

Abstract
The present study describes an optimized method for isolating peripheral blood circulating tumor cells (CTCs) and performing KRAS mutation analysis. The approach combines isolation of peripheral blood mononuclear cells and immunomagnetic labeling with CD45 and CD326 human microbeads with KRAS analysis performed with a Therascreen KRAS kit by quantitative PCR. KRAS mutations were detected in the CTCs of patients with metastatic colorectal cancer (mCRC). CTCs may represent an alternative to invasive procedures and their analysis may be representative of the current disease status of the patient. This proposed analysis may be performed in a daily clinical practice.
Keywords: circulating tumor cells, metastatic colorectal cancer, KRAS gene, peripheral blood, inmunomagnetic labeling
Introduction
Colorectal cancer (CRC) is one of the most significant worldwide public health problems (1). During the natural course of the disease, ~50% of patients may develop metastasis and up to 25% of these present with metastatic disease at the moment of diagnosis. Advances in the treatment of metastatic colorectal cancer (mCRC) include the development of new antitumoral agents, including epidermal growth factor receptor-targeted monoclonal antibodies (EGFR-mAbs) and tyrosine kinase inhibitors, and the use of biomarkers. The KRAS mutational status is currently the only biomarker that is predictive of the response to therapy using EGFR-mAbs that have been validated for clinical practice in mCRC and recommended by the National Comprehensive Cancer Network (NCCN) 2012 Guidelines (version 1, 2012) (2,3). However, not all mCRC patients with wild-type KRAS respond to EGFR-mAb treatment, which may be due to alterations in other genes, including BRAF, PIK3CA and PTEN(4,5). Furthermore, numerous studies have shown discordance in the KRAS mutational status between the primary tumor and the metastatic lesions (6–8). Thus, the study of KRAS in metastases may have clinical relevance and may, at least partly, explain the resistance to EGFR-mAbs in patients with KRAS wild-type primary tumors. Metastasis involves the concept of circulating tumor cells (CTCs) that are associated with the colonization of distant organs. Since the study by Ashworth in 1869, in which malignant cells that were similar to the primary tumor were identified to circulate in the peripheral blood (9), increased efforts, particularly in recent years, have focused on the development of reliable methods for the enrichment and identification of CTCs (10–12). The present study describes an easy, affordable procedure using magnetic labeling that allows the isolation of an enriched CTC fraction from peripheral blood and, furthermore, the analysis of the KRAS mutational status of the CTCs. A comparison between the KRAS mutational status in CTCs and the corresponding tumor tissue was performed.
Materials and methods
Patients
A total of 23 mCRC patients who were in remission or whose primary tumor KRAS mutational status was available in the patient clinical records were selected for this study. Ethylenediaminetetraacetic acid (EDTA)-anticoagulated peripheral blood (10 ml) was obtained from each patient, all of who exhibited disease progression. This study was approved by the ethics committee of the University Clinic of Navarra. All the participants provided their informed consent prior to blood sample extraction.
Isolation of peripheral blood mononuclear cells (PBMCs; time, 1.5 h)
When working with anticoagulated peripheral blood, PBMCs should be isolated using density gradient centrifugation. Ficoll-Paque™ Plus was used for this purpose (17-1440-03; GE Healthcare, Buckinghamshire, UK). The blood samples were diluted with saline serum (ratio, 1:1) and gently added on the top of the Ficoll-Paque Plus medium (ratio, 2/3 blood:1/3 Ficoll) taking care not to mix the two layers. Following centrifugation for 20 min at 400 × g, the PBMCs that were located at the interface layer were collected by inserting the pipette directly through the plasma (alternatively, the upper plasma layer may be removed and the PBMCs may be collected). The PBMCs were washed twice with saline serum and centrifuged for 8 min at 300 × g. Buffer A was added to the PBMCs, which were then centrifuged at 200 × g for 10–15 min. Buffer A consisted of phosphate-buffered saline (PBS; pH 7.2), 0.5% bovine serum albumin (BSA) and 2 mM EDTA). This step was repeated twice.
