

Abstract
CD8+ T lymphocyte responses are a critical arm of the immune response to respiratory virus infection and may play a role in the pathogenesis of interstitial lung disease. We have shown that CD8+ T cells induce significant lung injury in the absence of virus infection by adoptive transfer into mice with alveolar expression of a viral transgene. The injury is characterized by the parenchymal infiltration of host cells, primarily macrophages, which correlates with physiologic deficits in transgenic animals. CD8+ T cell–mediated lung injury can occur in the absence of perforin and Fas expression as long as TNF-α is available. Here, we show that the effect of TNF-α expressed by CD8+ T cells is mediated not exclusively by cytotoxicity, but also through the activation of alveolar target cells and their expression of inflammatory mediators. CD8+ T cell recognition of alveolar cells in vitro triggered monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-2 (MIP-2) expression in the targets, which was mediated by TNF-α. Antigen-dependent alveolar MCP-1 expression was observed in vivo as early as 3 hours after CD8+ T cell transfer and depended upon TNF-R1 expression in transgenic recipients. MCP-1 neutralization significantly reduced parenchymal infiltration after T cell transfer. We conclude that alveolar epithelial cells actively participate in the inflammation and lung injury associated with CD8+ T cell recognition of alveolar antigens.
This article may have been published online in advance of the print edition. The date of publication is available from the JCI website, http://www.jci.org. J. Clin. Invest. 106:R49–R58 (2000).
Introduction
CD8+ T lymphocyte responses represent an important arm of adaptive antiviral immunity. The mechanisms employed by these cells in viral clearance include both cytolytic and noncytolytic effector functions (1–3). Although respiratory dysfunction frequently accompanies respiratory virus infection, the relative contribution of the virus infection itself and the CD8+ T cell antiviral effector activities account for lung injury in this context is unclear. CD8+ T cells accumulate in the lung parenchyma in a variety of inflammatory and interstitial lung diseases, but the nature of their specific contribution to lung injury is also unclear (4–9). We have developed a model to examine the specific effects of CD8+ cytolytic T cells on lung injury, in the absence of virus infection. Activated antiviral T cells induce significant pulmonary inflammation and injury after adoptive transfer into transgenic animals expressing a viral antigen, influenza hemagglutinin (HA), on alveolar epithelial cells and in the absence of virus infection (10, 11). The injury results in considerable respiratory dysfunction and eventual death in a time frame that depends upon cell dose. We also demonstrated that CD8+ T cell–mediated lung injury occurs in the absence of perforin and Fas, but neutralizing Ab to TNF-α completely abrogates lung injury that occurs in the absence of both mediators (12). In vitro, alveolar epithelial-derived cells are sensitive to the cytotoxic effects of perforin and TNF-α expressed by CD8+ T cells but are insensitive to induction of apoptosis by Fas ligand, despite expression of functional Fas (12). These cells are also significantly less susceptible to cytolysis induced by soluble TNF-α than by TNF-α expressed by T lymphocytes (12).
CD8+ T cells predominantly express a transmembrane form of TNF-α (13–15), which may initiate injury through direct cytotoxic effects on alveolar epithelial cells. This may contribute to the observed respiratory dysfunction that evolves in HA-transgenic recipients. However, the inflammatory infiltration that ensues 3 to 4 days after adoptive transfer into HA-transgenic recipients consists largely of neutrophils, host lymphocytes, and (predominantly) activated macrophages; it is the presence of large numbers of these cells that correlates most strongly with the profound respiratory impairment observed after T cell transfer (11). Since TNF-α (in its soluble form) is known to induce expression of a variety of inflammatory mediators in respiratory epithelial cells (16–18), we hypothesized that there may be a noncytotoxic component of the effect of TNF-α in injury after T cell transfer. There are several chemokines that might participate in the recruitment of mononuclear phagocytes, and most have a number of cellular sources (19–21). In addition to the transferred T cells, a potential source of these chemokines may be the alveolar epithelium, triggered by T cell–receptor recognition and engagement, either individually or as a population. In this study we show that alveolar epithelial cells are triggered as a result of specific CD8+ T cell–antigen recognition to express the inflammatory chemokines monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-2 (MIP-2) and show that this induction is mediated by TNF-α. In particular, MCP-1 appears to play a major role in the parenchymal infiltration that ensues after T cell transfer. In addition we present evidence that MCP-1 expression by alveolar epithelium may occur as a result of direct induction as well as bystander activation, both of which are dependent upon specific antigen recognition by the T cell.
