypes
Above the apparent pKa for M accumulation, ESR and BR behave very similarly: (i) the light energy absorbed by retinal destabilizes the proton of the Schiff base; (ii) the proton, released by the Schiff base, is captured by the primary acceptor, Asp-85; (iii) the proton release occurs late in the photocycle48 similar to BR at pH below the pKa for proton release98 and in BR mutants lacking the native extracellular proton-releasing complex65–67,99; and (iv) the photocycle is finished when the Schiff base is reprotonated and the retinal is reisomerized.
Unlike in the higher pH region, ESR differs markedly (Figure 3) from BR below the apparent pKa for M accumulation. Specifically, transient accumulation of the M state, which is crucial for proton pumping, is apparently absent (Figures 3A and 4B). However, in spite of this, post-M states are formed (Figure 5C), and we, therefore, conclude that at low pH the ESR cycle does include the two crucial steps: (i) Schiff base deprotonation with proton transfer to Asp-85, and (ii) the subsequent Schiff base reprotonation by proton from a different donor, which constitute the general paradigm of proton pumps in all retinal proteins. This is consistent with the interpretation of the titration that revealed that Asp-85 is mostly unprotonated in this pH region48, and available, although less effectively than at higher pH, as proton acceptor. The detection of post-M states in the photocycle below the apparent pKa of M accumulation (Figure 5C) provides a rationale for our earlier observation48 that proton transport is by no means abolished in this pH region.
At the functional level ESR is different from BR in several important aspects, however. The first is the structure of the counterion to the chromophore, the Asp-85/His-57 complex48 with a sufficiently high pKa, of ~6.5 in the liposomes that creates an influence on the photocycle near physiological pH, which has no analogue in BR. The second is the altered pKa’s of both the Schiff base and that of the proton-donating group or the chain employed in the reprotonation of the Schiff base both in the initial state and in intermediates. The third is the timing of the retinal reisomerization, which does not allow formation of the all-trans conformation, associated with an O-like state, until the very end of the photocycle.
The familiar one-dimensional (1D) sequence of K-L-M-N-O intermediates of the BR photocycle is actually an oversimplification. It tracks predominantly the Schiff base dynamics, and in BR the particular sequence of steps is defined by the interplay of BR-specific rate-limiting steps and the transiently changed pKa’s of the acceptor and the donor12. However, when the transiently changed protonation status of the other two crucial groups, the primary acceptor and the primary donor, are explicitly taken into account, the full road-map of the paths for the possible photocycles is even more complex than that the 2D scheme presented above in discussion of modeling. It is three-dimensional (3D). The specific path employed in BR is just one of those many, and the one in ESR is different from it (see above). Thus, each of the first three sources of differences listed above could lead to an altered path, different from that of BR, creating a photocycle, which might run in full accord with the BR principles but nevertheless along a different path. In ESR the one mostly evident difference is that the initial (non-excited) state of the photocycle is different from that of BR in respect to the protonation state of the group/pool from which the proton, which reprotonates the Schiff base, originates. The primary donor, Asp-96, is initially protonated in BR but the group that constitutes its functional homologue in the wild-type ESR is initially deprotonated.
Additional complications for direct comparison between the ESR and the BR photocycles are the strongly altered spectral features of individual intermediates. First, the differential absorption of the M-like state in ESR is such that its formation could be noticed by absorption changes at and above 590 nm, i.e. in the region used for monitoring the red-shifted states. Similarly, the formation of the late red-shifted states leads to significant absorption changes at and below 420 nm where the blue-shifted M-like states are monitored. In both cases this cross-influence in ESR is stronger than in BR. Second, the strongly red-shifted absorption spectrum of the N-like state(s) in the visible has little in common with the N state in BR82 but rather mimics that of the O-like state. Third, the ESR photocycle is much more sensitive to the local environment -partial dehydration (data not shown), solubilization with detergents (compare48,85), or even incorporation in a different lipid100 – than that of BR. All these features contribute to creating an impression of a photocycle that seems to differ from that of BR much stronger than it actually does.
The apparent phenotype of ESR is shaped by its specific structural features. However, once those are properly accounted for, its proton-pumping mechanism seems to be in full accord with the general rules common to all retinal proteins, which function as proton pumps.
Conclusions
While the apparent pKa values are sensitive to the protein environment, the phenotype of the ESR photocycle described in this paper for the protein incorporated in the lipid environment is nearly indistinguishable (once the difference in pKa’s is accounted for) to that described for the DDM-48 and the LysoPG-85 solubilized samples, reflecting the intrinsic events of the photocycle not masked by the specific environment.
We report detection, separation and characterization of all the main expected intermediates of the ESR photocycle. Above the apparent pKa ~6.5 for M accumulation these are: (i) a pre-K state not photo-reconvertable even at 80 K; (ii) at least two distinct K-like states; (iii) the elusive L-like state; (iv) the blue-shifted M-like state; (v) the moderately red-shifted N1 state; followed by (vi) a strongly red-shifted N2 state, and later (vii) a weakly red-shifted N3 state. The last one, N3, is always present only as a minority contribution, and may or may not be an O-like state.