Enrichment of circulating tumor cells (time, 2 h)
CD45 Human MicroBeads (130-045-801; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) were used for the enrichment of the epithelial tumor cells from the peripheral blood by the depletion of the CD45+ cells. The cell number from the previous step was measured and following centrifugation at 300 × g for 10 min and complete aspiration of the supernatant, 80 μl buffer A and 20 μl CD45 MicroBeads per 107 total cells were added, mixed and refrigerated (4–8°C) for 15 min. Following a wash step with 2 ml buffer A per 107 cells (300 × g for 10 min), 500 μl buffer A was added (up to 108 cells). The cell suspension was loaded onto a separator column that was placed in a magnet (MiniMACS Starting kit; Miltenyi Biotec). The magnetically-labeled CD45+ cells were retained within the column. The unlabeled cell fraction (CD45− fraction) that was enriched in the epithelial tumor cells ran through.
CD326 epithelial cell adhesion molecule (EpCAM) Human MicroBeads are used for the enrichment of epithelial tumor cells
The cell number from the CD45− fraction that was obtained in the previous step was measured and following centrifugation at 300 × g for 10 min and complete aspiration of the supernatant, 300 μl buffer A and 100 μl FcR reagent (120-000-442; Miltenyi Biotec) per 5×107 total cells were added and mixed. A total of 100 μl CD326 Microbeads were added per 5×107 total cells, mixed well and refrigerated at 4–8°C for 30 min. Following a wash step with 5–10 ml buffer A per 5×107 cells (300 × g for 10 min), 500 μl buffer A was added up to 108 cells). The cell suspension was loaded onto a separator column placed in a magnet (MiniMACS Starting kit; Miltenyi Biotec). Subsequent to the labeling and separating procedures, three cell fractions were obtained, CD45+, CD45−/CD326− and CD45−/CD326+. A schematic of the procedure is shown in Fig. 1.
Figure 1.
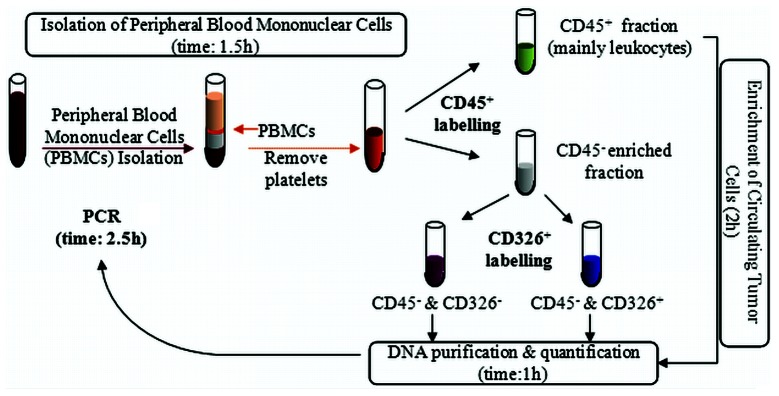
Schematic procedure of the KRAS mutational status analysis of peripheral blood isolated CTCs in mCRC patients. PCR, polymerase chain reaction; CTCs, circulating tumour cells; mCRC, metastatic colorectal cancer.
DNA purification and quantification (time, 1 h)
The QIAamp® Mini kit (51101; Qiagen GmbH, Hilden, Germany) was used for DNA purification from the cell fractions that were obtained previously. The procedure was performed as described in the QIAamp Mini kit instructions. DNA quantification was performed using the NaNoDrop-2000/2000c Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA), following the manufacturer’s instructions.
