

Abstract
While the cellular heat-shock response has been a paradigm for studying the impact of thermal stress on RNA metabolism and gene expression, the genome-wide response to thermal stress and its connection to physiological stress resistance remain largely unexplored. Here, we address this issue using an array-based exon expression analysis to interrogate the transcriptome in recently established Drosophila melanogaster stocks during severe thermal stress and recovery. We first demonstrated the efficacy of exon-level analyses to reveal a level of thermally induced transcriptome complexity extending well beyond gene-level analyses. Next, we showed that the upper range of both the cellular and physiological thermal stress response profoundly affected message expression and processing in D. melanogaster, limiting expression to a small subset of transcripts, many that share features of known rapidly responding stress genes. As predicted from cellular heat-shock research, constitutive splicing was blocked in a set of novel genes; we did not detect changes to alternative splicing during heat stress, but rather induction of intronless isoforms of known heat-responsive genes. We observed transcriptome plasticity in the form of differential isoform expression during recovery from heat shock, mediated by multiple mechanisms including alternative transcription and alternative splicing. This affected genes involved in DNA regulation, immune response, and thermotolerance. These patterns highlight the complex nature of innate transcriptome responses under stress and potential for adaptive shifts through plasticity and evolved genetic responses at different hierarchical levels.
Keywords: Drosophila, heat stress, thermal extremes, transcription, splicing
THE distribution of ectotherms including Drosophila species can often be linked to their physiological thermal tolerances (Addo-Bediako et al. 2000; Mitchell and Hoffmann 2010; Kellermann et al. 2012). Terrestrial Drosophila from a range of environments may exist close to their maximal range and be constrained to increase upper tolerance limits, posing a threat to persistence under climate warming (Kellermann et al. 2012). Elucidating the factors delimiting upper thermal limits depends on understanding how physiological responses link with the underlying molecular processes in an integrative framework. Limited progress toward this end has been made so far, despite the cellular reaction to heat stress being the most ubiquitous and well-characterized molecular stress response.
Seminal work exploiting the tightly controlled conditions of homogeneous cell lines has led to fine-scale molecular dissections of the heat-shock response in wide-ranging taxa including yeast, Drosophila, and humans. At the cellular level, heat shock triggers a dramatic reprogramming of gene expression to favor the rapid turnover of a class of molecular chaperones known as the Heat-shock proteins (Hsps) (Lindquist and Craig 1988; Yost et al. 1990). Apart from the selective activation of a subset of genes predominantly harboring heat-shock factor (HSF) sequence-binding elements, transcription is inhibited during heat shock due to reduced nucleosome mobility and RNA Polymerase II elongation (Birch-Machin et al. 2005; Guertin and Lis 2010; Gonsalves et al. 2011; Teves and Henikoff 2011). In eukaryotes, heat shock also inhibits pre-mRNA splicing whereby intron removal from the nascent transcript to form the mature messenger is blocked, a process bypassed in the majority of intron-lacking Hsps (Yost and Lindquist 1986; Bond 1988; Lindquist and Craig 1988). As well as protein thermoprotection, Hsps are implicated to play a role in splicing recovery. Pretreatments at moderately high temperatures have been shown to preserve splicing at more severe subsequent stresses, known as “splicing thermotolerance,” a process thought to occur at least in part because of the accumulation of Hsps (Yost and Lindquist 1986, 1991; Bond 1988; Corell and Gross 1992; Bracken and Bond 1999; Marin-Vinader et al. 2006;). More recent research suggests that splicing thermotolerance likely stems from “SRSF10 thermotolerance,” wherein phosphorylation of the splicing factor SRSF10 is maintained during heat stress modulated in part by Hsp27 (Shi et al. 2011). The tractability of the splicing machinery or “spliceosome” to recognize different splice-site sequences results in alternative splicing (AS) of different mRNAs from the same pre-mRNA (reviewed in Graveley 2001; Biamonti and Caceres 2009; Nilsen and Graveley 2010). Intriguingly, different mechanisms have been proposed to control constitutive splicing and modulate alternative splicing in heat-shocked human cells. Dephosphorylation of the splicing regulator SRSF10 affects the interaction of components of the spliceosome to bind to pre-mRNA to block constitutive splicing, while the recruitment of specific splicing factors away from active sites into nuclear stress bodies (nSBs) are proposed to initiate alternative splicing providing a model for heat-induced alternative splicing through 5′ splice-site selection and exon-skipping events (Denegri et al. 2001; Biamonti 2004; Biamonti and Caceres 2009).
Despite strong conservation of the response in the genes and transcripts tested so far, data are lacking both at the genome-wide level (Biamonti and Caceres 2009), and importantly, at the organismal level. Microarray studies at different Drosophila life stages have explored gene-level expression and temporal expression patterns in response to mild heat exposure at 36°–37° as well as in recovery (Leemans et al. 2000; Sorensen et al. 2005; Gonsalves et al. 2011; Zhou et al. 2012). However, with a focus on quantifying total transcriptional output, gene-level studies provide only a generalized picture of the transcriptome under stress and are limited in resolution to profile further levels of stress-induced complexity. Now the rule rather than the exception, mechanisms such as alternative splicing and alternative transcription underlie transcriptome plasticity and proteome diversity with wider implications in the control of gene expression contributing to phenotypic variation and plasticity, in human disease, and in response to stress (Faustino and Cooper 2003; Ali and Reddy 2008; Marden 2008; Nilsen and Graveley 2010; Mastrangelo et al. 2012). In the latter case, research in plants highlights that shifts in gene expression required to mount a stress response are regulated at different stages ranging from the transcriptional to post-translational level (Mastrangelo et al. 2012). Specifically, post-transcriptional modifications such as AS are central in this process and in generating transcripts and proteins with novel functions during environmental flux (reviewed in Mazzucotelli et al. 2008; Mastrangelo et al. 2012).
Here, we utilized a custom microarray platform to profile the transcriptome prior, during and in recovery from the upper range of both the cellular heat-shock response and physiological limits of wild-derived Drosophila melanogaster. For many genes encoding multiple transcripts, this unique platform allowed us to deconvolute gene-level expression for the first time as well as to directly compare different gene-level expression estimates by hybridizing two modules to one platform and universally correcting for nonspecific hybridization with probes designed to evaluate pure background. We predicted that exon-level analyses will more comprehensively profile the stress-mediated transcriptome because, in contrast to 3′-end biased analyses, exons more reliably measure gene expression across the whole transcript, as well as providing measures of individual exons for isoform sensitivity (Kapur et al. 2007; Lockstone 2011). Based on models of the molecular heat-shock response, we hypothesized that innate Drosophila transcriptional responses to high temperature would include selective activation of a subset of heat-activated transcripts, blocked constitutive splicing, and heat-activated alternative splicing resulting in changes to gene expression and splicing profiles in recovery. Our exon-level analyses reveal more transcriptome complexity than gene-level estimates during stress and recovery. We show that heat stress activated a specific subset of largely intronless or intron-poor transcripts, and, in a novel set of transcripts, caused a constitutive splicing blockade. While we did not observe evidence for alternative splicing during heat stress, we found isoform changes mediated by an array of RNA metabolic events during recovery for a number of genes. These data provide a genome-wide link between the heat-shock response and an organism’s response to thermal stress and provide the first evidence that stress elicits deeper levels of transcriptome complexity than previously documented.
Materials and Methods
D. melanogaster population and culture
D. melanogaster were sampled from three locations in Coffs Harbour, New South Wales, Australia (30 18′ 18′′S 153 07′ 48′′; 30 19′ 10′′S 153 05′ 20′′E; and 30 17′ 33′′S 153 08′ 13′′E), in 2010. The experimental population was founded at generation F2 of laboratory culture by pooling 10 virgin progeny of each sex from 20 isofemale lines from each location (1200 flies from 60 isofemales in total). The population was mass-bred for three generations prior to the first phenotypic assays. Flies were maintained at 25° under constant light on dextrose dead-yeast agar medium in multiple 500-ml bottles. At each generation, progeny were mixed and randomly allocated into new bottles of at least 300 flies each.
Assessing thermal limits with heat knockdown
Heat knockdown time (Hoffmann et al. 2002) was used to assess innate heat tolerance in 5-day-old generation F4 mass bred females. The experimental flies were reared under controlled density conditions by removing parents after 2 hr of oviposition. Imagoes were collected into mixed-sex cohorts until 24 hr prior to the assays where females were separated into vials using aspiration without CO2. Individual females were placed in 10 ml dry vials submerged in a water bath heated to either 38.5° or 39° and knockdown time was scored as the time taken (to the nearest second) for flies to become incapacitated. Survival curves were generated from three replicate trials of ∼100 flies each. Mortality after 48 hr on food at 25° was assessed in groups of 10 flies after 5, 10, 15, 20, and 32 min of heat exposure.
Thermal stress and recovery time-series sampling
The heat stress and recovery time-series sampling used for transcript quantification was determined from the survival curve data. We defined the population “thermal limits” as the average time taken to incapacitate the majority of flies without ensuing mortality within a 2-day window. This criterion permitted the assessment of severe thermal stress on transcript profiles that are not critically impaired by cellular degradation and/or apoptosis. From the survival curves, exposure to 39° resulted in consistent mortality 48-hr post-knockdown, while exposure for 31.5 min at 38.5° knocked down ∼70% of flies with a 100% survival rate at 25° for at least 48 hr. The latter conditions were therefore used for all sample collections.
For the microarray experiment, densities were standardized by placing 50 generation F5 eggs into vials. The emerging flies were collected and sorted as above. For the stress assay, groups of 20 5-day-old females were placed in 15 ml Bunzel cryotubes and heat stressed as above. Flies were sampled and snap frozen in liquid N2 according to the following treatments: immediately prestress (25°, on media); stress, 5, 10, 15, 25, and 31.5 min (38.5° no media); recovery (25°, on media) from severe stress (38.5°, 31.5 min) at 1, 4, 8, 12, 24, 36, and 48 hr. Real-time RT–PCR of known heat-responsive genes Hsp68 and hsrω were run on three replicates from each of the 13 time points to determine the efficacy of the thermal stress and to choose 8 final points for the microarray time series (data not shown). Based on this preliminary data, flies were tested prestress, 15 min into and at the end of the exposure (31.5 min), and after 4, 8, 12, 36, and 48 hr recovery from severe stress (38.5°, 31.5 min; Figure 1). Three replicates of 20 flies from 8 time points were assessed on the arrays for a total of 24 RNA extractions and hybridizations.
Figure 1.
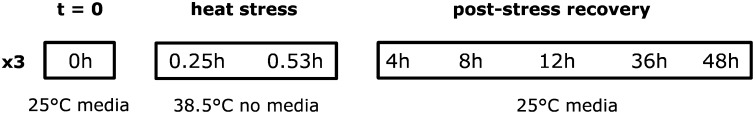
Treatment and sampling schematic of the thermal stress and recovery time series. Groups of 20 female flies were sampled at t = 0 (25°), at 15 and 31.5 min exposure to 38.5° (representing 0 and ∼70% knockdown, respectively), and in recovery (25°) from 31.5 min at 38.5°. The population upper thermal limits and microarray time series were determined by preliminary assessments using heat-knockdown survival curves and survival after 48-hr recovery.
RNA extraction
Total RNA was extracted from groups of 20 females using TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and then purified and DNAase treated using standard techniques (RNeasy kit; QIAGEN, Valencia, CA; TURBO DNase digestion, ABI). RNA was quantified on a Nanodrop Spectrophotometer (NanoDrop Technologies, Wilmington, DE) and integrity was assessed following both purification and DNase treatment steps with 1% agarose gel electrophoresis.