At pH below the pKa ~6.5 instead of the blue-shifted M-like state(s) an additional red-shifted intermediate was revealed by kinetic analysis.
The apparent dynamics of the crucial intermediate for proton pumping, the M-like state that indicates the deprotonated Schiff base, is affected in the wild-type ESR by two major pH-dependent transitions with pKa of ~6.5 and ~8.5. Both affect the apparent amplitudes of kinetic components. Paradoxically, neither of those is reflected in the virtually pH-independent apparent rate constants.
A curious and counterintuitive feature of the wild-type ESR photocycle is that in spite of the observed lack of accumulation of the M-like state(s) at pH below the pKa of ~6.5, IR spectra revealed the presence of post-M intermediates with protonated Asp-85 in this pH region, confirming our earlier hypothesis48 that M is still produced even when not accumulated at low pH.
Detection of post-M states below the apparent pKa ~6.5 of the transient M accumulation reveals that Asp-85 is able to undergo protonation. This is independent confirmation of our earlier proposal48 that prior to excitation Asp-85 is indeed mostly in the unprotonated state in this pH range.
The second half of the ESR photocycle is dominated by the presence of several red-shifted intermediates. Reassignment of these states from the O-like into the N-like ones helps to properly correlate the time-resolved spectroscopic changes with mechanistic understanding of underlying processes.
Unlike in BR, in ESR the proton that reprotonates the Schiff base during the M-to-N reaction is not provided by an initially protonated donor group inside the protein but rather a proton has to be taken up from the bulk on the time scale of M decay.
The phenotype of the K96A mutant is in full accord with the corresponding D96-like mutants in BR, and PR.
The presence of lysine in position 96 is crucial for the efficiency of the proton-transporting chain during reprotonation of the Schiff base, indicating that the ε-amino group of Lys-96 might be the source of the exchangeable proton that reprotonates the Schiff base.
Alternatively, this proton might rather originate from a water molecule in the half-channel connecting the Schiff base to the bulk, while the Lys-96 presence is required only for the maintenance of this water-filled half-channel.
In ESR the cis-to-trans re-isomerization in the second half of the photocycle is impeded, and seems to be the slowest process of the photocycle. As a result, a distinct O-like state, which is formed in BR after cis-to-trans re-isomerization, is either entirely absent or is present only as a minor contribution at the very end of the photocycle.
Although the apparent kinetics and pH-dependences of the photocycles of ESR and BR differ significantly, once the differences in the pKa’s of their functionally important groups are properly taken into account the phenotype of the ESR is shaped by the same fundamental mechanisms common to all the retinal protein proton pumps.
Acknowledgments
The authors acknowledge support from the National Institutes of Health (GM029498 to J.K.L) to fund research on mechanisms of functioning and FTIR spectroscopy, the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the DOE (DE-FG02-86ER13525 to J.K.L) to fund time-resolved spectroscopy in the visible, Federal Targeted Program on Scientific and Scientific-Pedagogical Staff of Innovative Russia (to L.E.P.) to fund construction and purification of mutants.
References
- 1.Oesterhelt D, Stoeckenius W. Rhodopsin-Like Protein from the Purple Membrane of Halobacterium Halobium. Nature – New Biology. 1971;233:149–152. doi: 10.1038/newbio233149a0. [DOI] [PubMed] [Google Scholar]
- 2.Oesterhelt D, Stoeckenius W. Functions of a New Photoreceptor Membrane. Proc Natl Acad Sci USA. 1973;70:2853–2857. doi: 10.1073/pnas.70.10.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spudich JL, Yang CS, Jung KH, Spudich EN. Retinylidene Proteins: Structures and Functions from Archaea to Humans. Annu Rev Cell Dev Biol. 2000;16:365–392. doi: 10.1146/annurev.cellbio.16.1.365. [DOI] [PubMed] [Google Scholar]