Methods
T lymphocyte clones.
CD8+ T cell clones, specific for the 210–219 epitope of A/Japan/57 HA were used in these experiments and were generated by limiting dilution, as described previously (22). They were restimulated weekly in vitro with irradiated syngeneic splenocytes that were infected with A/Japan/57 influenza and cultured in Iscove’s complete medium supplemented with 10 U/ml IL-2. On day 5 after in vitro stimulation, T cells were used in assays with target cells infected with A/Japan/57 influenza (for 30 minutes and washed) or loaded with synthetic peptide representing the 210–219 epitope of the A/Japan/57 HA (10). In some experiments, anti–IFN-γ mAb (R&D, Minneapolis, Minnesota, USA) was added to wells at a final concentration of 5 μg/ml, or anti–TNF-α Ab (Genzyme, Boston, Massachusetts, USA) was added at a final dilution of 1:100. For stimulation in the absence of antigen-presenting cells, plates were coated with anti-CD3 (PharMingen, San Diego, California, USA) for 1 hour and rinsed before addition of T cells, or T cells were stimulated by phorbol ester (PDB; Sigma, St. Louis, Missouri, USA) and ionomycin (Sigma).
Alveolar epithelial-derived cells.
MLE-15 cells (10) were transfected with the class I MHC molecule, Kd (11). For analysis of chemokine expression, 2 × 106 MLE-Kd cells were plated in 24-well plates and allowed to adhere overnight. The next day, peptide (where appropriate) and T cells were added to the culture. After incubation for a specified period, the supernatants (and nonadherent cells) were removed, centrifuged, and the cells were collected. Trypsin/EDTA was added to the remaining (adherent) cells, and these were removed, centrifuged, the cells collected, and combined with the nonadherent cells for total RNA extraction with Trizol (Life Technologies, Rockville, Maryland, USA). RNase protection assays were performed using probes for a panel of chemokine messages (PharMingen) in accordance with the manufacturer’s instructions. In most experiments 32P-labeled probes were used. In several experiments digoxigenin-labeled probes were used, followed by membrane transfer and blotting with peroxidase-coupled antidigoxigenin Ab’s (Roche, Indianapolis, Indiana, USA). For effector/target-cell separation, anti–CD8-coupled magnetic beads were used (Dynal, Lake Success, New York, USA) in accordance with the manufacturer’s instructions. After 4 hours of incubation, supernatants were removed, and the adherent cells were rinsed twice with media before the addition of trypsin/EDTA. The cells were removed and incubated with the beads for 15 minutes, and the beads were removed. All experimental samples were subject to two rounds of magnetic bead separation in order to remove all contaminating T cells from the epithelial cell sample.
Adoptive transfer.
HA-transgenic mice (H-2d) expressing the A/Japan/57 influenza HA under the transcriptional control of the surfactant protein C (SPC) promoter were used for in vivo studies (10). Transgene-negative littermates were used as control recipients in all experiments. Animals subject to adoptive transfer were used at 10–12 weeks of age (18–22 g). Mice deficient in TNF receptor-1 (TNF-R1) expression (p55–/–) were obtained from the Jackson Laboratories (Bar Harbor, Maine, USA) and were bred to express the HA transgene for some experiments. The offspring were screened for the H-2d haplotype (by flow cytometry) and the HA transgene (by PCR) as described (10), as well as for the TNF-R1–deficient genotype by three-primer PCR (23). On day 5 after stimulation, CD8+ T cell clones were separated from stimulators by density-gradient centrifugation and injected into the tail vein of appropriate recipient animals. In some experiments animals received Ab to MCP-1 (PharMingen) or isotype control by tail vein injection at the time of T cell transfer. Lungs were harvested at appropriate times for histology or homogenized in Trizol (Life Technologies Inc.), after which the RNA was extracted for analysis.
Histology and in situ hybridization.