Gene and exon identification with a custom array platform
The custom Drosophila array on the Affymetrix platform UFL Custom Dros_snpa520726F was used in this study. The chip design is described in detail in Yang et al. (2011). The two D. melanogaster expression modules for the perfect match 3′ expression probes (3′ IVT probes) of the Affymetrix GeneChip Drosophila Genome 2.0 array (900531, 900532, and 900533) and the probes of the Affymetrix Drosophila Tiling 2.0 Array (901021) that map uniquely to exonic sequence were analyzed. The 3′ IVT probes are biased to the 3′-end of the gene, while probes in the Tiling Array module generally span the entire gene. For multi-transcript genes, the 3′ IVT probesets tend to span an exon common to all transcripts, while the Tiling Array probesets can target alternative exons. For the Tiling Array module, exons with alternative start/end sites in the same genome region were labeled as a single unique exonic region, but are called exons for simplicity. Each “exon” corresponds to a unique probeset, although the majority of exonic regions designate a single exon. Probe sequences correspond to the genome build of Flybase v. R5.11 (Yang et al. 2011). Hereafter, the 3′ IVT and Tiling Array modules are referred to as the 3′ IVT module and exon module, respectively. Tiling Array probesets that target a constitutive exon are referred to as constitutive exon probesets.
Sample labeling, fragmentation, and array hybridizations
The RNA was concentrated by precipitating in 0.5 vol 7.5 M ammonium acetate at −70° for 1 hr and resuspending in 10 ul DEPC-treated H20. Targets were prepared for array hybridization using Affymetrix reagents following the no-amplification protocol of the GeneChip WT double-stranded target assay (Yang et al. 2011). Briefly, first- and second-strand cDNA was synthesized using a random primer from 10 µg of RNA (WT double-stranded DNA synthesis kit, GeneChip Sample Cleanup Module). Purified dsDNA (7.5 µg) was fragmented and labeled using the WT double-stranded DNA terminal labeling kit. The labeled cDNA was hybridized to the arrays following the manufacturer’s protocol with the Fluidics Station 450 protocol (FS450_0001) and scanned with an Affymetrix 7G scanner. The hybridizations and scanning were performed at the Ramaciotti Centre (University of New South Wales, Sydney, NSW, Australia).
Signal quantification, probeset summation, and quality control
Signals were quantified and normalized for the D. melanogaster 3′ expression and exon modules (Yang et al. 2011). Briefly, raw intensity values were extracted using the Affymetrix Power Tools apt-cel-extract program. Individual probes were matched to their corresponding GC bin control probes by the number of G/C bases in the 25mer probe. Each probe was background corrected and normalized by subtracting the median intensity of the corresponding GC bin control. Probe set summation for probe set I, Yi, was estimated as Yi = , where Xij is the intensity for probe j in set i; GCj is the average intensity for the corresponding GC bin control and Ni is the number of probes in probe set i (Yang et al. 2011). Kernel density estimates for each array were used to assess probe-level signal distributions across all modules. Kappa statistics were used to examine raw signal intensity agreement of individual probes, the normalized signals of probesets, and gene expression-level estimates.
Individual probe signals and normalized probesets were compared between slides of replicate treatments, while probesets corresponding to the same gene were compared between the 3′ IVT and exon modules by individual slide. Reliability of gene expression estimates for probesets targeting 11 heat-shock protein genes was also assessed using both Kappa statistics and Bland–Altman plots for each module individually between slides and between the 3′ IVT and exon modules on the same slide (McIntyre et al. 2011; Yang et al. 2011). Array processing, probeset summation, and quality control were performed using SAS software v. 9.3 (SAS Institute, Cary, NC).
cDNA synthesis and real-time PCR
Two micrograms of purified DNAse-treated total RNA was used to generate cDNA with the following protocol in a 20-µl reaction volume: 2 µl 50-ng random hexamers (Bioline) and 4 µl 2.5 mM dNTPs were heated to 70° for 5 min then cooled on ice. 2 µl 10× M-MuLV buffer and 1 µl M-Mulv reverse transcriptase 200 U/ul was added and the samples were incubated at 42° for 60 min; then the enzyme was deactivated at 90° for 10 min. The cDNA was diluted 1:5 in water. Real-time PCR was performed on the LightCycler 480 (Roche) using SYBR Green in a 2× universal buffer containing the following: 50 mM MgCl2, 10× NEB Buffer, 2× Roche high resolution melt, 25 mM dNTPs, Immolase Taq 5 U/µl. A typical 10-µl PCR reaction contained 5 µl universal buffer, 4 µl 1 µM primer mix, and 1 µl cDNA. Three biological replicates were run for each time point, with two technical replicates per cDNA in a 384-well plate format. All transcripts corresponding to the same gene/treatment were run on the same plate with biological replicates run on separate plates. Mature, processed mRNAs were assessed using exon-junction primers while unprocessed transcripts were assessed using an exon and intron primer pair. Transcript and transcript isoform primer sequences were designed using Primer-BLAST (NCBI), QuantPrime, and GETPrime (Arvidsson et al. 2008; Gubelmann et al. 2011) and are shown in Supporting Information, Table S1.
Statistical analyses
Temporal changes in genes:
The temporal effect of thermal stress on overall gene expression was examined in the 3′ IVT module (n =12,217 genes), for constitutive exons in the exon module (n =13,301 genes), and for all exons in the exon module irrespective of exon type (13,523 genes).
The fixed-effects ANOVA model
| (1) |
was fit for each gene, where Yin is the signal for probeset i and replicate n, µ is the overall mean, ti is the fixed effect of time point (before, during, and after recovery from severe heat shock), and εin is the error. All probes in a probeset were used and probesets targeting the same gene and/or exon were averaged. The null hypothesis that flies had equal gene expression before, during, and after stress was tested using an F-test, and P-values were corrected for multiple tests using a false discovery rate (FDR) approach (Benjamini and Hochberg 1995). An FDR level of 0.2 was used to determine statistical significance.
To partition the temporal effect of thermal stress and recovery on the expression of different transcript isoforms, probesets targeting multiple alternative exons of a gene or at least one alternative and one constitutive exon were identified from the exon module (n = 2009 genes, 15,782 probesets). Constitutive exons represent expression signals from all transcript isoforms, providing an estimate of overall or “composite” gene expression, while alternative exons represent either single or subset alternative isoforms.
For all exons of the 2009 multi-transcript genes, the fixed effects ANOVA model
| (2) |
was fit for each gene with exon type xi and time point tj as fixed effects. Probesets targeting multiple constitutive exons were classified as one exon type while probesets targeting alternative exons were considered different exon types. The main effect of exon or time point and the exon-by-time-point interaction represent the average of probesets across exon types. To test if isoforms are differentially expressed over time, the significance of the interaction between exon type and time point (xitj) was tested using an F-test with FDR correction (McIntyre et al. 2006; Telonis-Scott et al. 2009).
While model (2) tests for differences in the transcript abundance of exon types (xi) and tests whether the gene is heat responsive over time (tj), the power to detect the interaction term (xitj) is reduced by the relationship between whole-gene and alternative isoform measures. Where signal differences are large enough and the variances small enough, the expression patterns of the constitutive exons should reflect the predominantly expressed alternative isoform/s. To increase the power and precision to detect differences in alternative exon expression over the time course, the within-group error variance was reduced by fitting a model with the constitutive exons as a covariate (Sokal and Rohlf 1995). For genes with at least one constitutive exon and two alternative exons (n = 1094), the model
| (3) |
was fit, where c is the average of the constitutive exons at each time point for each gene. The means of the alternative exons at each time point were adjusted by the covariate using linear regression. Where the slopes for each exon type are parallel, the test for equality of the Y-intercepts is equivalent to testing for differences in means between the exons. Parallelism of the regression lines was first tested with the interaction of the covariate with exon and type point. Analyses were conducted with SAS software v. 9.3.
We also fit a model on only the alternative exons for genes with at least two alternative exons and examined the interaction (not shown). All genes with a significant interaction between exon and time point were detected in models (2) and (3).
Comparisons of thermally responsive genes across probe modules: whole gene vs. exon-level detection:
To compare the temporal effect of thermal stress on the overall expression of all genes between probes on the 3′ IVT and exon modules, genes represented on both modules were identified (n = 11,530). The proportion of significant heat-responsive genes from all 11,530 genes was compared between the 3′ IVT probesets and the exon constitutive probesets and across all exons from model (1). To compare the relative detection levels of thermally responsive multi-transcript genes across the different probe types and modules, the number of overlapping 2009 multi-transcript genes [model (2)] were identified for the 3′ IVT probesets, constitutive exon probesets, and exon probesets (n = 1,721 genes). All outcomes were corrected for the new test numbers for each probeset type by rerunning FDR at the cutoff of 0.2. The proportions of significant genes across the module/probeset types were compared using χ2 goodness-of-fit tests.
Temporal profiling using clustering and STEM:
For cluster analysis, the data were filtered to include genes/transcripts showing evidence of temporal modulation (i.e., significant time-point response) and/or a significant interaction between different isoform types over time [exon-by-time-point interaction, models (2) and (3)]. First, the relationships between modules across time points were visualized using hierarchical clustering by average linkage (Spearman rank correlation) on the average signals of each time point for the significant genes of each module from model (1) (MultiExperiment viewer v. 4.81). Next, coexpression patterns of genes [constitutive exon probes, model (1)] and transcripts [time point, exon-by-time-point interaction, models (2) and (3)] were profiled using the short time-series miner (STEM) (Ernst and Bar-Joseph 2006). As STEM normalizes data relative to time point zero with a log2 transformation, the probesets were resummarized using a log2 transformation in place of the natural log. A constant was added to the background subtracted signals to avoid negative or zero signal values, and all probes were analyzed. For transcripts absent or on the extremely low end of the signal distribution (i.e., where background subtracted signals are low or <0), the STEM normalization method tended to increase noise and/or inflate expression relative to time zero; therefore, the probesets were again resummarized without the addition of a constant where GC band background estimates are greater than signals [Yi = ]. Negative values were then excluded from the clustering when present in two or more replicates of a time point. The STEM clustering method was implemented with the following settings: m = 50, c = 3 (constitutive probes), and c = 2 (alternative exons); advanced options = minimum absolute expression change = 0.7 with change based on maximum–minimum; all permutations per transcript; FDR < 0.05.
Gene annotation enrichment modules:
Significant genes/transcripts were assessed for annotation enrichment using DAVID (Dennis et al. 2003; Huang et al. 2009). Flybase IDs were converted to DAVID IDs (D. melanogaster background) and analyzed with the following settings: functional categories = cog ontology, SP_PIR_keywords, UP_seq_features; gene ontology, GOTERM_BP_FAT (biological process), GOTERM_MP_FAT (molecular process), GOTERM_CC_FAT (cellular component); protein domains, INTERPRO, PIR_SUPERFAMILY, SMART; pathway, KEGG pathways. Probability values (EASE scores) were determined using a modified Fisher’s exact test (Dennis et al. 2003; Huang et al. 2009) and were corrected for multiple testing with an FDR threshold of 0.1. To highlight group term enrichment of related genes, annotation terms were summarized into modules using the following custom settings: simterm overlap = 3; similarity threshold = 0.5; initial group membership = 5; final group membership = 5; multiple linkage threshold = 0.5. The enrichment score is the −log-transformed geometric mean of all the EASE scores of each annotation term in the group (Dennis et al. 2003; Huang et al. 2009).
Real-time PCR:
Transcript abundance was calculated relative to the thermally and temporally stable “housekeeping” gene RpL11 [where relative expression of transcript of interest (TOI) = 2^(RpL11–TOI)]. RpL11 abundance was tested using one-way ANOVA with the fixed effect of time point. Two-way ANOVAs were run for each gene with the fixed factors of transcript and time point and transcript-by-time-point interaction. Given the large number of transcripts and time points tested, for simplicity and to correct for multiple testing, relative abundances of each transcript were compared to nonstress conditions for using one-way ANOVAs with Dunnett’s tests. Two-way ANOVAs with planned contrasts for a subset of genes showed similar results to the Dunnett’s tests. All data were log2 transformed to improve the residual distributions.
Results
Quality control
The quality control evaluations revealed no hybridization anomalies and high expression reliability between replicate arrays at the probe and probeset level, as well as between probe modules summarized at the gene level (File S1, Table S2, and Figure S1).