- 4.Brown LS, Jung KH. Bacteriorhodopsin-Like Proteins of Eubacteria and Fungi: The Extent of Conservation of the Haloarchael Proton-Pumping Mechanism. Photochem Photobiol Sci. 2006;5:538–546. doi: 10.1039/b514537f. [DOI] [PubMed] [Google Scholar]
- 5.Lanyi JK. Bacteriorhodopsin. Annu Rev Physiol. 2004;66:665–688. doi: 10.1146/annurev.physiol.66.032102.150049. [DOI] [PubMed] [Google Scholar]
- 6.Lanyi JK. Proton Transfers in the Bacteriorhodopsin Photocycle. Biochim Biophys Acta. 2006;1757:1012–1018. doi: 10.1016/j.bbabio.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Schobert B, Lanyi JK. Halorhodopsin is a Light-Driven Chloride Pump. J Biol Chem. 1982;257:10306–10313. [PubMed] [Google Scholar]
- 8.Lozier RH, Bogomolni RA, Stoeckenius W. Bacteriorhodopsin: A Light-Driven Proton Pump in Halobacterium Halobium. Biophys J. 1975;15:955–962. doi: 10.1016/S0006-3495(75)85875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartmann R, Oesterhelt D. Bacteriorhodopsin-Mediated Photophosphorylation in Halobacterium Halobium. Eur J Biochem. 1977;77:325–335. doi: 10.1111/j.1432-1033.1977.tb11671.x. [DOI] [PubMed] [Google Scholar]
- 10.Birge RR, Cooper TM. Energy Storage In The Primary Step of the Photocycle of Bacteriorhodopsin. Biophys J. 1983;42:61–69. doi: 10.1016/S0006-3495(83)84369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birge RR, Cooper TM, Lawrence AF, Masthay MB, Zhang C-F, Zidovetzki R. Revised Assignment of Energy Storage in the Primary Photochemical Event in Bacteriorhodopsin. J Am Chem Soc. 1991;113:4327–4328. [Google Scholar]
- 12.Dioumaev AK. Infrared Methods for Monitoring the Protonation State of Carboxylic Amino Acids in the Photocycle of Bacteriorhodopsin. Biochemistry (Moscow) 2001;66:1269–1276. doi: 10.1023/a:1013135419529. [DOI] [PubMed] [Google Scholar]
- 13.Braiman MS, Mogi T, Marti T, Stern LJ, Khorana HG, Rothschild KJ. Vibrational Spectroscopy of Bacteriorhodopsin Mutants: Light- Driven Proton Transport Involves Protonation Changes of Aspartic Acid Residues 85, 96, and 212. Biochemistry. 1988;27:8516–8520. doi: 10.1021/bi00423a002. [DOI] [PubMed] [Google Scholar]
- 14.Gerwert K, Hess B, Soppa J, Oesterhelt D. Role of Aspartate-96 in Proton Translocation by Bacteriorhodopsin. Proc Natl Acad Sci USA. 1989;86:4943–4947. doi: 10.1073/pnas.86.13.4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braiman MS, Bousché O, Rothschild KJ. Protein Dynamics in the Bacteriorhodopsin Photocycle: Submillisecond Fourier Transform Infrared Spectra of the L, M, And N Photointermediates. Proc Natl Acad Sci USA. 1991;88:2388–2392. doi: 10.1073/pnas.88.6.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fahmy K, Weidlich O, Engelhard M, Tittor J, Oesterhelt D, Siebert F. Identification of the Proton Acceptor of Schiff Base Deprotonation in Bacteriorhodopsin: A Fourier-Transform Infrared Study of the Mutant Asp85 --> Glu in its Natural Lipid Environment. Photochem Photobiol. 1992;56:1073–1083. [Google Scholar]
- 17.Dioumaev AK, Brown LS, Shih J, Spudich EN, Spudich JL, Lanyi JK. Proton Transfers in the Photochemical Reaction Cycle of Proteorhodopsin. Biochemistry. 2002;41:5348–5358. doi: 10.1021/bi025563x. [DOI] [PubMed] [Google Scholar]
- 18.Miranda MR, Choi AR, Shi L, Bezerra AG, Jr, Jung KH, Brown LS. The Photocycle and Proton Translocation Pathway in a Cyanobacterial Ion-Pumping Rhodopsin. Biophys J. 2009;96:1471–1481. doi: 10.1016/j.bpj.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waschuk SA, Bezerra AG, Jr, Shi L, Brown LS. Leptosphaeria Rhodopsin: Bacteriorhodopsin-Like Proton Pump From a Eukaryote. Proc Natl Acad Sci USA. 2005102:6879–6883. doi: 10.1073/pnas.0409659102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer U, Oesterhelt D. Chromophore Equilibria in Bacteriorhodopsin. Biophys J. 1979;28:211–230. doi: 10.1016/S0006-3495(79)85172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramaniam S, Marti T, Khorana HG. Protonation State of Asp (Glu)-85 Regulates the Purple-to-Blue Transition in Bacteriorhodopsin Mutants Arg-82-->Ala and Asp-85-->Glu: The Blue Form is Inactive in Proton Translocation. Proc Natl Acad Sci USA. 1990;87:1013–1017. doi: 10.1073/pnas.87.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metz G, Siebert F, Engelhard M. High-Resolution Solid State 13C NMR of Bacteriorhodopsin: Characterization of [4-13C]Asp Resonances. Biochemistry. 1992;31:455–462. doi: 10.1021/bi00117a022. [DOI] [PubMed] [Google Scholar]