KRAS mutational status analysis (time, 2.5 h)
All DNA was analyzed for a set of seven KRAS point mutations, 12-Ala, Asp, Arg, Cys, Ser, Val and 13-Asp, using the Therascreen KRAS kit (870011; Qiagen GmbH). The analysis was performed on a quantitative PCR instrument (Rotor-Gen Q; Qiagen GmbH).
Results and discussion
Blood samples were studied from 23 mCRC patients whose primary tumor KRAS mutational status was available. Following the enrichment of the CTCs, three fractions were obtained, CD45+, CD45−/CD326− and CD45−/CD326+. The KRAS Mutation kit detected seven specific KRAS mutations using a region of the KRAS exon-4 as a control. The CD45+ cell fraction contained mainly leukocytes, thus, a signal appeared in the control assay that corresponds to the KRAS exon-4, but no KRAS mutation was detectable, since somatic mutations were only present in the fraction that contains the CTCs, if detectable. The CD45−/CD326− and CD45−/CD326+ cell fractions exhibited a signal of the KRAS exon-4 control due possibly to the residual mononuclear cells; however, the amplification signal appeared at a higher cycle compared with the corresponding CD45+ since the expected mononuclear cells amount was low. The CTCs were identified in the CD45−/CD326+ cell fraction and, therefore, the KRAS mutations, if detectable by the kit, appear in this fraction. The present study identified a correspondence between the primary tumor-KRAS status and the CTC-KRAS status in 17 patients (15 wild-type and two mutated). In five cases, the tumor-KRAS mutation was not detected in the CTCs. In those patients, a low concentration of CTCs explained the results as their DNA concentration was below the kit detection limits. Notably, another discrepancy was identified in one patient (case CR6), who demonstrated a KRAS wild-type in the primary tumor and a KRAS-12Ala mutation in the CTC enriched cell fraction (Fig. 2). In this case, another scenario should be proposed. It is known that in a minority of cases (5–10%) the KRAS mutational status is heterogeneous and may vary between the primary tumor and the metastasis (13). In a recent study, Bossard et al defined a KRAS mutational mosaicism, with 4 out of 18 patients showing KRAS status discordances between the primary colorectal cancer and the metastasis (14). The biological significance of this mosaicism is unclear, but indicates diverse metastatic potentials in various populations of tumor cells and/or the acquirement of mutations during or following metastasis. Bouchahda et al proposed that KRAS mutations may be acquired late during the metastatic phase of colorectal cancer more frequently than currently recognized (15). Furthermore, since CTCs may be considered as an intermediate step in colonization, their metastatic potential depends on a number of genetic abnormalities that may include the acquisition of a KRAS mutation. Therefore, the CTC-KRAS-Ala mutation that was identified in a patient from the present study may lead to an improved adaptation to adverse conditions, migration and colonization of distant tissues. By the end of the present study, the CR6 patient developed hepatic metastasis (tissue from metastases not available) and a change from anti-EGFR to Irinotecan-based treatment was considered. Future studies examining large series and examining KRAS, BRAF, PTEN and PI3K may provide valuable insight into the carcinogenesis and metastasizing patterns of mCRC, thus guiding treatment options for patients.
Figure 2.

Image of the CTC-KRAS mutational status for patient CR6. The CD45+ cell fraction (mainly leukocytes) provides a positive signal for the control assay corresponding to KRAS exon-4. The CD45−/CD326− cell fraction exhibits a positive signal of the control assay corresponding to KRAS exon-4. The CD45−/CD326+ cell fraction exhibits a positive signal of the control assay corresponding to KRAS exon-4 and a signal for the KRAS-12Ala mutation. CTC, circulating tumour cell.
In summary, the present study indicated that the isolation and analysis of CTCs from the peripheral blood of mCRC patients, using a minimally invasive, relatively economical and optimized method, may have high clinical relevance. There are limitations to the procedure, since during epithelial to mesenchymal transition, the expression of epithelial markers on CTCs, such as EpCAM may be downregulated and become undetectable (16). CTC studies represent an alternative to invasive procedures and their analysis may become a prognostic factor, acting as a ‘liquid biopsy’. CTCs are able to survive chemotherapy and may indicate a lack of therapeutic efficacy, allowing the early end of ineffective treatments and providing a representation of the current disease status of the patient.