Gene-level analysis: detection of more thermally responsive genes with the constitutive exon probesets at FDR < 0.05
Gene expression before, during, and in recovery from extreme heat stress was examined for all genes in the 3′ IVT expression module, for genes with probesets specific to constitutive exons in the exon module, and across all exon types for genes in the exon module [ANOVA, model (1)]. The overall proportion of differentially expressed genes detected with one-way ANOVAs (main effect of time point) was similar between the modules ranging from 8.7 to 9.7% (FDR level 0.2; Table 1). The raw P-values, FDR-corrected P-values, and average signals for each of the eight time points and modules are given in Table S3. When genes common to all modules were compared (n = 11,530), adjusted proportions of differentially expressed genes ranged from 9 to 9.9% (Figure 2). More genes were detected with the constitutive exon probes compared to the 3′ IVT and all exon probes (borderline significance for the group comparison, χ2, d.f. = 2, P = 0.055). Moreover, a higher proportion of genes was detected at the more stringent FDR threshold of 0.05 for the exon module [∼70% compared to ∼50% in the 3′ IVT module, χ2, d.f. = 2, P < 0.001 (Table 1 and Figure 2)]. The thermally responsive genes detected with the 3′ IVT module and from the constitutive exons of the exon module were assessed for overall patterns of enriched gene function using functional annotation clustering with the DAVID software (Dennis et al. 2003). A total of 1078 and 1273 genes from the 3′ IVT and exon modules, respectively, mapped to DAVID identifiers and were included in the analysis. The data were summarized into 13 and 17 nonredundant clusters for the 3′ IVT and constitutive exon probesets, respectively, with enrichment scores ranging from 9.77 to 2.31 (equivalent to P < 0.0001 and 0.02 on the non-log scale; Table S5). Ten clusters overlapped between the two modules indicating a high degree of functional similarity including genes involved in heat shock, signaling, innate immunity, defense, proteolysis, cytochrome P450s, drug metabolism, egg production, and aspects of metabolism (Table S5). Clusters that differed include genes enriched for odorant binding and egg chorion in the 3′ IVT module and symporter activity and epidermal growth factor genes (EGFs) in the exon module (Table S5).
Table 1. Results for model (1) ANOVAs testing for expression changes over time following heat stress (time-point term) on all genes from the 3′ IVT module and exon modules, and genes with constitutive probes from the exon module, shown at three FDR levels.
| Exon module | |||
|---|---|---|---|
| FDR | 3′ IVT module (n = 12,217) | All probes (n = 13,523) | Constitutive probes (n = 13,301) |
| <0.05 | 554 | 843 | 918 |
| <0.1 | 243 | 153 | 156 |
| <0.2 | 317 | 177 | 211 |
Figure 2.

Proportion of 11,530 genes common to the 3′ IVT module, constitutive probes from the exon module, and for all probes in the exon module that were differently expressed over the time series following severe thermal stress. FDR is shown at 0.05, 0.1, and 0.2.
Gene-level temporal profiling: paucity of early responding genes and enrichment of innate immunity defense responses in mid-recovery from severe thermal stress
Gene expression patterns across the time series were examined in more detail using STEM. STEM implements an algorithm designed specifically for short time series with permutation testing to profile significantly overenriched coexpression clusters (Ernst and Bar-Joseph 2006). The significant genes from both the 3′ IVT module and exon module constitutive exon probesets were clustered and found to give similar results for the majority of genes (Figure S3 and Figure 3, respectively). Gene-level estimates using all exons were not examined given the similarity to the constitutive exons from hierarchical clustering (Figure S2), and for simplicity the constitutive exon data are presented. Of the 1273 thermally responsive genes, 1112 passed the filtering criteria (see Materials and Methods), and 854 (77%) clustered into six significantly overrepresented expression profiles (FDR < 0.05; Figure 3A). Notably during heat stress, all enriched profiles showed either no change or downregulated expression relative to time zero (Figure 3A). Expression responses tended to be delayed until midrecovery, alternatively increasing by 12-hr recovery (profiles 9 +26, Figure 3A) or by decreasing at 8- to 12-hr recovery (profile 25, Figure 3A) with peak expression at 36-hr late recovery (Figure 3A). Genes grouped in profile 43 were very highly expressed at 8 and 12 hr recovery, with average normalized expression relative to zero up to fourfold higher (Figure 3A). Fold changes ranged between 8- and 16-fold for individual genes such as defense and immunity genes IM1, IM2, IM3, IM4, IM23, AttC, and AttA, a number remained elevated 2 days post-stress (Figure 3A and Table S4). Similarly, genes grouped in profile 35 were upregulated by 8-hr recovery, although average peak expression was seen at 12- to 36-hr recovery, with the largest signal increase of 32-fold at 12-hr recovery involving an unknown allergen-related protein coded by CG13905 (Figure 3A and Table S4). Genes clustered in profile 40 were similar to profile 35 with average expression peaking at 12 and 36 hr, although average signals were slightly more elevated during early recovery (Figure 3A). In this group, the highest responding gene TotX (bacterium and heat defense) was upregulated over 16-fold relative to time zero by 12-hr recovery (Table S4). Patterns of GO enrichment by temporal profile revealed early–mid-recovery activation of signaling genes (profiles 40, 43, and 35, Table S5), strong defense responses including microbial immunity (profiles 43 and 35, Table S5), as well as glutathione transferase activity, enzyme inhibition (profile 43, Table S5), and sugar and carbohydrate metabolism (profile 35, Table S5). The mid- to late expression profiles revealed a second wave of signaling and stress-response genes including redox, cytochrome P450 expression, ion transport, and maintenance of chemical homeostasis (profiles 9+26, Table S5). The highest average signal increases ranged from 5- to 6-fold relative to time zero, including genes such as TotM (heat and bacterium defense), sodh-1 (oxidation–reduction processes), and CG34219 [unknown protein (profiles 9+26, Figure 3B and Table S5)]. Gene expression of loci involved in metabolic function, vitellogenin, and lipid transport was also modulated relatively late in recovery, with the highest responding glucose metabolism genes Mal-A1 and Mal-A2 and nutrient reservoir gene Lsp2 over 4- and 11-fold, respectively, by 36 hr (profile 25, Table S4 and Table S5). Profile 26 was enriched for genes involved in signaling, protein and sugar disassembly (proteolysis, glycosidases), and starch and sugar metabolism (Table S4 and Table S5).
Figure 3.

Short time-series expression miner (STEM) profiles of 1112 genes identified from the constitutive probes of the exon module with ANOVA. Average normalized (relative to time zero) signals of each profile/profile cluster are shown on the y-axis, and time in hours is shown on the x-axis. The dashed line indicates thermal stress sampled at 15 and 31.5 min at 38.5°, solid line indicates recovery up to 48 hr post-stress at 25°. (A) Profiles enriched for mid- to late-recovery-expressed genes with peak expression at 12 or 36 hr. Rapidly heat-responsive genes (B) are under enriched with peak at either at late stress/early recovery.
Given the specificity of stimuli required for immune pathway activation (Davies et al. 2012) the genes enriched for innate immune defense responses during recovery from thermal stress were explored in greater detail. First, genes involved in any aspect of fly immunity were identified from the CV term report generated by searching “immune system process, GO ID GO:0002376” (Flybase version 2013_03). This list was then filtered to include only those genes that were heat responsive from the ANOVA analysis (65 genes, Table S6) to more closely examine gene function and expression patterns. Note that the less conservative significance threshold of <0.05 (uncorrected ANOVA P-value) was used given that only 9 genes missed the FDR cutoff but were clearly differentially expressed and therefore likely to represent type II error. Strikingly, genes spanning the repertoire of Drosophila innate defense mechanisms were upregulated during recovery from severe thermal stress. Drosophila immune challenge elicits a number of interconnected defense modules that can be broadly divided into humoral [i.e., antimicrobial peptide (AMP), production in the fat body] or cellular (phagocytosis and encapsulation in the hemolymph) responses (reviewed in Lemaitre and Hoffmann 2007). The humoral response to infection is regulated at the transcriptional level to produce AMPs mediated by the Toll (gram positive microbes, fungi) and IMD (gram negative microbes) pathways (Kaneko and Silverman 2005; Lemaitre and Hoffmann 2007; Valanne et al. 2011). Upstream of these signaling cascades, the peptidoglycan receptor proteins (PGRPs) sense specific forms of bacterial cell-wall peptidoglycans and activate the immune pathways to regulate the expression of AMP genes that function in humoral immunity (IMD, Toll) or wound healing and stress defense (IMD, via the JNK pathway). Here, five PGRPs were upregulated, including PGRP-LC (required to activate the IMD pathway) and PGRP-SD (required for Toll signaling) (Table S6 and Figure S4) in addition to several key members of the IMD and Toll pathways (bsk, Dif, IM23, IM3, kay, Myd88, nec, and SPE). Most notably, at least one gene member from all seven classes of AMPs was expressed (Lemaitre and Hoffmann 2007), including gram-negative active DiptericinB, Attacin A, Attacin B, and Attacin C, Drosocin and CecropinC, gram-positive bacteria active Defensin, and anti-fungals Drosomycin and Metchnikowin (Figure S4). In addition to IMD- and Toll-regulated AMP upregulation, the primary defense genes regulated by the JAK/STAT pathway TepII (antimicrobial) and Turandot (stress genes) were also upregulated (Figure S4) (Lemaitre and Hoffmann 2007). Finally, several genes that function both in the humoral and cellular immune response (phagocytosis and engulfment) were present, including crq, NimC1, PGRP-LC¸ Sr-CI, and TepII in addition to wound-healing genes (Bc, melanisation) and the primary plasmtocyte-specific gene required for clotting (hemolectin).
The temporal expression patterns of the IMD- and Toll-specific PGRPs corresponded with the induction of target AMPs; i.e., the IMD PGRPs LC and LF were upregulated early in recovery (4–8 hr; Figure S4A) while the gram −ve AMPS peaked at 8–12 hr (Figure S4B). In comparison, peak PGRF-SD expression was delayed until 12 hr (Figure S4A), followed by the corresponding shift in the gram +ve AMP Def induction to 12–36 hr (Figure S4C), while the antifungals were induced by 8 hr but peaked at 12- to 36-hr recovery (Figure S4D). The IMD and Toll signaling genes tended to be induced by 8-hr recovery with peak expression at 12-hr recovery, although IM23 and IM3 (Toll signaling) were induced to a much greater degree (Figure S4B). Finally, peak expression of the JAK/STAT-regulated stress genes were delayed until 12–36 hr (Figure S4E).
Preponderance of intron-lacking and nascent transcripts during severe thermal stress
Of the 1112 genes profiled using STEM <60 (∼5%) were “early” responders (i.e., expressed during heat stress or by 4-hr recovery). Although the early genes clustered into three nonsignificant coexpression profiles, this group comprised the most highly inducible genes and were functionally enriched for Hsps (Figure 3B and Table S5). Expression was high within 15 min of heat stress and peaked at 4-hr recovery, with fold changes ranging from 23- to 120-fold for Hsp70ba, Hsp23, and Hsp68, respectively (Figure S1 and Figure 3B). While the exon and 3′ IVT module expression profiling showed comparable results for the majority of genes, slightly more genes clustered as early responders in the 3′ IVT module resulting in statistically significant enrichment of profile 38 (Figure S3 and Figure 3B).
Given the radical impact of severe heat shock on gene expression programs at both the transcriptional and RNA processing levels in cell cultures, expression of the transcripts encoded by the early responding genes was considered in more detail. The majority of thermally transcribed Hsps bypass RNA splicing prior to nuclear transport due to a lack of introns. For genes encoding transcripts that require splicing, thermal stress may have several effects on expression, including repressed or induced transcription, inhibited RNA processing and transport, or combinations of the above. If the splicing of newly transcribed or highly constitutively expressed transcripts is blocked, then transcripts accumulated during thermal stress should remain in their nascent state. The early responding genes coexpressed with the Hsps were first examined for intron paucity, where the pre-mRNA intron, mRNA, and CDs sequence lengths were compared among the genes with the most robust temporal patterns (Flybase version 2012_04, Table 2). Over half of the genes coded for short intronless transcripts (29/51 genes), with a median mRNA length of 874 bp and 666-bp coding region. This includes 12 genes with known roles in the heat-shock response (Hsps inclusive, Table 2). The intron-containing genes were characterized by few, short introns, median intron number 2 and median intron length 61 bp, and longer transcript lengths (median length 1550 bp, CDs 1293, Table 2).