- 23.Balashov SP, Imasheva ES, Govindjee R, Ebrey TG. Titration of Aspartate-85 in Bacteriorhodopsin: What it Says About Chromophore Isomerization and Proton Release. Biophys J. 1996;70:473–481. doi: 10.1016/S0006-3495(96)79591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Száraz S, Oesterheit D, Ormos P. pH-Induced Structural-Changes in Bacteriorhodopsin Studied by Fourier-Transform Infrared-Spectroscopy. Biophys J. 1994;67:1706–1712. doi: 10.1016/S0006-3495(94)80644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braiman MS, Dioumaev AK, Lewis JR. A Large Photolysis-Induced pKa Increase of the Chromophore Counterion in Bacteriorhodopsin: Implications For Ion Transport Mechanisms of Retinal Proteins. Biophys J. 1996;70:939–947. doi: 10.1016/S0006-3495(96)79637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zscherp C, Schlesinger R, Tittor J, Oesterhelt D, Heberle J. In Situ Determination of Transient pKa Changes of Internal Amino Acids of Bacteriorhodopsin By Using Time-Resolved Attenuated Total Reflection Fourier-Transform Infrared Spectroscopy. Proc Natl Acad Sci USA. 1999;96:5498–5503. doi: 10.1073/pnas.96.10.5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balashov SP, Imasheva ES, Boichenko VA, Anton J, Wang JM, Lanyi JK. Xanthorhodopsin: A Proton Pump With a Light-Harvesting Carotenoid Antenna. Science. 2005;309:2061–2064. doi: 10.1126/science.1118046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otto H, Marti T, Holz M, Mogi T, Lindau M, Khorana HG, Heyn MP. Aspartic Acid-96 Is The Internal Proton Donor in the Reprotonation of the Schiff Base of Bacteriorhodopsin. Proc Natl Acad Sci USA. 1989;86:9228–9232. doi: 10.1073/pnas.86.23.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butt H-J, Fendler K, Bamberg E, Tittor J, Oesterhelt D. Aspartic Acids 96 and 85 Play a Central Role in the Function of Bacteriorhodopsin as a Proton Pump. EMBO J. 1989;8:1657–1663. doi: 10.1002/j.1460-2075.1989.tb03556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imasheva ES, Balashov SP, Wang JM, Dioumaev AK, Lanyi JK. Selectivity of Retinal Photoisomerization in Proteorhodopsin is Controlled by Aspartic Acid 227. Biochemistry. 2004;43:1648–1655. doi: 10.1021/bi0355894. [DOI] [PubMed] [Google Scholar]
- 31.Smith SO, Lugtenburg J, Mathies RA. Determination of Retinal Chromophore Structure in Bacteriorhodopsin with Resonance Raman Spectroscopy. J Mem Biol. 1985;85:95–109. doi: 10.1007/BF01871263. [DOI] [PubMed] [Google Scholar]
- 32.Fodor SPA, Ames JB, Gebhard R, Van Den Berg EMM, Stoeckenius W, Lugtenburg J, Mathies RA. Chromophore Structure in Bacteriorhodopsin’s N Intermediate: Implications for the Proton-Pumping Mechanism. Biochemistry. 1988;27:7097–7101. doi: 10.1021/bi00418a064. [DOI] [PubMed] [Google Scholar]
- 33.Sasaki J, Lanyi JK, Needleman R, Yoshizawa T, Maeda A. Complete Identification of C=O Stretching Vibrational Bands of Protonated Aspartic Acid Residues in the Difference Infrared Spectra of M and N Intermediates Versus Bacteriorhodopsin. Biochemistry. 1994;33:3178–3184. doi: 10.1021/bi00177a006. [DOI] [PubMed] [Google Scholar]
- 34.Smith SO, Pardoen JA, Lugtenburg J, Curry B, Mathies RA. Chromophore Structure in Bacteriorhodopsin’s O640 Photointermediate. Biochemistry. 1983;22:6141–6148. [Google Scholar]
- 35.Otto H, Marti T, Holz M, Mogi T, Stern LJ, Engel F, Khorana HG, Heyn MP. Substitution of Amino Acids Asp-85, Asp-212, and Arg-82 in Bacteriorhodopsin Affects the Proton Release Phase of the Pump and the pKa of the Schiff Base. Proc Natl Acad Sci USA. 1990;87:1018–1022. doi: 10.1073/pnas.87.3.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drachev LA, Kaulen AD, Skulachev VP. Time Resolution of the Intermediate Steps in the Bacteriorhodopsin-Linked Electrogenesis. FEBS Lett. 1978;87:161–167. doi: 10.1016/0014-5793(78)80157-4. [DOI] [PubMed] [Google Scholar]