At appropriate times after adoptive transfer, animals were sacrificed, their trachea exposed, and their airways perfused with 50% embedding media (Fisher Scientific Co., Pittsburgh, Pennsylvania, USA) in PBS at a pressure of 25 cm H2O; the lungs were excised and snap-frozen. Sections were cut and stained with hematoxylin-eosin (H&E) or with peroxidase-labeled Ab to a macrophage marker (F4/80; Caltag Laboratories, Burlingame, California, USA) and counterstained with hematoxylin. Semiquantitative histologic analysis was performed by averaging the total cell counts in five high-power fields (hpf’s) under oil immersion (×100) from each of two animals (in each group). For in situ hybridization, lungs were perfused through the trachea with 10% formalin. Tritiated riboprobes were prepared as described previously (10) using an MCP-1 cDNA (kindly provided by Barrett Rollins, Dana Farber Cancer Institute, Boston, Massachusetts, USA). In situ hybridization was performed with a 2-to-4–week exposure before analysis, as described (10).
ELISA.
MLE-Kd cells (2 × 106) were plated in 6-well plates with equal numbers of CD8+ T cells and incubated in 0.5 ml media. Supernatants were removed and assays for MCP-1 and TNF-α were performed using a sandwich ELISA (PharMingen) in accordance with the manufacturer’s instructions. The matrix metalloproteinase inhibitor KB8301 (PharMingen) was added at a final concentration of 5 μM to appropriate wells for the duration of the incubation period.
Statistical analysis.
Significant differences were determined by Student’s unpaired t test or through one-way ANOVA.
Results
CD8+ T cell recognition of an alveolar epithelial antigen leads to chemokine expression in the lung.
CD8+ T lymphocytes are found in the alveolar walls and spaces in a variety of inflammatory lung diseases, often in association with other inflammatory cells such as neutrophils and macrophages (4, 5, 8, 9). Using a murine model of interstitial pneumonitis, we have shown previously that CD8+ T cell recognition of a specific antigen expressed on alveolar epithelial cells is sufficient to induce progressive and, ultimately, lethal lung injury, the time course of which is dependent upon cell dose (11). Although specific recognition by CD8+ T cells is required to initiate pneumonitis, the inexorable progression of lung injury in HA-transgenic mice is characterized primarily by the dramatic accumulation of host inflammatory cells in the lung parenchyma, particularly macrophages, as shown in Figure 1. Sections of lung tissue harvested from SPC-HA–transgenic mice 5 days after transfer of clone CD8+ T cell 40-2 exhibited marked interstitial inflammatory infiltration predominantly by macrophages, as indicated by immunoperoxidase staining with a macrophage-specific mAb. Other cells recruited into the interstitium 3 to 5 days after CD8+ T cell transfer include host CD8+ and CD4+ lymphocytes and, to a lesser extent, neutrophils (data not shown). Such infiltration was not observed in nontransgenic recipients, whose lung parenchyma appears nearly normal by comparison after adoptive transfer of clone 40-2. This phenomenon was observed after transfer of other CD8+ T cell clones, as well as after transfer of short-term bulk cultures of HA-specific cytotoxic T lymphocytes (11). The dramatic inflammatory infiltration observed in the HA-transgenic animals correlates with significant pulmonary function abnormalities, such as reduced lung volume and diffusing capacity, as well as eventual death. Since the inflammatory influx occurs only in the transgenic animals and the expression of the transgene is limited to alveolar epithelial cells (10), we hypothesize that soluble factors, such as chemokines, are produced at the epithelial surface as a direct result of T cell receptor (TCR) engagement of peptide/MHC complex on the alveolar cell. These chemokines may contribute to the recruitment of inflammatory cells into the lung parenchyma, which considerably amplifies the injury. Soluble factors may be produced by the T cells or the epithelial target cells (or both) upon TCR engagement. As shown in Figure 2, whole-lung homogenates demonstrate expression of several chemokine mRNA species as early as 6 hours after adoptive transfer of CD8+ T cells, including lymphotactin (Ltn), RANTES, MIP-1β, MIP-1α, MIP-2, IFN-γ–inducible protein 10 (IP-10), and MCP-1 messages, to varying degrees. The largest band represents MCP-1 mRNA, expression of which appeared to decline somewhat by 72 hours after cell transfer. In contrast, RANTES, MIP-1α, and MIP-1β appeared to increase slightly over time. There were faint bands in the nontransgenic control lungs, most evident at the earliest time point (6 hours) and diminishing considerably by 24 hours after cell transfer. One or more of these chemokine messages may result in translation products that are responsible for, or contribute to, the inflammatory infiltrates observed in transgenic mice after T cell transfer.