Table 2. Transcript structure of the early responding genes from the constitutive exon analysis.
| Genea | Intron/s (bp) | mRNA (bp) | CDs (bp) | CDs interrupted by intron? |
|---|---|---|---|---|
| Acp54A1 | — | 293 | 140 | — |
| CG32198 | — | 470 | 411 | — |
| CG15357 | — | 485/560 | 347 | — |
| CG14966 | — | 587 | 422 | — |
| Robl37BC | — | 617 | 348 | — |
| obst-I | — | 661 | 659 | — |
| CG30487 | — | 728 | 597 | — |
| CG18125 | — | 744 | 719 | — |
| CG6974 | — | 756 | 755 | — |
| Acp29AB | — | 767 | 704 | — |
| GstE7 | — | 769 | 672 | — |
| GstE1 | — | 831 | 675 | — |
| CG4461b | — | 864 | 603 | — |
| Hsp23b | — | 849/884 | 561 | — |
| fan | — | 912 | 656 | — |
| CG32271 | — | 925 | 747 | — |
| Hsp26b | — | 958 | 627 | — |
| Hsp67Bcb | — | 987 | 600 | — |
| CG32302 | — | 1079 | 941 | — |
| Hsp27b | — | 1220 | 642 | — |
| CG2887b | — | 1234 | 1029 | — |
| CR17024 | — | 1288 | NA | NA |
| CG15904 | — | 1899 | 1899 | — |
| CR40546 | — | 1944 | NA | NA |
| CG11619 | — | 2100 | 2099 | — |
| Hsp68b | — | 2228 | 1908 | — |
| Hsp70Bc b | — | 2386 | 1926 | — |
| Hsp70Ba b | — | 2475 | 1926 | — |
| CG6000/CR44907 b | −/144 | 588/539 | 446/NA | −/NA |
| DnaJ-1b | −/245 | 2161/1916 | 1005 | −/N |
| Hsrωb | −/711;711;644 | 21,520;14,086/1176;1670;1223 | NA | NA |
| stvb | 65;78;118/65;443;25;78/2553;65;78;430 | 2491/2570;2496/3279;3357;382 | 1551/1629;1551;1551/830;1908;1833 | Y |
| CG31287b | 53;56 | 777 | 699 | Y |
| CG14227 | 67;65;67 | 933 | 861 | Y |
| CG10264 | 212;74 | 1022 | 811 | Y |
| Gr85a | 52 | 1194 | 1194 | Y |
| CG14011 | 54;59;62 | 1482/1477/1474 | 1167 | N |
| aur | 63;61 | 1573 | 1236 | Y |
| CG12267 | 68;62;56;57;57;49;68 | 1654 | 1569 | Y |
| Cyp6T3 | 60 | 1694 | 1506 | Y |
| Ugt86DE | 65 | 1735 | 1584 | Y |
| CG3281 | 52 | 1866 | 1617 | Y |
| Cyp6G2 | 50 | 1989 | 1560 | Y |
| CG15326 | 52;270 | 2004 | 2001 | Y |
| CG5608 | 67;62;66;51;60;61;66;52 | 2244 | 2064 | Y |
| CG6785 | 91;74 | 3985 | 1716 | Y |
| CG31659c | 62;48;60;60;48 | 632/695 | 579/528 | Y |
| CG15449c | 224;101 | 765 | 396 | Y |
| CG14342c | 99;74;58 | 776 | 426 | Y |
| CG13813c | 59 | 1527 | 1293 | Y |
| CG6792c | 55 | 2009 | 1410 | Y |
| ref(2)Pc | 631 | 2223/2417 | 1800 | Y |
| CG5204c | 205;56 | 2364 | 2244 | Y |
Pre-mRNA intron, mRNA and coding sequence (CDs) lengths are shown (/ indicates transcript isoforms).
List derived from the clustering output showing genes coregulated during thermal stress and may not include some thermally upregulated genes with different overall expression patterns.
Known role in response to heat including chaperones and protein folding, either from experimental data or inferred from sequence similarity.
Not detected as upregulated until 4-hr recovery at 25°.
Next, for a subset of the genes with introns, the extent of intron splicing during thermal stress was approximated using real-time PCR with exon–intron and exon/junction primer pairs to distinguish between pre-mRNA and mRNAs, respectively. Six genes coding for varying transcript, intron number, and size were chosen, including genes activated by HSF and adjacent to Hsp70BA (CG12267 and CG5608) and adjacent to Hsp70Aa, and Ab (aur) (in Figure 4A, note that the average log2 signals are shown without error bars for ease of comparison with Figure 4B, and untransformed data with error bars are shown in Figure S5). We also included Hsp83 as a control¸ which is heat induced but not processed in Drosophila cells and whole animal extracts in the upper temperature range of 38° due to the blocked splicing of a 1131-bp intron (Yost and Lindquist 1986).
Figure 4.

Detailed transcript analyses of early responding genes confirm RNA splicing inhibition during heat shock. Dashed lines indicate thermal stress at 38.5°; solid lines indicate recovery up to 48 hr post-stress at 25°. Exon profiles from the microarray analyses (constitutive exon set), shown as average normalized expression on the y-axis. (B) Real-time PCR profiling using exon-junction primers to target mature mRNA levels (solid line) and intron/exon primers to target pre-mRNA levels (shaded line) relative to RpL11 (y-axis). Negligible Gr85a and CG10264 mRNA suggests that these genes may be transcriptionally active during heat shock. Hsp83 is shown to confirm nascent transcript accumulation during heat stress. All data were log2 transformed for visual clarity and nontransformed data with error bars (±SE of the mean) are shown in Figure S5.
Barring CG32187, the genes exhibited differences in pre-and mRNA temporal patterns of expression/accumulation/abundance (Figure 4B), reflected in significant effects of time, transcript type, and time-by-transcript-type interactions in the two-way ANOVAs (P < 0.01–0.0001, Table S7). For each transcript type, relative transcript abundances during heat stress and recovery were compared to prestress conditions using one-way ANOVAs with Dunnett’s tests. In contrast to the exon array data, no changes were observed during heat stress for any mRNAs (Figure 4, A and B, and Table S8). During recovery, however, CG32187 and Hsp83 mRNAs were more highly expressed at 4- and 8-hr recovery (P < 0.01–0.0001, Figure 4, A and B, and Table S8). Although at quite low abundances relative to RpL11, pre-mRNA levels were highly elevated at hyperthermia compared to basal conditions (Figure 4B, P < 0.0001, Table S8). While pre-mRNA levels decreased over the time course, the relative abundances remained significantly different from prestress levels into recovery for some genes (Figure 4B, P < 0.0001–0.05, Table S8). Notably, while mRNA levels of Gr85a and CG10264 were at the lowest end of the PCR range indicating negligible expression (>35 cycles), pre-mRNA abundances increased dramatically during stress and comprised the majority of early detected transcripts. Although all RNA samples were DNase treated to remove genomic DNA, residual DNA present in RNA preparations may be selectively amplified using exon–intron primers. As a control, mock cDNAs (reverse transcriptase minus) were prepared from three random thermal stress samples and showed no amplification for any transcript after 40 PCR cycles (data not shown).
At the gene level, we demonstrated that exon-level analysis is a more powerful way than 3′-biased gene estimates to assess stress-regulated transcriptional output. The complex gene expression dynamics revealed that severe thermal stress elicited several modes of defense regulated at the transcriptional level. The strongest transcriptional signatures mapped to major defense modes including the immediate heat-shock response. This was accompanied by rapid but incomplete transcriptional inhibition and likely a global block in RNA processing- which is bypassed by the intron-lacking Hsps and other similar transcripts. Upon restoration to ambient temperature, Drosophila females mounted a broad-spectrum immune response encompassing genes associated with multiple innate biotic (pathogen) and abiotic (stress) defense responses. This was evident from the top down, beginning with the upregulation of stress sensing and recognition proteins to initiate signaling cascades that induce the expression of immune effectors, as well as downstream signaling involved in wound healing and other stress responses such as heat and oxidative stress.
Exon module analysis: partitioning temporal gene-level expression to the isoform-level during thermal stress and recovery
For multi-transcript genes, the time-series data were modeled in several ways to account for the complex structure of the genes and transcript specific probes (identified by exon type). First, two-way ANOVAs with the fixed effects of exon type, time point, and exon-type-by-time-point interaction were fit for probesets targeting alternative exons [2009 genes, model (2)]. The P-values, FDR-corrected P-values, and average log signals for the eight time points are given in Table S10. There was a strong effect of exon type, with >93% of genes showing average signal intensity differences between transcripts at FDR 0.05 (Table 3). Over 17% of genes showed evidence of temporal regulation at FDR 0.05, increasing to 23% at FDR 0.2 (Table 3). The majority of multi-transcript genes included probesets targeting at least one constitutive exon providing a gene-level expression measure. For many genes, the expression patterns of the more responsive alternative exons were captured by the constitutive exons, lowering power to detect an exon-type-by-time-point interaction (Table 3 and Table S10). This was significantly improved by including the mean normalized signal of the constitutive exons for each gene as a covariate in the model, thereby increasing the power to detect more subtle differences between isoforms over the time course by almost an order of magnitude [model (3), Table 3, and Table S9]. The data were filtered to retain genes with probesets targeting a minimum of two alternative exons and a constitutive exon, resulting in almost 10% (100/1094 genes) showing differential alternative exon expression over time (Table 3). The data were first tested for parallelism of slopes by testing for interactions between the covariate with the main terms of exon type and time point (see Materials and Methods). Significant covariate/main term interactions were negligible and not among the significant genes (data not shown).
Table 3. ANOVA results for the main effects of exon-type and time-point and the interaction term to test for differential expression of constitutive and/or alternative exons over the time course from 2009 multi-transcript genes [model (2)].
| Main effects [model (2)] | Interaction (exon type × time point) | |||
|---|---|---|---|---|
| FDR level | Exon type | Time point | Model (2) | Model (3)a |
| <0.05 | 1873 | 345 | 9 | 56 |
| <0.1 | 28 | 38 | 1 | 20 |
| <0.2 | 23 | 75 | 3 | 24 |
Model (3) was fit with the constitutive exons as a covariate for 1094 genes with constitutive exon(s) and at least two alternative exons (interaction term of interest shown).
Significance thresholds are based on type III SS. All model terms for (3) are given in Table S9.
A final master list of 493 significant genes was compiled from the statistical models incorporating the time-point term from model (2) (458 genes) and exon-type-by-time-point interactions from models 2 and 3 (13 and 100 genes, respectively; Table S9 and Table S10). The model (2) time-point term provides a summary of the thermally responsive multi-transcript genes as well as providing some isoform-specific information, i.e., where isoform representation is incomplete for a gene owing to different aspects of the probe annotations (gene model changes/ambiguities or overlapping exons classed as a single exon for simplicity). The time-point term may also indicate temporally coregulated isoforms. Information regarding isoform expression may also be indirectly inferred from the constitutive exon probeset signals where nonresponsive isoform/s are represented but the responsive isoform/s are omitted. For those genes with differential isoform expression over the time series, four genes from model (2) interactions overlapped with significant model (3) interactions, while the remainder were filtered from model (3) (i.e., without constitutive probes or with a constitutive probe and only one alternative exon).