- 37.Mowery PC, Lozier RH, Chae Q, Tseng YW, Taylor M, Stoeckenius W. Effect of Acid pH on the Absorption Spectra and Photoreactions of Bacteriorhodopsin. Biochemistry. 1979;18:4100–4107. doi: 10.1021/bi00586a007. [DOI] [PubMed] [Google Scholar]
- 38.Liu SY. Light-Induced Currents from Oriented Purple Membrane. 1. Correlation of the Microsecond Component (B2) with the L-M Photocycle Transition. Biophys J. 1990;57:943–950. doi: 10.1016/S0006-3495(90)82614-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dioumaev AK, Wang JM, Bálint Z, Váró G, Lanyi JK. Proton Transport by Proteorhodopsin Requires That the Retinal Schiff Base Counterion Asp-97 Be Anionic. Biochemistry. 2003;42:6582–6592. doi: 10.1021/bi034253r. [DOI] [PubMed] [Google Scholar]
- 40.Miller A, Oesterhelt D. Kinetic Optimization of Bacteriorhodopsin by Aspartic Acid 96 as an Internal Proton Donor. Biochim Biophys Acta. 1990;1020:57–64. [Google Scholar]
- 41.Brown LS, Lanyi JK. Determination of the Transiently Lowered pKa of the Retinal Schiff Base During the Photocycle of Bacteriorhodopsin. Proc Natl Acad Sci USA. 1996;93:1731–1734. doi: 10.1073/pnas.93.4.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kandori H. Role of Internal Water Molecules in Bacteriorhodopsin. Biochim Biophys Acta. 2000;1460:177–191. doi: 10.1016/s0005-2728(00)00138-9. [DOI] [PubMed] [Google Scholar]
- 43.Furutani Y, Shibata M, Kandori H. Strongly Hydrogen-Bonded Water Molecules in the Schiff Base Region of Rhodopsins. Photochem Photobiol Sci. 2005;4:661–666. doi: 10.1039/b416698a. [DOI] [PubMed] [Google Scholar]
- 44.Garczarek F, Gerwert K. Functional Waters in Intraprotein Proton Transfer Monitored by FTIR Difference Spectroscopy. Nature. 2006;439:109–112. doi: 10.1038/nature04231. [DOI] [PubMed] [Google Scholar]
- 45.Freier E, Wolf S, Gerwert K. Proton Transfer Via a Transient Linear Water-Molecule Chain in a Membrane Protein. Proc Natl Acad Sci USA. 2011;128:11435–11439. doi: 10.1073/pnas.1104735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Béjà O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich SB, Gates CM, Feldman RA, Spudich JL, et al. Bacterial Rhodopsin: Evidence For a New Type of Phototrophy in the Sea. Science. 2000;289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- 47.Luecke H, Schobert B, Stagno J, Imasheva ES, Wang JM, Balashov SP, Lanyi JK. Crystallographic Structure of Xanthorhodopsin, the Light-Driven Proton Pump With a Dual Chromophore. Proc Natl Acad Sci USA. 2008;105:16561–16565. doi: 10.1073/pnas.0807162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balashov SP, Petrovskaya LE, Lukashev EP, Imasheva ES, Dioumaev AK, Wang JM, Sychev SV, Dolgikh DA, Rubin AB, Kirpichnikov MP, et al. Aspartate-Histidine Interaction in the Retinal Schiff Base Counterion of the Light-Driven Proton Pump of Exiguobacterium Sibiricum. Biochemistry. 2012;51:5748–5762. doi: 10.1021/bi300409m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergo VB, Sineshchekov OA, Kralj JM, Partha R, Spudich EN, Rothschild KJ, Spudich JL. His-75 in Proteorhodopsin, a Novel Component in Light-Driven Proton Translocation by Primary Pumps. J Biol Chem. 2009;284:2836–2843. doi: 10.1074/jbc.M803792200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hempelmann F, Hoelper S, Verhoefen M-S, Woerner AC, Fiedler S-A, Pfleger N, Wachtveitl J, Glaubitz C. His75-Asp97 Cluster in Green Proteorhodopsin. J Am Chem Soc. 2011;133:4645–4654. doi: 10.1021/ja111116a. [DOI] [PubMed] [Google Scholar]
- 51.Petrovskaya LE, Lukashev EP, Chupin VV, Sychev SV, Lyukmanova EN, Kryukova EA, Ziganshin RH, Spirina EV, Rivkina EM, Khatypov RA, et al. Predicted Bacteriorhodopsin from Exiguobacterium Sibiricum Is a Functional Proton Pump. FEBS Lett. 2010;584:4193–4196. doi: 10.1016/j.febslet.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 52.Luecke H, Schobert B, Richter H-T, Cartailler JP, Lanyi JK. Structure of Bacteriorhodopsin at 1.55 Å Resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 53.Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. Untangling Genomes From Metagenomes: Revealing an Uncultured Class of Marine Euryarchaeota. Science. 2012;335:587–590. doi: 10.1126/science.1212665. [DOI] [PubMed] [Google Scholar]