Figure 1.

Histologic sections from lungs harvested 4 days after adoptive transfer of 5 × 106 CD8+ T cell clone 40-2. (a) The benign appearance of lung parenchyma harvested from a nontransgenic littermate, at low power. (b) The pattern of injury in the SPC-HA–transgenic animals, also at low power (a and b were stained with H&E). (c and d) Immunoperoxidase staining of lung sections with an antimacrophage Ab (F4/80), at low power (c) and high power (d).
Figure 2.

CD8+ T cell transfer results in antigen-dependent chemokine expression. RNase protection assays were performed on RNA extracted from whole-lung homogenates after adoptive transfer of 5 × 106 CD8+ T cell clone 40-2. Transgenic and nontransgenic animals were sacrificed at 6, 24, 48, and 72 hours and assayed for chemokine expression.
CD8+ T cell recognition of alveolar epithelial-derived cells in vitro triggers chemokine expression in a peptide-dependent manner.
To further analyze chemokine expression occurring directly as a result of T cell recognition of epithelial cells, we took advantage of an in vitro system developed to study T lymphocyte cytotoxicity of alveolar epithelial cells (11, 12). The MLE-Kd cell is a type II alveolar epithelial cell line (24) transfected with the cDNA for the class I MHC molecule Kd, which we and others have demonstrated to be an excellent model of primary type II pneumocytes (12, 24, 25). We showed previously that recognition of influenza HA peptide antigens on this epithelial cell by Kd-restricted CD8+ T cells in vitro results in considerable epithelial cytolysis after influenza infection or exogenous peptide loading, to a similar degree with each. However, this was observed at effector-to-target (E:T) ratios that appear to be nonphysiologic (11); therefore, the in vivo relevance of this is still under investigation. However, in order to ask whether CD8+ T cell recognition of alveolar epithelial cells might result in expression of proinflammatory chemokines in a pattern similar to that observed in vivo, we performed RNase protection assays on RNA extracts from mixed cultures, coincubated for 4 hours. As shown in Figure 3, RNA extracted from MLE-Kd cells and CD8+ T cells (clone 40-2), cultured together in the presence of peptide antigen, demonstrated expression of several chemokine genes, including Ltn, RANTES, MIP-1β, MIP-1α, and MCP-1, as well as faint expression of MIP-2 and IP-10. Expression was dependent upon peptide dose: a signal was evident at concentrations as low as 10–10 M, but lost at 10–12 M, peptide concentration, except for faint levels of RANTES and MIP-1β message.
Figure 3.

CD8+ T cell recognition of antigen presented by alveolar epithelial-derived cells results in peptide-dependent chemokine expression. RNase protection assays were performed on RNA extracted from in vitro cocultures of CD8+ T cells and MLE-Kd cells. Varying doses of antigenic peptide (10–6, 10–8, 10–10, and 10–12 M) were added to the cultures and incubated for 6 hours. With the exception of slight expression of RANTES and MIP-1β, chemokine expression becomes undetectable at 10–12 M peptide.
Labeled CD8+ T cells traffic into the lung parenchyma as early as 3 hours after adoptive transfer and become undetectable by approximately 48 hours thereafter (R.I. Enelow et al., unpublished observations). The inflammatory influx is minimal at this time point but becomes exuberant by 72 to 96 hours after T cell transfer (11). Therefore, it was of particular interest to investigate whether another cellular source of the chemoattractant signals, other than the transferred CD8+ T lymphocyte, might contribute to the inflammatory infiltration. The alveolar epithelial cell target cell represents an important candidate, since it is the first cell encountered by the activated T cell in an antigen-specific fashion (the early contribution of endothelial cells cannot be formally excluded, although in the absence of epithelial transgene expression lung infiltration and injury is not observed). To assess the pattern of alveolar cell chemokine expression in the absence of T cells, soluble TNF-α was used to stimulate MLE-Kd cells in vitro, and RNA was extracted at several time points. As shown in Figure 4, RNase protection assays indicate that RANTES, MIP-2, and MCP-1 expression was induced by TNF-α treatment of MLE-Kd cells, consistent with observations of others (16–18). The dose used was saturating (12), so it is unclear whether such expression might occur under physiologic conditions. It is also not clear that these cells would represent the exclusive source of these chemokine species in the context of a T cell-alveolar cell interaction in vivo.