Gene-level analyses miss complex transcriptome dynamics during thermal stress and recovery
By hybridizing two probe modules on one platform and universally correcting for nonspecific hybridization with probes designed to evaluate pure background (Affymetrix 2007), 3′-bias gene expression signals (3′ IVT probesets) were directly comparable to whole-gene and isoform-specific signals (exon probesets). To compare the efficacy of the different probe modules and models to detect more complex transcriptomic responses to thermal stress, the 2009 multi-transcript genes were reduced to 1721 genes common between the modules and probe types and compared for the time-point term. FDRs were readjusted in all cases for 1721 tests. Thermally responsive multi-transcript genes were highly enriched in the exon module [model (2), taking into account exon type] (Figure 5, χ2, d.f. = 1, P < 0.0001 for individual comparison, χ2, d.f. = 3, P < 0.0001 for group comparison). Significance levels were lowest in the 3′ IVT module and when the exon module was analyzed using model (1) (irrespective of exon type) (Figure 5, χ2, d.f. = 1, P < 0.0001 for individual comparisons). This is particularly evident in the 3′ IVT module with only 1.5% of genes detected as significant at FDR 0.05 (Figure 5). More genes were detected with the constitutive exon probesets across the time series for model (1) analysis, although this was still only about half of the genes compared to the exon probesets analyzed with model (2) (Figure 5).
Figure 5.

Proportion of 1721 multi-transcript genes common to the 3′ IVT module, exon-constitutive probes, and exon module for model (1) and all probes in the exon module for model (2) that were differentially expressed over the time series following severe thermal stress. FDR is shown at 0.05, 0.1, and 0.2.
Temporal profiling at the isoform level: underrepresentation of early responding isoforms and enrichment of mid- to late-recovery expression following severe thermal stress
The alternative exon/exon subsets (excluding constitutive exons) from the 493 master list genes from model (2) were grouped according to their temporal expression patterns using STEM (Ernst and Bar-Joseph 2006). The probesets were first resummarized with a log2 transformation then filtered to remove high-variance, low-expression transcripts (negative or zero background-subtracted values were not retained by adding a positive constant as in the statistical analysis; see Materials and Methods).
Of the 1330 transcript/subsets, 876 passed the filtering criteria (see Materials and Methods), and 616 (70%) clustered into six significantly overrepresented profiles (FDR < 0.05, Figure 6A). During heat stress, most enriched profiles were characterized by either stable or downregulated expression relative to time zero, with average peak expression at either 12- or 36-hr recovery (Figure 6A). The majority of transcripts were detected by 12-hr recovery, with expression peaking at 36 hr and decreasing to prestress levels by 48 hr (profile 27, combined profiles 11+18, Figure 6A). Transcripts with the highest expression at 36 hr also tended to be induced by 4-hr recovery (profiles 41 and 34, Figure 6A), although expression resumed prestress levels by 48 hr in profile 34 but remained elevated in profile 41 (Figure 6A). Gene-enrichment annotation by coexpression profile showed an overabundance of early to mid-expressed transcripts coded by immunoglobulin-like, transmembrane, and ribonucleotide binding genes (profiles 40 and 14, Table S11 and Table S12). Expression normalized to time zero ranged from 2- to 10-fold increases from 4- to 12-hr recovery in genes such as heat-shock cognate Hsc70-3 (heat shock), CA-P60A (calcium homeostasis), CG10924 (gluconeogenesis), Dgp-1 (GTP binding), TepII (antibacterial humoral response), and CG32103 (transporter) (Table S10, Table S11, and Table S12).
Figure 6.

STEM profiles of 876 transcripts/transcript subsets from 493 heat-responsive multi-transcript genes identified from the exon module with ANOVA. Average normalized (relative to time zero) signals are shown on the y-axis; time in hours is shown on the x-axis. The dashed line indicates thermal stress up to 31.5 min at 38.5°; solid line indicates recovery up to 48-hr post-stress at 25°. (A) Profiles enriched for mid- to late-recovery transcripts with peak expression at 12 or 36 hr. Early responding transcripts (B) are underenriched and more highly expressed, peaking at either late stress or early recovery.
Mid- to late-recovery transcripts were coded by genes enriched for phosphoproteins (i.e., CG5288), transmembrane function (CG15096), muscle-cell function, and actin binding (profiles 27 and 34, Table S12). Transcripts suppressed or downregulated until late recovery formed the largest temporal profile, reflected in the wider array of enriched gene functions including calcium ion binding, actin binding, immunoglobulins, ion transport, transmembrane, homeostatic processes, and neuron recognition (profiles 11+18, Table S12).
In agreement with the gene-level analysis, early responding transcripts were rare, comprising 4.1% of the temporally regulated transcripts/transcript subsets. These transcripts were summarized into three nonenriched profiles, with peak expression at late stress (profile 49, Figure 6B), 4 hr (profiles 21+45, Figure 6B), or both (profile 48, Figure 6B). The three profiles were pooled for gene annotation enrichment analysis, which together showed an abundance of genes for neuron development and differentiation, as well as transmembrane function (Table S12).
Over 90% of multi-transcript genes exhibited differences in isoform/subset expression levels, and almost a quarter were heat responsive [ANOVA model (2)]. When the constitutive exons were taken into account [ANOVA model (3)], almost 10% of genes had differential isoform/subset expression over the time series. Exon-specific analyses revealed more thermally induced expression complexity than gene-level analyses; i.e., exon analyses detected an order of magnitude more thermally responsive multi-transcript genes than 3′−end gene estimates at FDR = 0.05. Similar to gene-level analyses, temporal profiling of isoform/subsets revealed a paucity of expression during high temperature with transcripts regulated either early to mid- or mid- to late recovery. Early to mid-recovery transcripts mapped to genes with enriched functions associated with immunoglobulins, phosophoproteins, ribonucleotide, and transmembrane binding, while late expressed transcripts mapped to genes involved in immunoglobulins, ion transport, and homeostatic processes.
Heat stress affects gene expression programs via complex RNA processing
The two-way ANOVA and coexpression analyses revealed a number of multi-transcript genes varying in degrees of structural complexity and differential isoform expression. Characteristic of exon-level analyses, a number of exons/subsets showed subtle expression changes over time, which can be difficult to both identify and interpret (Lockstone 2011). To demonstrate the breadth of events underlying transcriptome complexity in response to heat stress, a subset of genes with clear expression patterns was focused on in more detail, and while certainly not exhaustive, they present an array of transcriptional and RNA processing examples.
During thermal stress, several of the most highly detected alternative exons mapped to genes identified as heat responsive either through experimental evidence or electronic annotation (McQuilton et al. 2012). Isoform generation included events such as alternative splicing (Hsrω, DnaJ-1, CG6000), alternative start exons, transcription and/or alternative donor sites (stv, Hsc70-3, Hs70-4), and intron retention (stv). During recovery from heat stress, differential isoform expression was greatest between 4- and 12-hr recovery in a number of genes with transcripts generated by alternative transcription (kay, Dgp-1, Dip-B) alternative splicing of cassette exons (TepII), and combinations thereof (CG10924, srp, Xrp1).
Hsrω (nuclear speckle organization), DnaJ-1, and CG6000 (protein folding) each code for intronless transcript/s that were stress regulated in the exon array data (Figure 7A, left box, B and C bottom box). Heat-induced expression was observed for the intron-lacking ncRNA subset Hsrω F:B (formerly the nuclear N transcript), peaking at 4-hr recovery (Figure 7A, left box). Owing to overlapping exon structure between all transcripts, the intron containing ncRNA subset (Hsrω-A;D;E formerly the cytoplasmic or C transcript, 644/711-bp intron) was not included in the array analysis. Real-time PCR was employed to assess the relative abundances of the intron-lacking F:B subset of the processed A:D:E subset (Figure 7A, right box). Two-way ANOVA revealed significant terms for transcript, time point, and time-point-by-transcript interaction (P < 0.0001–0.05); one-way ANOVAs with Dunnett’s tests confirmed expression during heat stress relative to time zero in the F:B subset (P < 0.05) and showed that expression was delayed until early recovery in the A:D:E subset (P < 0.01).
Figure 7.

Heat-elicited intronless isoforms of known heat-responsive alternatively spliced genes. (A) Hsrω: gene model showing intronless nuclear transcript subset RB:RF and spliced cytoplasmic subset RD:RA:RE (top); average normalized log2 exon expression for the RB:RF subset and constitutive exons (left); and real-time PCR data confirming early RB:RF expression relative to RpL11 and delayed expression of the RD:RA:RE subset (right). The PCR data were log2 transformed for visual clarity and nontransformed data with error bars (±SE of the mean) are shown in Figure S6. (B) Dnaj-1: gene model showing intronless transcript RB and spliced isoform RA (top); average normalized log2 exon expression showing strong induction of the RB transcript relative to both transcripts during heat stress (bottom). (C) CG6000/CR449007: gene model showing intronless RC transcript and spliced RB/CR44907 isoform and proximity to Hsp68 (top); average normalized log2 exon expression showing strong induction of both transcripts.
The intronless isoform DnaJ-1-RB increased in abundance over 55-fold prestress to 31.5 min at 38.5°, while probesets targeting exons common to both the RA (245-bp intron containing) and RB isoforms showed only a 2.8-fold increase (Figure 7B). Consistent with the modENCODE Drosophila temporal expression data (Graveley et al. 2011a), the processed isoform is constitutively highly expressed in adult females, while the exon data captured the rapid accumulation of intronless isoform during heat stress (Figure 7B).
Transcript abundances at the CG6000 locus (adjacent to Hsp68) increased during heat stress (Figure 7C). Genome annotations prior to FlyBase version FB2012_05 documented two CG6000 isoforms; CG6000-RA (intron lacking) and CG6000-RB (144-bp intron). However, from FB2012_05, CG6000-RB was reclassified as the noncoding gene CR44097 and ncRNA CR44097-RA, and CG6000-RA was renamed CG6000-RC (McQuilton et al. 2012). The exon probesets were blasted to the updated annotation and fell into unique regions targeting CG6000-RC and CR44097-RA, as well as a common region of both. Both transcripts showed peak expression at late stress, but differed in temporal patterns (exon-type-by-time-point interaction FDR < 0.0001) and while CG6000-C was moderately expressed under nonstress conditions consistent with modENCODE data, CR44097-RA abundance increased from very low/absent prestress to low expression peaking by late stress (Figure 7C).
The exon data revealed differential expression patterns including high-stress induction of isoforms/subsets at the stv and the heat-shock cognate Hsc70-3 and Hsc70-4 loci (exon-by-time-point interaction FDR < 0.0001). In all cases, the responsive isoforms derive from alternative start exons coded from the intron of the primary transcript (Figure 8, A, B, and C, top). Given this transcript structure, two scenarios may account for the elevated alternative exon signals: (1) hybridization of accumulated pre-mRNA intronic sequence resulting from blocked splicing or (2) hybridization of alternative start exon sequence resulting from heat-induced alternative transcription. To test this, real-time PCR was employed to compare the relative abundances of the unprocessed primary transcripts (intronic primer close to the first start exon subset) and mRNAs (exon-junction primers). Of the seven annotated stv transcripts (chaperone binding, proteolysis), alternative exons targeting the smaller RB:RC:RG subset and RD transcript showed strong elevation during thermal stress (40- and 16-fold relative to nonstress, respectively), while the long RA:RE:RF subset only very marginally increased by 4-hr recovery (Figure 8A, middle). Consistent with the array data, the two-way real-time PCR ANOVA was significant for all terms (time, transcript, and time-by-transcript interaction, P < 0.0001). However, the PCR data for the processed RB:RC:RG subset revealed only a slight, nonsignificant high-temperature increase, with the largest increase delayed until 4-hr recovery (Figure 8A, bottom). Instead, pre-RA:RE:RF levels were significantly higher by late stress (P < 0.05), while the processed RA:RE:RF transcript set was moderately highly expressed at a general level but did not change during stress or recovery (Figure 8A, bottom). Similar to the array data, the processed RD transcript was detected in low abundance but, unlike the array data expression, was outside the range of PCR detection until 4-hr recovery (Figure 8A, bottom). For Hsc70-4 (chaperone binding), the large RA transcript is extremely highly expressed across Drosophila development and maturity (Graveley et al. 2011a), consistent with the exon array data. Exon probsets revealed that RA was both thermally and temporally stable, in contrast to the smaller transcripts with alternative exons nested in the RA intron, which showed marked signal intensity elevation during thermal stress up to 4-hr recovery (Figure 8B, top and middle). Real-time PCR confirmed the stable expression of the RA transcript, but showed that only the unprocessed pre-RA transcript accumulated during high temperature and early recovery (Dunnett’s test, P < 0.05), with transcript abundances overlapping with the thermally and temporally stable RD mRNA (Figure 8B, bottom). Finally, the Hsc70-3 (RNA interference, centrosome duplication) short and long alternative exons represented by the exon probes showed differential expression patterns during thermal stress, with the short-form RC increasing during stress and the long-form RB increasing during recovery to peak at 12-hr post-stress (Figure 8C, top, middle). Owing to low complexity sequence at the exon junctions, transcript-specific primer design was not successful with several software options, even with relaxed stringency conditions. Instead, an intron–exon primer pair was designed flanking the RB start exon to assess the unprocessed RNA patterns. One-way ANOVA revealed a significant effect of time (P < 0.01), while Dunnett’s tests showed that expression relative to nonstress conditions was significantly higher at early stress and recovery (P < 0.05, <0.01, respectively, Figure 8C, bottom) as well as 8- and 12-hr recovery (P < 0.01, Figure 8C, bottom).