- 54.Imasheva ES, Balashov SP, Wang JM, Lanyi JK. pH-Dependent Transitions in Xanthorhodopsin. Photochem Photobiol. 2006;82:1406–1413. doi: 10.1562/2006-01-15-RA-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodrigues DF, Ivanova N, He Z, Huebner M, Zhou J, Tiedje JM. Architecture of Thermal Adaptation in an Exiguobacterium Sibiricum Strain Isolated From 3 Million Year Old Permafrost: A Genome and Transcriptome Approach. BMC Genomics. 2008;9:547–56. doi: 10.1186/1471-2164-9-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dioumaev AK, Lanyi JK. Two Bathointermediates of the Bacteriorhodopsin Photocycle, From Time-Resolved Nanosecond Spectra in the Visible. J Phys Chem B. 2009;113:16643–16653. doi: 10.1021/jp907393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dioumaev AK. Evaluation of Intrinsic Chemical Kinetics and Transient Product Spectra from Time-Resolved Spectroscopic Data. Biophys Chem. 1997;67:1–25. doi: 10.1016/s0301-4622(96)02268-5. [DOI] [PubMed] [Google Scholar]
- 58.Chizhov I, Chernavskii DS, Engelhard M, Müller K-H, Zubov BV, Hess B. Spectrally Silent Transitions in the Bacteriorhodopsin Photocycle. Biophys J. 1996;71:2329–2345. doi: 10.1016/S0006-3495(96)79475-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dioumaev AK, Lanyi JK. Infrared Monitoring of Interlayer Water in Stacks of Purple Membranes. Photochem Photobiol. 2009;85:598–608. doi: 10.1111/j.1751-1097.2008.00512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dioumaev AK, Lanyi JK. Bacteriorhodopsin Photocycle at Cryogenic Temperatures Reveals Distributed Barriers of Conformational Substates. Proc Natl Acad Sci USA. 2007;104:9621–9626. doi: 10.1073/pnas.0703859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dioumaev AK, Lanyi JK. Switch from Conventional to Distributed Kinetics in the Bacteriorhodopsin Photocycle. Biochemistry. 2008;47:11125–11133. doi: 10.1021/bi801247e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dioumaev AK, Wang JM, Lanyi JK. Low-Temperature FTIR Study of Multiple K Intermediates in the Photocycles of Bacteriorhodopsin and Xanthorhodopsin. J Phys Chem B. 2010;114:2920–2931. doi: 10.1021/jp908698f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Larkin M, Brown NP, Chenna R, Mcgettigan PA, Mcwilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, et al. Clustal W and Clustal X Version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 64.Dunn RJ, Mccoy J, Simsek M, Majumdar A, Chang SH, Rajbhandary UL, Khorana HG. The Bacteriorhodopsin Gene. Proc Natl Acad Sci USA. 1981;78:6744–6748. doi: 10.1073/pnas.78.11.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dioumaev AK, Brown LS, Needleman R, Lanyi JK. Fourier Transform Infrared Spectra of a Late Intermediate of the Bacteriorhodopsin Photocycle Suggest Transient Protonation of Asp- 212. Biochemistry. 1999;38:10070–10078. doi: 10.1021/bi990873+. [DOI] [PubMed] [Google Scholar]
- 66.Brown LS, Sasaki J, Kandori H, Maeda A, Needleman R, Lanyi JK. Glutamic Acid 204 Is the Terminal Proton Release Group at the Extracellular Surface of Bacteriorhodopsin. J Biol Chem. 1995;270:27122–27126. doi: 10.1074/jbc.270.45.27122. [DOI] [PubMed] [Google Scholar]
- 67.Balashov SP, Imasheva ES, Ebrey TG, Chen N, Menick DR, Crouch RK. Glutamate-194 to Cysteine Mutation Inhibits Fast Light-Induced Proton Release in Bacteriorhodopsin. Biochemistry. 1997;36:8671–8676. doi: 10.1021/bi970744y. [DOI] [PubMed] [Google Scholar]
- 68.Dioumaev AK, Richter H-T, Brown LS, Tanio M, Tuzi S, Saitô H, Kimura Y, Needleman R, Lanyi JK. Existence of a Proton Transfer Chain in Bacteriorhodopsin: Participation of Glu-194 in the Release of Protons to the Extracellular Surface. Biochemistry. 1998;37:2496–2506. doi: 10.1021/bi971842m. [DOI] [PubMed] [Google Scholar]
- 69.Balashov SP. Protonation Reactions and their Coupling in Bacteriorhodopsin. Biochim Biophys Acta. 2000;1460:75–94. doi: 10.1016/s0005-2728(00)00131-6. [DOI] [PubMed] [Google Scholar]