Figure 4.

Alveolar epithelial-derived cells express chemokine message upon exposure to soluble TNF-α. RNase protection assays were performed on RNA extracted from MLE-Kd cells incubated in vitro with 10,000 U (saturating dose) of soluble rmTNF-α. The predominant bands represent MCP-1, MIP-2, and RANTES, expression of which was evident 2 hours after addition of TNF-α.
To ask whether the CD8+ T cells themselves are capable of expressing RANTES, MIP-2, or MCP-1, T cell clones were triggered in the absence of antigen-presenting cells, with either PDB plus ionomycin, or with varying concentrations of plate-bound anti-CD3. As shown in Figure 5, CD8+ T cells predominantly expressed message for Ltn, MIP-1α, and MIP-1β upon maximal stimulation. Low levels of T cell activation gene-3 (TCA-3), RANTES, and IP-10 were expressed, the latter two constitutively (particularly RANTES). However, no expression of MIP-2 or MCP-1 was observed, even after significant overexposure of the gel. Taken together, the data from these two experiments strongly suggest that the sole source of MIP-2 and MCP-1 expression, triggered by specific CD8+ T cell recognition of alveolar cells, are the alveolar epithelial target cells. Though this does not demonstrate directly target-cell expression triggered by T cell recognition, it does suggest that there may be a contribution of both the T cells and the epithelial target cells to the chemokine milieu of the lung parenchyma that results from CD8+ T cell recognition and that both are dependent on specific TCR engagement of epithelial-cell peptide/MHC complexes.
Figure 5.

CD8+ T cells express chemokines upon activation, but not MCP-1 or MIP-2. RNase protection assays were performed on RNA extracted from CD8+ T cells after 4-hour stimulation with PDB and ionomycin or plate-bound anti-CD3 at varying concentrations. With the exception of RANTES (and possibly IP-10), there was no constitutive chemokine expression by T cells without stimulation. MIP-1α, MIP-1β, and Ltn are most prominently expressed by the T cells upon stimulation, as well as faint levels of IP-10, TCA-3, and RANTES message. No MIP-2 or MCP-1 expression was evident, even with significant overexposure (not shown).
TCR engagement triggers target-cell chemokine expression, which is mediated by TNF-α.
It has been observed previously that epithelial cells are capable of expressing a variety of inflammatory mediators upon incubation with soluble factors such as TNF-α or IL-1, and our data are consistent with this. However, target-cell cytokine or chemokine expression has not been examined in response to specific T cell recognition. Furthermore, the canonical fate of a virus-infected (or peptide-treated) target cell upon specific recognition by cytolytic T lymphocytes is apoptotic cell death (26–28), which would theoretically preclude transcriptional activity. However, it has become apparent in recent years that the in vivo susceptibility of target cells to T cell–mediated cytolysis may be a more complex and regulated process than presumed since the identification of cellular regulators of protection against perforin/granzyme–mediated cytotoxicity (29–31) and TNF-mediated cytotoxicity (32). Furthermore, whether or not target-cell recognition results in apoptosis or activation upon recognition by CD8+ T cells, there is also the possibility that bystander activation might result in peptide-dependent T cell–triggered epithelial chemokine expression. To directly assess the transcriptional contribution of epithelial target cells to the inflammatory milieu triggered by T cell recognition, we incubated MLE-Kd cells with CD8+ T cells for 4 hours, and then separated the two cell populations using anti-CD8 coupled to magnetic beads. As demonstrated in Figure 6, MCP-1 and MIP-2 expression was evident exclusively in the target-cell fraction, whereas MIP-1α and Ltn expression was found exclusively in the T cell fraction. RANTES and MIP1-β were most abundantly expressed in the T cell fraction, but there was trace expression in the target-cell fraction (possibly due to contamination). There was no difference in expression between peptide-loaded or influenza-infected target cells. Furthermore, anti–TNF-α ablated target-cell MCP-1 and MIP-2 expression, whereas there was no such effect on T cell chemokine expression. No effect was observed with anti–IFN-γ (or with anti-Fas ligand; not shown). These data suggest that specific CD8+ T cell recognition of antigen on epithelial target cells triggers induction of alveolar cell chemokine message expression, mediated by TNF-α, which may play an important role in the amplification of T cell–mediated lung inflammation. MCP-1 expression, in particular, may contribute to lung injury initiated by CD8+ T cells in our in vivo system, since the inflammatory infiltrates consist predominantly of macrophages and, to a lesser extent, lymphocytes, both of which may be recruited in response to MCP-1 expression (20, 21, 33).