Figure 8.

Elucidating expression complexity of alternatively transcribed genes upregulated during thermal stress in the exon data. (A) stv: gene model showing the long and short transcript subsets derived by alternative transcription and alternative splicing (top); average normalized log2 exon expression for the RA:RE:RF, RB:RC:RG subsets and RD and constitutive exons (middle); and real-time PCR data relative to RpL11 showing no expression variation of the RA:RE:RF subset, accumulation of the primary transcript during heat stress, and induction of the RB:RC:RG subset and RD isoform by alternative transcription only during recovery. (B) Hsc70-4: gene model showing isoforms derived by alternative transcription (top); average normalized log2 exon expression of the RA, and RD isoforms, and RB:RF subset (middle); and real-time PCR data showing only accumulation of the primary transcript during heat stress (bottom). (C) Hsc70-3: gene model showing isoforms derived by alternative transcription (top); average normalized log2 exon expression of long (RB, recovery induced), and short (RC, stress induced) (middle); and real time PCR data for only the primary transcript showing accumulation during heat stress (bottom). The PCR data were log2 transformed for visual clarity and nontransformed data with error bars (±SE of the mean) are shown in Table S7.
During severe thermal stress, several of the transcripts with highest signal intensities from exon array data were mapped to known heat-responsive genes. Hsrω, DnaJ-1, and CG6000 showed increased expression of intronless isoforms during high temperature although temporal expression patterns were gene specific. The long noncoding HsrωRB:RF RNA was weakly heat induced, with peak expression at early recovery, returning to prestress levels by 48 hr. Similar to Hsps, the chaperones Dnaj-1 and CG6000 were strongly induced by thermal stress although they differed in peak expression at 4-hr recovery and late stress, respectively. However, the intron-containing noncoding RNA at the CG6000 locus was highly induced during stress before returning to prestress levels by 12-hr recovery. The exon data also showed elevated probe signals in the alternative start exons of stv, Hsc70-4, and Hsc70-3. Further examination with real-time PCR revealed that the signals were likely due to accumulated primary transcript, indicative of blocked RNA processing of transcripts during heat stress. This was supported by delayed elevation of processed isoforms of Hsrω, stv, and Hsc70-3 until recovery.
Multiple modes of alternative exon expression during recovery from thermal stress: alternative transcription, alternative splicing, and intron retention
Several genes with complex transcript structures exhibited distinct patterns of differential alternative exon expression during stress recovery in isoforms produced by alternative splicing, or by combinations of splicing mechanisms. For example, srp (transcription-factor-specific DNA binding, immune inducibility) codes for six transcripts as well as a noncoding RNA (CR44133) (Figure 9A, top). The exon probesets target four transcript subsets of which two showed evidence of differential temporal variation [model (3) interaction term FDR< 0.0001, STEM profiles 40 and 16, Tables S10, S11]. Signal intensity of the RB:RE subset increased by 8-hr recovery, while increases in the RA:RB subset were delayed until 12-hr recovery (Figure 9A, middle). The RA:RB transcripts differ in a cassette exon that is common to RB and RE¸ while RB and RE differ in start exons and retained intronic sequence (Figure 9A, top). Exon-junction priming confirmed these expression trends (time term P < 0.01, transcript term P < 0.0001, Figure 9A bottom).
Figure 9.

Examples of differentially expressed isoforms derived from combinations of alternative transcription and splicing during recovery. (A–C, top) Gene models showing the different transcript isoforms. (A) srp: average normalized log2 exon expression of the RA:RB, RA:RD:RF:RG, RB:RE, RD:RG:RE:ncRNA subsets and constitutive exons (middle), and average expression relative to RpL11 confirming the different temporal profiles of the RA:RB and RB:RE subsets (bottom). (B) Xrp1: average normalized log2 exon expression of the RB, RD, and constitutive exons and RA:RE:RF:RG and RA:RB:RC:RE:RG subsets; and average expression relative to RpL11 confirming the early recovery induction of the short RD isoform but similar profiles of the RB and RD isoforms at 8 and 12 hr. (C) TepII; average normalized log2 exon expression all alternative plus constitutive exons showing differential induction of all isoforms during recovery from thermal stress (bottom). The PCR data were log2 transformed for visual clarity and nontransformed data with error bars (±SE of the mean) are shown in Table S7.
The Xrp1 locus (DNA binding, protein dimerization) codes for seven transcripts differing in transcriptional start sites and cassette exons (Figure 10B, top). Probesets targeting the alternative start exons of the longest and shortest isoforms RB and RD (respectively) exhibited differential temporal expression where RD began to accumulate by 4-hr recovery peaking at 8-hr recovery, while the long-form RD exhibited later induction with peak expression at 12-hr recovery (Figure 9B, middle, STEM profiles 40 and 14, Table S11). Real-time PCR confirmed the early expression of RD relative to RB, but did not differentiate between the 8- and 12-hr recovery expression peaks as in the array data (Figure 9B lower).
Figure 10.

Examples of differentially expressed isoforms from alternative transcription during recovery from thermal stress. (A–C, top) Gene models showing the different transcript isoforms. (A) CG10924: average normalized log2 expression of the RA, RB, and constitutive exons (middle); average expression relative to RPL11 confirming recovery induction of the RA isoform and differential expression of the RB isoform, although this was out of the PCR range during stress. (B) kay: average normalized log2 expression of all isoform exons and constitutive exons (middle); average expression relative to RpL11 confirming the different temporal profiles of the RB and RF isoforms (bottom). (C) Dgp-1; average normalized log2 expression showing the strong induction of the short RB isoform by 8-hr recovery and the delay in the longer RA isoform until 12-hr recovery (bottom). (D) Dip-B: average normalized log2 expression of the RA:RC subset and constitutive exons illustrating the induction of the shorter RB isoform, which can be inferred from the constitutive exon profile (bottom). The PCR data were log2 transformed for visual clarity and nontransformed data with error bars (±SE of the mean) are shown in Table S7.
All five TepII (defense, antibacterial immune response) isoforms were modulated during recovery with peak expression at 12-hr recovery (Figure 9C, bottom). The isoforms differ by an alternatively spliced cassette exon (Figure 9C, top) and despite similar expression peaks, there were variations in the overall temporal patterns of expression [model (3) interaction term P < 0.0001, STEM profiles 41, 14, and 40, Tables S10, S11].
Alternative start site usage during recovery from thermal stress
During recovery, the exon data revealed differential temporal expression of several genes with isoforms generated by alternative transcriptional start sites (CG10924, kay, Dgp-1, Dip-B, Figure 10, top). Probesets targeting the short CG10924 isoform RB (GTP binding, gluconeogenesis) showed signal increases from extremely low/absent expression by early stress up to 12-hr recovery, while the long RA isoform increased from 4-hr recovery with peak intensity at 8- and 12-hr recovery (Figure 10A, middle). Real-time PCR with exon junction primers confirmed significant differences in the temporal patterns of the two isoforms (exon-type-by-time-point interaction FDR < 0.0001, Figure 10B, bottom). While the expression levels of the RB isoform were not detected with PCR until 4-hr recovery, the temporal patterns of the two isoforms remained significantly different when compared from 4 hr onward (exon-type-by-time-point interaction P < 0.0001). The short RB isoform was detected by 4-hr recovery and maximally expressed at 8-hr recovery, with a sharp decrease after 12-hr recovery, while the magnitude of change was less pronounced for the RA isoform (Figure 10A, bottom). Expression patterns during stress, however, require further investigation given the extremely low level of detection using both platforms.
The kay locus (RNA pol II transcription factor, JNK cascade) codes for four alternatively transcribed isoforms and showed differential expression in the exon data [model (3) interaction term P < 0.0001, Figure 10B, middle]. Coexpression analyses grouped the two shortest isoforms RB and RF into profiles 40 and 14, respectively (Table S11), where RB showed elevated expression early in recovery, while RF expression was delayed until 12 hr (Figure 10B, middle). Real-time PCR confirmed the overall temporal patterns of all four transcripts including the early expression of RB (Figure 10B, bottom; transcript term P < 0.0001, time term P < 0.01), although the interaction term was not significant given the higher variance in the PCR data.
Dgp-1 (Elongation factor-GTP binding domain) and Dip-B (tri/di-ptidyl-peptidase, proteolysis) code for isoforms via alternative start exons in the untranslated regions (UTRs, Figure 9, C and D, top). Alternative exon probesets targeting the truncated isoforms of both genes showed elevated expression by 8-hr recovery from thermal stress (Figure 10, C and D, bottom). Signal intensity of the less-abundant Dgp-1 RB exon rose to match RA abundance between 4- and 8-hr recovery, and remained elevated at 12-hr recovery, whereas increases in RA isoform abundance were delayed until 12-hr recovery [Figure 10C, bottom, model (3) ANOVA exon-type-by-time-point interaction term FDR < 0.05, STEM profiles 40 and 14, Table S11].
Dip-B illustrates where the expression of a strongly responsive alternative exon not represented on the array can be inferred from the constitutive exon signals [model (2) ANOVA exon-type-by-time-point-interaction FDR < 0.0001, Table S10]. Probesets targeting the RA:RC showed no change over the time series, while probesets targeting RA:RB:RC exons strongly increased in signal intensity by 8–12 hr, indirectly highlighting RB as the thermally responsive isoform (Figure 10D, bottom).
While intronless isoforms of heat-responsive genes were preferentially induced during thermal stress, differential expression of isoforms generated by multiple events was observed during recovery at basal temperatures when processing is reestablished. This included alternative transcription, alternative splicing (including intron retention and cassette exon splicing) and combinations thereof. Exposure to high temperature therefore affected gene expression at multiple modes of regulation, generating noncoding transcripts, transcripts differing in 5′-UTRs but coding for the same protein as well as transcript isoforms coding for different proteins. The differentially regulated isoforms mapped to genes important in thermotolerance, gene regulation, and immunity providing a link to the strongest expression signatures observed from the gene-level analyses.
Discussion
In this study we analyzed the Drosophila transcriptome during severe heat stress and recovery using exon expression. We employed a thermal regime at the upper range of the heat-shock response and the physiological limits of a natural fly population. To our knowledge, this is the first global analysis of the effect of heat stress on innate RNA metabolism at the whole organism level. Severe heat stress significantly affected the modulation of gene expression, including RNA transcription and processing events such as constitutive and alternative splicing.
As predicted, additional thermally responsive loci were detected using exon-level analyses when directly compared to 3′-end biased analyses. Gene-level detection was slightly higher with constitutive exons than when analyzed across all exons; this was likely due in part to more reliable signal estimates from “core” (high-certainty existence) annotated exons and/or lower signal intensities of alternatively expressed exons (Lockstone 2011).