- 70.Garczarek F, Brown LS, Lanyi JK, Gerwert K. Proton Binding Within a Membrane Protein by a Protonated Water Cluster. Proc Natl Acad Sci USA. 2005102:3633–3638. doi: 10.1073/pnas.0500421102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maeda A. Application of FTIR Spectroscopy to the Structural Study on the Function of Bacteriorhodopsin. Israel J Chem. 1995;35:387–400. [Google Scholar]
- 72.Bagley K, Dollinger G, Eisenstein L, Singh AK, Zimányi L. Fourier Transform Infrared Difference Spectroscopy of Bacteriorhodopsin and Its Photoproducts. Proc Natl Acad Sci USA. 1982;79:4972–4976. doi: 10.1073/pnas.79.16.4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Balashov SP, Ebrey TG. Trapping and Spectroscopic Identification of the Photointermediates of Bacteriorhodopsin at Low Temperatures. Photochem Photobiol. 2001;73:453–462. doi: 10.1562/0031-8655(2001)073<0453:tasiot>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 74.Rothschild KJ, Marrero H. Infrared Evidence That the Schiff Base of Bacteriorhodopsin is Protonated: Br570 and K Intermediates. Proc Natl Acad Sci USA. 1982;79:4045–4049. doi: 10.1073/pnas.79.13.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weidlich O, Siebert F. Time Resolved Step Scan FTIR Investigations of the Transition from KL to L in the Bacteriorhodopsin Photocycle. Identification of Chromophore Twists by Assigning Hydrogen Out Of Plane (HOOP) Bending Vibrations. Appl Spectrosc. 1993;47:1394–1400. [Google Scholar]
- 76.Sasaki J, Yuzawa T, Kandori H, Maeda A, Hamaguchi H. Nanosecond Time-Resolved Infrared Spectroscopy Distinguishes Two K Species in the Bacteriorhodopsin Photocycle. Biophys J. 1995;68:2073–2080. doi: 10.1016/S0006-3495(95)80386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dioumaev AK, Braiman MS. Two Bathointermediates in the Bacteriorhodopsin Photocycle, Distinguished by Nanosecond Time-Resolved FTIR Spectroscopy. J Phys Chem B. 1997;101:1655–1662. [Google Scholar]
- 78.Siebert F, Mäntele W. Investigation of the Primary Photochemistry of Bacteriorhodopsin by Low-Temperature Fourier-Transform Infrared Spectroscopy. Eur J Biochem. 1983;130:565–573. doi: 10.1111/j.1432-1033.1983.tb07187.x. [DOI] [PubMed] [Google Scholar]
- 79.Hashimoto K, Choi AR, Furutani Y, Jung KH, Kandori H. Low-Temperature FTIR Study Of Gloeobacter Rhodopsin: Presence of Strongly Hydrogen-Bonded Water and Long-Range Structural Protein Perturbation upon Retinal Photoisomerization. Biochemistry. 2010 doi: 10.1021/bi100184k. [DOI] [PubMed] [Google Scholar]
- 80.Zscherp C, Heberle J. Infrared Difference Spectra of the Intermediates L, M, N, and O of the Bacteriorhodopsin Photoreaction Obtained by Time-Resolved Attenuated Total Reflection Spectroscopy. J Phys Chem B. 1997;101:10542–10547. [Google Scholar]
- 81.Siebert F, Rödig C. Introduction To Step-Scan FTIR. 1. Bruker Optik GmBH; Rudolf-Plank-Str. 23, D76275 Ettingen, Germany: 2000. pp. 45–52. Chapter H. [Google Scholar]
- 82.Hessling B, Souvignier G, Gerwert K. A Model-Independent Approach to Assigning Bacteriorhodopsin’s Intramolecular Reactions to Photocycle Intermediates. Biophys J. 1993;65:1929–1941. doi: 10.1016/S0006-3495(93)81264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gergely C, Zimányi L, Váró G. Bacteriorhodopsin Intermediate Spectra Determined over a Wide pH Range. J Phys Chem B. 1997;101:9390–9395. [Google Scholar]
- 84.Chen WG, Braiman MS. Kinetic Analysis of Time-Resolved Infrared Difference Spectra of the L Intermediates and M Intermediates of Bacteriorhodopsin. Photochem Photobiol. 1991;54:905–910. [Google Scholar]
- 85.Maeda A, Sasaki J, Shichida Y, Yoshizawa T, Chang M, Ni B, Needleman R, Lanyi JK. Structures of Aspartic Acid-96 in the L and N Intermediates of Bacteriorhodopsin: Analysis by Fourier Transform Infrared Spectroscopy. Biochemistry. 1992;31:4684–4690. doi: 10.1021/bi00134a022. [DOI] [PubMed] [Google Scholar]
- 86.Balashov SP, Petrovskaya LE, Imasheva ES, Lukashev EP, Dioumaev AK, Wang JM, Sychev SV, Dolgikh DA, Rubin AB, Kirpichnikov MP, et al. Breaking the Carboxyl Rule: Lysine-96 Facilitates Reprotonation of the Schiff Base in the Photocycle of a Retinal Protein From Exiguobacterium Sibiricum. J Biol Chem. 2013 doi: 10.1074/jbc.M113.465138. DOI-code: dx.doi.org/10.1074/jbc.M113.465138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cao Y, Váró G, Klinger AL, Czajkowsky DM, Braiman MS, Needleman R, Lanyi JK. Proton Transfer From Asp-96 to the Bacteriorhodopsin Schiff Base Is Caused by a Decrease of the pKa of Asp-96 which Follows a Protein Backbone Conformational Change. Biochemistry. 1993;32:1981–1990. doi: 10.1021/bi00059a015. [DOI] [PubMed] [Google Scholar]