Acknowledgements
The authors would like to thank all the patients that participated in this study.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 3.Jimeno A, Messersmith WA, Hirsch FR, Franklin WA, Eckhardt SG. KRAS mutations and sensitivity to epidermal growth factor receptor inhibitors in colorectal cancer: practical application of patient selection. J Clin Oncol. 2009;27:1130–1136. doi: 10.1200/JCO.2008.19.8168. [DOI] [PubMed] [Google Scholar]
- 4.Sartore-Bianchi A, Bencardino K, Cassingena A, et al. Therapeutic implications of resistance to molecular therapies in metastatic colorectal cancer. Cancer Treat Rev. 2010;36(Suppl 3):S1–S5. doi: 10.1016/S0305-7372(10)70012-8. [DOI] [PubMed] [Google Scholar]
- 5.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 6.Artale S, Sartore-Bianchi A, Veronese SM, et al. Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J Clin Oncol. 2008;26:4217–4219. doi: 10.1200/JCO.2008.18.7286. [DOI] [PubMed] [Google Scholar]
- 7.Kosakowska EA, Stec R, Charkiewicz R, Skoczek M, Chyczewski L. Molecular differences in the KRAS gene mutation between a primary tumor and related metastatic sites - case report and a literature review. Folia Histochem Cytobiol. 2010;48:597–602. doi: 10.2478/v10042-010-0078-z. [DOI] [PubMed] [Google Scholar]
- 8.Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010;16:790–799. doi: 10.1158/1078-0432.CCR-09-2446. [DOI] [PubMed] [Google Scholar]
- 9.Ashworth T. A case of cancer in which cells similar to those in the tumours were seen in blood after death. Aust Med J. 1869;14:146–149. [Google Scholar]
- 10.Gerges N, Rak J, Jabado N. New technologies for the detection of circulating tumour cells. Br Med Bull. 2010;94:49–64. doi: 10.1093/bmb/ldq011. [DOI] [PubMed] [Google Scholar]
- 11.Alunni-Fabbroni M, Sandri MT. Circulating tumour cells in clinical practice: Methods of detection and possible characterization. Methods. 2010;50:289–297. doi: 10.1016/j.ymeth.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 12.Lustberg M, Jatana KR, Zborowski M, Chalmers JJ. Emerging technologies for CTC detection based on depletion of normal cells. Recent Results Cancer Res. 2012;195:97–110. doi: 10.1007/978-3-642-28160-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baas JM, Krens LL, Guchelaar HJ, Morreau H, Gelderblom H. Concordance of predictive markers for EGFR inhibitors in primary tumors and metastases in colorectal cancer: A review. Oncologist. 2011;16:1239–1249. doi: 10.1634/theoncologist.2011-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bossard C, Küry S, Jamet P, et al. Delineation of the infrequent mosaicism of KRAS mutational status in metastatic colorectal adenocarcinomas. J Clin Pathol. 2012;65:466–469. doi: 10.1136/jclinpath-2011-200608. [DOI] [PubMed] [Google Scholar]
- 15.Bouchahda M, Karaboué A, Saffroy R, et al. Acquired KRAS mutations during progression of colorectal cancer metastases: Possible implications for therapy and prognosis. Cancer Chemother Pharmacol. 2010;66:605–609. doi: 10.1007/s00280-010-1298-9. [DOI] [PubMed] [Google Scholar]
- 16.Wicha MS, Hayes DF. Circulating tumor cells: not all detected cells are bad and not all bad cells are detected. J Clin Oncol. 2011;29:1508–1511. doi: 10.1200/JCO.2010.34.0026. [DOI] [PubMed] [Google Scholar]