Exon-level analyses provided >16% greater sensitivity to detect multi-transcript genes with at least one heat-responsive transcript compared to gene-level analyses and revealed a greater degree of transcriptome plasticity during heat stress and recovery than previously profiled with gene-level analyses (i.e., Leemans et al. 2000; Sorensen et al. 2005; Nielsen et al. 2006). Our results support findings that isoform-level analyses reveal transcriptome complexity that is more comprehensive than analyses restricted to the gene level (Howard and Heber 2010) and, importantly, highlight gaps in understanding the impact of stress on gene expression programs.
Gene-level analyses
Congruent with heat-inhibited transcription, only a small set of genes showed elevated signals during heat stress, while most changes were delayed for up to 8 hr into recovery. We anticipate, however, that the number of genes transcriptionally active during heat stress were likely underestimated, as hybridization-based profiling may be less sensitive to detect rapid transcriptional changes than direct methods such as mapping Pol II bound to DNA (Teves and Henikoff 2011). Regardless, heat-induced transcription was evident for over a quarter of the genes here known to be heat activated largely by HSF binding (Guertin and Lis 2010; Gonsalves et al. 2011; McQuilton et al. 2012).
As predicted, the most highly inducible genes were Hsps coding for intronless transcripts. Hsps have evolved a lack of intervening sequences to allow newly synthesized transcripts to bypass the splicing block and accumulate in the cytoplasm for rapid, functional protein synthesis (Yost et al. 1990). Moreover, intron paucity is a general feature of rapidly stress responsive genes across eukaryotic lineages, where the energetic costs of transcription are proportional to intron lengths (Jeffares et al. 2008). Hsps comprised 12 of the 31 genes coding for intronless transcripts, while the remainder were mostly protein-coding genes not previously associated with thermal induction. For a number of the latter genes, the Drosophila ModENCODE treatment data revealed evidence for varying degrees of expression in response to temperature extremes and/or different chemical exposures (Graveley et al. 2011b), suggesting that at least some of the genes may be stress responsive (i.e., moderate to very high heat-shock induction of GstE1, GstE7, CR40546, CG32198). Further, several of the genes were either targets of or regulated by HSF including CG18125, GstE7, GstE1, and CG14966, with the latter being adjacent to Hsp83 (Birch-Machin et al. 2005; Jensen et al. 2008; Gonsalves et al. 2011).
Of the “early expressed” genes containing introns, several were known targets of HSF or proximal to members of the Hsp70 gene family (Birch-Machin et al. 2005; Jensen et al. 2008; McQuilton et al. 2012), i.e., stv, aur, CG12267, CG3281, CG5608, CG6785, CG6792. At least some of these genes are likely transcriptionally heat activated, although genome-wide studies demonstrate that thermally induced transcripts are not exclusively HSF regulated; likewise genes with HSF-bound promoters are not exclusively thermally transcribed (Guertin and Lis 2010; Gonsalves et al. 2011).
Given the uncertainty of the global impact of heat stress on transcript processing despite the splicing blockade being one of the best characterized stress responses (Biamonti and Caceres 2009), we investigated splicing in a novel subset of early responding transcripts. Consistent with heat-induced splicing inertia, we showed that mRNA abundances did not increase at 38.5° compared to nonstress conditions, in contrast to all but one of the pre-mRNAs. Moreover, the genes reflected a range of expression differences in response to heat stress. For genes that were moderate to very highly expressed under nonstress conditions (i.e., constitutively expressed genes with regulatory roles such as aur, Hsp83, CG12267), the elevated high-temperature exon array signals comprised a mixture of pre-and mature mRNAs. For genes with lower basal signal intensities, the elevated exon signals were composed predominantly of primary transcripts with peak abundances at either late stress or early recovery. Only Hsp83 and CG31287 mRNAs were significantly elevated during recovery, suggesting an increase in transcription with delayed processing of mature transcripts in response to heat stress, while aur and CG12267 appear to reflect only blocked splicing of constitutively expressed transcripts. Conversely, Gr85a and CG10264 appeared to be thermally transcribed consistent with low expression here and lack of expression in adult tissues under basal conditions (Graveley et al. 2011a).
The gene-level temporal dynamics revealed that thermally challenged Drosophila mounted several defense responses downstream of the initial heat-shock response, but for brevity we limit our consideration to the largest immune response. While the general impact of the heat-shock response on expression programs during hyperthermia is better elucidated, less clear is the role of immune defense during recovery. Emerging evidence increasingly supports molecular “cross-talk” between innate immune (specifically AMP expression) and other stress responses such as oxidative, osmotic, and nutrient stress (reviewed in Davies et al. 2012), as well as hypothermia (Sinclair et al. 2013). Aside from cross-talk or shared stress/immunity pathways, heat stress may elicit the immune response due to increased bacterial load resulting from thermal mismatching between the fly and flora. Given the upregulation of genes encoding AMPs across the entire spectrum of activity (gram −ve, +ve bacteria, yeast, and fungi), heat stress may cause increased susceptibility to increased commensal load (flies are allowed to recover on fresh standard medium), elicit a general stress response that is manifested in the “broad spectrum” immune response, and/or deplete essential proteins required for innate immunity that are replenished during recovery. At low temperatures, the immune response is inducible via tissue damage (i.e., wound-healing responses, or response to the release of gut flora into the hemocoel; Sinclair et al. 2013). Hyperthermia may induce similar scenarios despite flies recovering from our thermal regime.
Isoform/isoform subset-level analyses
For genes encoding multiple transcripts, exon-level analyses revealed pervasive differences in isoform/isoform subset abundances. Almost a quarter of genes were heat responsive from alternative exon analysis in contrast to gene-level estimates that ranged from only 7% (3′-end biased and across all exons) to 12% (exons targeting all transcripts). This deficit highlights the limitations of relying on total or partial (i.e., 3′-end biased) transcriptional output to understand the transcriptional response to thermal stress. Our data provide the first evidence of deeper levels of innate expression complexity in a thermally challenged organism, in the form of alterative exon expression in up to 10% of genes tested. Furthermore, the true extent of this complexity is likely underestimated given the short temporal sampling and modestly underpowered statistical analyses, as well as a simplified annotation that combined overlapping exons with alternative 3′ and/or 5′ sites into a single unique exonic region resulting in transcript subsets and omitted novel (i.e., unannotated) splicing events.
Temporal profiling of the full set of genes with either time responsive or differential exon expression revealed a paucity of heat-expressed transcripts similar to the gene-level analyses.
In this study, the exons most stably heat regulated tended to map to known thermally responsive genes and showed stress specificity, although not reflective of the proposed heat-induced alternative splicing model. Instead, intronless isoforms of the molecular chaperones Dnaj-1, CG6000, and Hsrω were induced, suggesting a bypass in splicing under high temperature. While an area of intensive research in human cell lines, the direct mechanisms mediating the splicing blockade and AS regulation during hyperthermia remain obscure. Blocked splicing likely results from diminished splicing efficiency due to the destruction of spliceosome components, the small nuclear ribonucleoprotein (snRNP) complexes, and reduced U1snRNP splice site recognition via interactions between the splicing repressor SRSF10 and U1snRP (Biamonti and Caceres 2009; Velichko et al. 2013). While we have demonstrated splicing inhibition and transcriptome plasticity in thermally challenged Drosophila, the degree of conservation of RNA-processing mechanisms affecting the spliceosome under hyperthermia in mammals and invertebrates remains to be tested.
Heat-shocked Drosophila cells do, however, assemble comparable structures to the nuclear stress bodies or “stress granules” that are proposed to affect AS regulation in human cells. The Hsrω gene codes for different transcript types that share the same 5′ end but are derived by alternative polyadenylation, a 3′-mRNA processing event that also gives rise to transcript and proteomic diversity (reviewed in Shi 2012). Under heat stress, the noncoding nuclear (intronless) or hsrω-n transcripts regulate RNA processing through the formation of nuclear “speckles” that recruit hnRNPs (heterogeneous nuclear ribonucleoproteins that associate with pre-mRNAs) (Prasanth et al. 2000; Jolly and Lakhotia 2006). During recovery, critical levels of hsrω-n are thought to be important in the spatial restoration of RNA processing factors (Lakhotia et al. 2012), while elevated cytoplasmic hsrω-c (intron containing) transcript levels during both heat and cold recovery suggest a further role of this gene in thermotolerance (Johnson et al. 2011). Here, both transcript sets were heat responsive, although expression of hsrω-c was delayed until recovery consistent with blocked splicing (Bendena et al. 1989). The modulation of Hsrω transcripts provides an obvious candidate mechanism for splicing regulation in natural Drosophila populations during heat shock and recovery.
It has been proposed that heat shock modulates alternative transcript profiles of genes that function in cell recovery (Biamonti and Caceres 2009). We clearly show that thermal stress affected alternative transcript expression on restoration to ambient temperatures over a 24-hr recovery period. We anticipate that the temporal expression profiles were unlikely confounded with circadian transcriptional rhythms as wild-type flies become arrhythmic under constant light (Konopka et al. 1989), and moreover, while isoforms of some genes are regulated by the period gene, circadian time affects isoform expression to a much lesser extent (Hughes et al. 2012).
While exhaustive analyses of all transcripts and genes are beyond the scope of this study, we highlight several examples representing the diversity of RNA metabolic events during recovery. Different transcript and/or protein isoforms may be favored during and/or following stress owing to greater transcriptional or translational efficiency, particularly in the latter case for transcripts differing only in their UTRs (Barrett et al. 2012). In the former, the translation of alternative transcript abundances to alternative polypeptide abundances and, further, the extent to which these relate to different functions, if at all, should become the purpose of future research. We highlighted differential expression of several genes with regulatory roles that could conceivably play a role in recovery mediated gene expression (i.e., srp, Xrp1, kay), particularly in terms of innate immunity. Moreover, the antibacterial gene TepII contains a central hypervariable region corresponding to specific complement factors that bind to pathogen surfaces in vertebrates (Bou Aoun et al. 2011). Alternative splicing of all five TepII isoforms during recovery presents an intriguing case to further investigate links between immune challenge and environmental stress.
Finally, we demonstrate for the first time that short isoforms derived from a complex combination of alternative transcription and splicing drive the elevated expression of the chaperone-binding stv gene during recovery from thermal stress. The stv locus codes for Bcl-2-associated anthanogene (BAG) proteins that can regulate the function of Hsp70, is a target of HSF, and has been shown to be thermally responsive (Jensen et al. 2008; Colinet and Hoffmann 2010; Gonsalves et al. 2011), although expression patterns have so far been described only at the gene level.
Our results highlight how total transcriptional output, gene-level studies miss the true complexity that underlies organismal responses to environmental stress. We have shown in an animal model that diverse mechanisms such as alternative splicing, alternative transcription, and the splicing block all contribute to the transcriptional complexity both during and in recovery from thermal stress. Future studies need to take this complexity into account when attempting to link genomic-level to organismal-level responses to stress.
Supplementary Material
Acknowledgments
We thank Lauren McIntyre and the McIntyre lab for bioinformatic support. We are grateful to two anonymous reviewers whose comments improved the manuscript. This work was funded by the Australian Research Council via its DECRA, Fellowship and Discovery Grant schemes, and by the Science and Industry Endowment Fund.