- 88.Cao Y, Váró G, Chang M, Ni BF, Needleman R, Lanyi JK. Water Is Required for Proton Transfer from Aspartate-96 to the Bacteriorhodopsin Schiff Base. Biochemistry. 1991;30:10972–10979. doi: 10.1021/bi00109a023. [DOI] [PubMed] [Google Scholar]
- 89.Dioumaev AK, Savransky VV, Tkachenko NV, Chukharev VI. Qunatum Yield and Extinction Measurements in Strongly Overlapping Reactant and Photoproduct Absorption Bands. II: Bathointermediate Formation in Bacteriorhodopsin Photocycle at Room Temperature. J Photochem Photobiol B:Biology. 1989;3:397–410. [Google Scholar]
- 90.Zimányi L, Lanyi JK. Deriving the Intermediate Spectra and Photocycle Kinetics from Time-Resolved Difference Spectra of Bacteriorhodopsin. The Simpler Case of the Recombinant D96N Protein. Biophys J. 1993;64:240–251. doi: 10.1016/S0006-3495(93)81360-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brown LS, Needleman R, Lanyi JK. Interaction of Proton and Chloride Transfer Pathways in Recombinant Bacteriorhodopsin with Chloride Transport Activity: Implications for the Chloride Translocation Mechanism. Biochemistry. 1996;35:16048–16054. doi: 10.1021/bi9622938. [DOI] [PubMed] [Google Scholar]
- 92.Váró G, Brown LS, Lakatos M, Lanyi JK. Characterization of the Photochemical Reaction Cycle of Proteorhodopsin. Biophys J. 2003;84:1202–1207. doi: 10.1016/S0006-3495(03)74934-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ames JB, Mathies RA. The Role of Back-Reactions and Proton Uptake During the N->O Transition in Bacteriorhodopsin’s Photocycle: A Kinetic Resonance Raman Study. Biochemistry. 1990;29:7181–7190. doi: 10.1021/bi00483a005. [DOI] [PubMed] [Google Scholar]
- 94.Váró G, Lanyi JK. Photoreactions of Bacteriorhodopsin at Acid pH. Biophys J. 1989;56:1143–1151. doi: 10.1016/S0006-3495(89)82761-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kobayashi T, Ohtani H, Iwai J-I, Ikegami A, Uchiki H. Effect of pH on the Photoreaction Cycles of Bacteriorhodopsin. FEBS Lett. 1983;162:197–200. [Google Scholar]
- 96.Zimányi L, Lanyi JK. Transient Spectroscopy of Bacterial Rhodopsins with an Optical Multichannel Analyzer. 2. Effects of Anions on the Halorhodopsin Photocycle. Biochemistry. 1989;28:5172–5178. doi: 10.1021/bi00438a039. [DOI] [PubMed] [Google Scholar]
- 97.Schobert B, Brown LS, Lanyi JK. Crystallographic Intermediates of Structures of the M and N Bacteriorhodopsin: Assembly of a Hydrogen-Bonded Chain of Water Molecules Between Asp-96 and the Retinal Schiff Base. J Mol Biol. 2003;330:553–570. doi: 10.1016/s0022-2836(03)00576-x. [DOI] [PubMed] [Google Scholar]
- 98.Wang T, Sessions AO, Lunde CS, Rouhani S, Glaeser RM, Duan Y, Facciotti MT. Deprotonation of D96 in Bacteriorhodopsin Opens the Proton Uptake Pathway. Structure. 2013;21:290–297. doi: 10.1016/j.str.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zimányi L, Váró G, Chang M, Ni B, Needleman R, Lanyi JK. Pathways of Proton Release in the Bacteriorhodopsin Photocycle. Biochemistry. 1992;31:8535–8543. doi: 10.1021/bi00151a022. [DOI] [PubMed] [Google Scholar]
- 100.Balashov SP, Govindjee R, Kono M, Imasheva ES, Lukashev EP, Ebrey TG, Crouch RK, Menick DR, Feng Y. Effect of the Arginine-82 to Alanine Mutation in Bacteriorhodopsin on Dark Adaptation, Proton Release, and the Photochemical Cycle. Biochemistry. 1993;32:10331–10343. doi: 10.1021/bi00090a008. [DOI] [PubMed] [Google Scholar]
- 101.Lyukmanova EN, Shenkarev ZO, Khabibullina NF, Kopeina GS, Shulepko MA, Paramonov AS, Mineev KS, Tikhonov RV, Shingarova LN, Petrovskaya LE, Dolgikh DA, et al. Lipid-Protein Nanodisks for Cell-Free Production of Integral Membrane Proteins in a Soluble and Folded State: Comparison with Detergent Micelles, Bicelles and Liposomes. Biochim Biophys Acta. 2011;1818:349–358. doi: 10.1016/j.bbamem.2011.10.020. [DOI] [PubMed] [Google Scholar]