Figure 6.

MCP-1 and MIP-2 expression by alveolar cells is triggered by CD8+ T cell recognition, and induction is mediated by TNF-α. RNase protection assays were performed on RNA extracted from in vitro cocultures of CD8+ T cells and MLE-Kd cells incubated in the presence of 10–8 M peptide or after influenza virus infection of the MLE-Kd cells. After 4 hours of incubation, all cells were removed and incubated with anti–CD8-coupled magnetic beads twice in order to fully separate the two cell populations. MCP-1 and MIP-2 expression was observed exclusively in the target-cell fraction and was inhibited by the addition of anti–TNF-α to the media during incubation.
To confirm that alveolar epithelial cells express MCP-1 in vivo in response to specific CD8+ T cell recognition, in situ hybridization was performed on sections of lung harvested from SPC-HA–transgenic (and nontransgenic) recipients of adoptive transfer of CD8+ clone 40-2. As shown in Figure 7, sections taken from HA+ animals 24 hours after adoptive transfer of CD8+ T cells demonstrate expression of MCP-1 message, particularly near the junctions of alveolar septae, in a pattern similar to that of HA expression described previously (10). No such expression was evident in lungs taken from nontransgenic control recipients. Expression in HA+ recipients was evident as early as 3 hours after CD8+ T cell transfer (not shown). We have observed previously that the lung parenchyma is histologically unremarkable 24 hours after adoptive transfer, and the only inflammatory cells that are detectable at this time point are the transferred CD8+ T cells (if they are fluorescent labeled), which are observable in the alveolar interstitium at the junctions of the septae (11).
Figure 7.
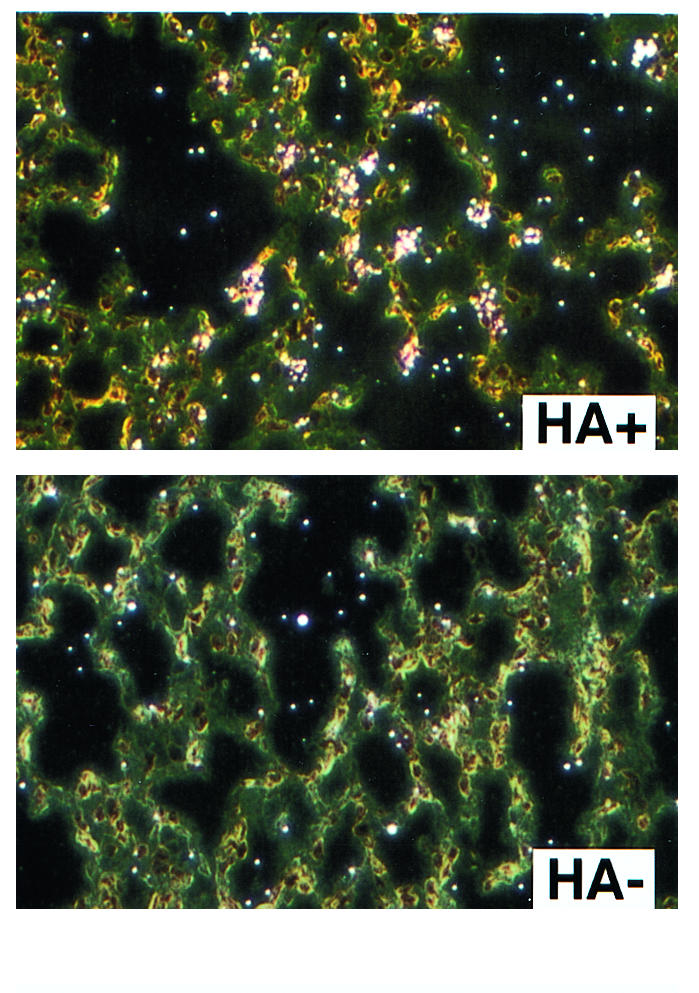
Alveolar epithelial MCP-1 expression is triggered by CD8+ T cell recognition in vivo. In situ hybridizations were performed with labeled probe for MCP-1 expression in transgenic and nontransgenic lung sections harvested 24 hours after adoptive transfer of 5 × 106 CD8+ T cell clone 40-2. MCP-1 expression in transgenic recipients was localized to the alveolar septae, most prominently at the junctions of the alveolar walls. No expression was evident in the nontransgenic recipients.