Footnotes
Communicating editor: J. A. Birchler
Literature Cited
- Addo-Bediako A., Chown S. L., Gaston K. J., 2000. Thermal tolerance, climatic variability and latitude. Proc. R. Soc. Lond. B Biol. Sci. 267: 739–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affymetrix, 2007 Quality assessment of exon and gene arrays. http://www.affymetrix.com/support/technical/whitepapers.affx
- Ali, G. S., and A. S. N. Reddy, 2008 Regulation of alternative splicing of pre-mRNAs by stresses, pp. 257–275 in Nuclear Pre-mRNA Processing in Plants Springer-Verlag, Berlin. [DOI] [PubMed] [Google Scholar]
- Arvidsson S., Kwasniewski M., Riano-Pachon D. M., Mueller-Roeber B., 2008. QuantPrime: a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics 9: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett L. W., Fletcher S., Wilton S. D., 2012. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 69: 3613–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendena W. G., Garbe J. C., Traverse K. L., Lakhotia S. C., Pardue M. L., 1989. Multiple inducers of the Drosophila heat shock locus 93D (hsr omega): inducer-specific patterns of the three transcripts. J. Cell Biol. 108: 2017–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y., 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300 [Google Scholar]
- Biamonti G., 2004. Nuclear stress bodies: A heterochromatin affair? Nat. Rev. Mol. Cell Biol. 5: 493–498 [DOI] [PubMed] [Google Scholar]
- Biamonti G., Caceres J. F., 2009. Cellular stress and RNA splicing. Trends Biochem. Sci. 34: 146–153 [DOI] [PubMed] [Google Scholar]
- Birch-Machin I., Gao S., Huen D., McGirr R., White R. A., et al. , 2005. Genomic analysis of heat-shock factor targets in Drosophila. Genome Biol. 6: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond U., 1988. Heat-shock but not other stress inducers leads to the disruption of a subset of SNRNPS and inhibition of invitro splicing in hela-cells. EMBO J. 7: 3509–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bou Aoun R., Hetru C., Troxler L., Doucet D., Ferrandon D., et al. , 2011. Analysis of thioester-containing proteins during the innate immune response of Drosophila melanogaster. J. Innate Immun. 3: 52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken A. P., Bond U., 1999. Reassembly and protection of small nuclear ribonucleoprotein particles by heat shock proteins in yeast cells. RNA: a Publication of the RNA Society 5: 1586–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colinet H., Hoffmann A., 2010. Gene and protein expression of Drosophila Starvin during cold stress and recovery from chill coma. Insect Biochem. Mol. Biol. 40: 425–428 [DOI] [PubMed] [Google Scholar]
- Corell R. A., Gross R. H., 1992. Splicing thermotolerance maintains pre-mRNA transcripts in the splicing pathway during severe heat shock. Exp. Cell Res. 202: 233–242 [DOI] [PubMed] [Google Scholar]
- Davies S. A., Overend G., Sebastian S., Cundall M., Cabrero P., et al. , 2012. Immune and stress response ‘cross-talk’ in the Drosophila Malpighian tubule. J. Insect Physiol. 58: 488–497 [DOI] [PubMed] [Google Scholar]
- Denegri M., Chiodi I., Corioni M., Cobianchi F., Riva S., et al. , 2001. Stress-induced nuclear bodies are sites of accumulation of pre-mRNA processing factors. Mol. Biol. Cell 12: 3502–3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis, G., B. T. Sherman, D. A. Hosack, J. Yang, W. Gao et al., 2003 DAVID: database for annotation, visualization, and integrated discovery. Genome Biology 4: R60. [PubMed] [Google Scholar]
- Ernst J., Bar-Joseph Z., 2006. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics 7: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino N. A., Cooper T. A., 2003. Pre-mRNA splicing and human disease. Genes Dev. 17: 419–437 [DOI] [PubMed] [Google Scholar]
- Gonsalves S. E., Moses A. M., Razak Z., Robert F., Westwood J. T., 2011. Whole-genome analysis reveals that active heat shock factor binding sites are mostly associated with non-heat shock genes in Drosophila melanogaster. PLoS ONE 6: e15934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley B. R., 2001. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 17: 100–107 [DOI] [PubMed] [Google Scholar]
- Graveley B. R., Brooks A. N., Carlson J. W., Duff M. O., Landolin J. M., et al. , 2011a The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley, B. R., G. May, A. N. Brooks, J. W. Carlson, L. Cherbas et al., 2011b The D. melanogaster transcriptome: modENCODE RNA-Seq data for differing treatment conditions, Personal communication to FlyBase, FBrf0213503.
- Gubelmann C., Gattiker A., Massouras A., Hens K., David F., et al. , 2011. GETPrime: a gene- or transcript-specific primer database for quantitative real-time PCR. Databasebar040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin M. J., Lis J. T., 2010. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 6: 1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A. A., Anderson A., Hallas R., 2002. Opposing clines for high and low temperature resistance in Drosophila melanogaster. Ecol. Lett. 5: 614–618 [Google Scholar]
- Howard B. E., Heber S., 2010. Towards reliable isoform quantification using RNA-SEQ data. BMC Bioinformatics 11(Suppl3):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Lempicki R. A., 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Hughes M. E., Grant G. R., Paquin C., Qian J., Nitabach M. N., 2012. Deep sequencing the circadian and diurnal transcriptome of Drosophila brain. Genome Res. 22: 1266–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffares D. C., Penkett C. J., Bahler J., 2008. Rapidly regulated genes are intron poor. Trends Genet. 24: 375–378 [DOI] [PubMed] [Google Scholar]
- Jensen L. T., Nielsen M. M., Loeschcke V., 2008. New candidate genes for heat resistance in Drosophila melanogaster are regulated by HSF. Cell Stress Chaperones 13: 177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. K., Cockerell F. E., McKechnie S. W., 2011. Transcripts from the Drosophila heat-shock gene hsr-omega influence rates of protein synthesis but hardly affect resistance to heat knockdown. Mol. Genet. Genomics 285: 313–323 [DOI] [PubMed] [Google Scholar]
- Jolly C., Lakhotia S. C., 2006. Human sat III and Drosophila hsr omega transcripts: a common paradigm for regulation of nuclear RNA processing in stressed cells. Nucleic Acids Res. 34: 5508–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T., Silverman N., 2005. Bacterial recognition and signalling by the Drosophila IMD pathway. Cell. Microbiol. 7: 461–469 [DOI] [PubMed] [Google Scholar]
- Kapur K., Xing Y., Ouyang Z., Wong W. H., 2007. Exon arrays provide accurate assessments of gene expression. Genome Biol. 8(5):R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermann V., Overgaard J., Hoffmann A. A., Fløjgaard C., Svenning J.-C., et al. , 2012. Upper thermal limits of Drosophila are linked to species distributions and strongly constrained phylogenetically. Proc. Natl. Acad. Sci. USA 109: 16228–16233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka R. J., Pittendrigh C., Orr D., 1989. Reciprocal behaviour associated with altered homeostasis and photosensitivity of Drosophila clock mutants. J. Neurogenet. 6: 1–10 [DOI] [PubMed] [Google Scholar]
- Lakhotia S. C., Mallik M., Singh A. K., Ray M., 2012. The large noncoding hsromega-n transcripts are essential for thermotolerance and remobilization of hnRNPs, HP1 and RNA polymerase II during recovery from heat shock in Drosophila. Chromosoma 121: 49–70 [DOI] [PubMed] [Google Scholar]
- Leemans R., Egger B., Loop T., Kammermeier L., He H. Q., et al. , 2000. Quantitative transcript imaging in normal and heat-shocked Drosophila embryos by using high-density oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 97: 12138–12143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B., Hoffmann J., 2007. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25: 697–743 [DOI] [PubMed] [Google Scholar]
- Lindquist S., Craig E. A., 1988. The heat-shock proteins. Annu. Rev. Genet. 22: 631–677 [DOI] [PubMed] [Google Scholar]
- Lockstone H. E., 2011. Exon array data analysis using Affymetrix power tools and R statistical software. Brief. Bioinform. 12: 634–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marden J. H., 2008. Quantitative and evolutionary biology of alternative splicing: how changing the mix of alternative transcripts affects phenotypic plasticity and reaction norms. Heredity 100: 111–120 [DOI] [PubMed] [Google Scholar]
- Marin-Vinader L., Shin C., Onnekink C., Manley J. L., Lubsen N. H., 2006. Hsp27 enhances recovery of splicing as well as rephosphorylation of SRp38 after heat shock. Mol. Biol. Cell 17: 886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrangelo A. M., Marone D., Laido G., De Leonardis A. M., De Vita P., 2012. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 185: 40–49 [DOI] [PubMed] [Google Scholar]
- Mazzucotelli E., Mastrangelo A. A., Crosatti C., Guerra D., Stanca A. M., et al. , 2008. Abiotic stress response in plants: when post-transcriptional and post-translational regulations control transcription. Plant Sci. 174: 420–431 [Google Scholar]
- McIntyre L. M., Bono L. M., Genissel A., Westerman R., Junk D., et al. , 2006. Sex-specific expression of alternative transcripts in Drosophila. Genome Biol. 7: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre L. M., Lopiano K. K., Morse A. M., Amin V., Oberg A. L., et al. , 2011. RNA-seq: technical variability and sampling. BMC Genomics 12: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuilton P., St Pierre S. E., Thurmond J., 2012. FlyBase 101: the basics of navigating FlyBase. Nucleic Acids Res. 40: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K. A., Hoffmann A. A., 2010. Thermal ramping rate influences evolutionary potential and species differences for upper thermal limits in Drosophila. Funct. Ecol. 24: 694–700 [Google Scholar]
- Nielsen M. M., Sorensen J. G., Kruhoffer M., Justesen J., Loeschcke V., 2006. Phototransduction genes are up-regulated in a global gene expression study of Drosophila melanogaster selected for heat resistance. Cell Stress Chaperones 11: 325–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen T. W., Graveley B. R., 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth K. V., Rajendra T. K., Lal A. K., Lakhotia S. C., 2000. Omega speckles: a novel class of nuclear speckles containing hnRNPs associated with noncoding hsr-omega RNA in Drosophila. J. Cell Sci. 19: 3485–3497 [DOI] [PubMed] [Google Scholar]
- Shi Y., 2012. Alternative polyadenylation: new insights from global analyses. RNA 18: 2105–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Nishida K., Di Giammartino D. C., Manley J. L., 2011. Heat shock-induced SRSF10 dephosphorylation displays thermotolerance mediated by Hsp27. Mol. Cell. Biol. 31: 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair B. J., Ferguson L. V., Salehipour-Shirazi G., MacMillan H. A., 2013. Cross-tolerance and cross-talk in the cold: relating low temperatures to desiccation and immune stress in insects. Integr. Comp. Biol. 24: 24. [DOI] [PubMed] [Google Scholar]
- Sokal R. R., Rohlf F. J., 1995. Biometry. W.H. Freeman, New York [Google Scholar]
- Sorensen J. G., Nielsen M. M., Kruhoffer M., Justesen J., Loeschcke V., 2005. Full genome gene expression analysis of the heat stress response in Drosophila melanogaster. Cell Stress Chaperones 10: 312–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis-Scott M., Kopp A., Wayne M. L., Nuzhdin S. V., McIntyre L. M., 2009. Sex-specific splicing in Drosophila: widespread occurrence, tissue specificity and evolutionary conservation. Genetics 181: 421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves S. S., Henikoff S., 2011. Heat shock reduces stalled RNA polymerase II and nucleosome turnover genome-wide. Genes Dev. 25: 2387–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valanne S., Wang J. H., Ramet M., 2011. The Drosophila Toll signaling pathway. J. Immunol. 186: 649–656 [DOI] [PubMed] [Google Scholar]
- Velichko A. K., Markova E. N., Petrova N. V., Razin S. V., Kantidze O. L., 2013. Mechanisms of heat shock response in mammals. Cell. Mol. Life Sci. 30: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Graze R. M., Walts B. M., Lopez C. M., Baker H. V., et al. , 2011. Partitioning transcript variation in Drosophila: abundance, isoforms, and alleles. G3: Genes, Genomes, Genetics 1: 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost H. J., Lindquist S., 1986. RNA splicing is interrupted by heat-shock and is rescued by heat-shock protein synthesis. Cell 45: 185–193 [DOI] [PubMed] [Google Scholar]
- Yost H. J., Lindquist S., 1991. Heat-shock proteins affect RNA processing during the heat-shock response of Saccharomyces cerevisiae. Mol. Cell. Biol. 11: 1062–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost H. J., Petersen R. B., Lindquist S., 1990. RNA metabolism- strategies for regulation in the heat-shock response. Trends Genet. 6: 223–227 [DOI] [PubMed] [Google Scholar]
- Zhou S. S., Campbell T. G., Stone E. A., Mackay T. F. C., Anholt R. R. H., 2012. Phenotypic plasticity of the Drosophila transcriptome. PLoS Genet. 8:e1002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.