Cytokine and chemokine protein expression are regulated by a variety of factors aside from message level, such as mRNA stability (34). We therefore assayed MCP-1 protein production and secretion triggered by CD8+ T cell recognition using ELISA. As demonstrated in Figure 8, MCP-1 protein was evident in cocultures of MLE-Kd cells and CD8+ T cells (40–2) as early as 2 hours after the start of incubation, but only in the presence of peptide. Furthermore, this expression was completely inhibited by anti–TNF-α (and by soluble TNF receptor-1 [TNFR-1]; not shown). MCP-1 production was not detectable in supernatants from T cells stimulated with anti-CD3 for 6 hours (not shown).
Figure 8.

MCP-1 protein production by alveolar cells is triggered by CD8+ T cell recognition, and induction is mediated by TNF-α. ELISAs were performed on supernatants of combined cultures at varying times in the presence (filled and hatched bars) or absence (open bars) of 10–8 M peptide. Hatched bars indicate wells in which Ab to TNF-α was added. MCP-1 protein expression was detectable by 4 hours and was significantly induced after 6 hours of incubation of MLE-Kd cells with CD8+ T cells and peptide. Protein production was inhibited by anti–TNF-α. AP < 0.05, BP < 0.01 compared with the level with anti-TNF.
The most likely explanation for this phenomenon is that specific recognition of antigen on an epithelial cell by a CD8+ T cell results in apoptosis of the recognized target cell (via perforin or membrane-bound TNF-α), as well as secretion of soluble TNF-α by the T cells (by enzymatic cleavage of transmembrane TNF-α). Soluble TNF-α may in turn trigger the remaining target cells to express MCP-1. However, an additional mechanism might involve the direct induction of MCP-1 expression by the target cells that are specifically recognized, possibly because the threshold for induction of target-cell chemokine expression is lower than the threshold for induction of cell death. To address the issue of whether transmembrane TNF-α on a CD8+ T cell, in the absence of soluble TNF-α, may trigger MCP-1 expression by alveolar epithelial target cells, T cells (clone 40-2) were treated with the matrix metalloproteinase inhibitor KB8301, which blocks proteolytic processing of membrane-bound TNF-α to its soluble form (35–37). As shown in Table 1, KB8301 inhibition of production of soluble TNF-α by 40-2 is extremely efficient, such that the amount of soluble TNF-α produced after 6 hours of incubation with peptide-treated MLE-Kd cells was below the limits of detection of a very sensitive ELISA. Nevertheless, the amount of MCP-1 produced in the cultures was considerable, though lower than that produced in the absence of KB8301 (P < 0.05). To rule out the possibility that small amounts of soluble TNF-α (below the limits of ELISA detection) may have been produced in the presence of KB8301 in a quantity sufficient to induce production of MCP-1, the supernatant from that culture was removed and added to a fresh culture of MLE-Kd cells. No additional MCP-1 production was observed after another 6-hour incubation; i.e., the level was the same as in the initial supernatant (not shown). A further control for the efficiency of the metalloproteinase inhibitor was accomplished by stimulating CD8+ T cells (40–2) with plate-bound anti-CD3 for 6 hours in the absence of antigen-presenting cells. Soluble TNF-α was undetectable in the presence of the inhibitor (though very abundant in its absence), and when MLE-Kd cells were incubated in these supernatants, no MCP-1 production was detectable after 6 hours (not shown). These data would suggest that specific recognition by CD8+ T cells might induce expression of MCP-1, both by production of soluble TNF-α, which activates bystander target cells, and by induction of expression in epithelial targets by membrane-bound TNF-α. The latter appears to play a predominant role in target-cell activation, though the relative contribution of each mechanism in vivo is still under investigation.
Table 1.
Metalloproteinase inhibition completely inhibits soluble TNF-α production but minimally abrogates target-cell MCP-1 production triggered by T cell recognition

Lung MCP-1 expression in vivo is critically dependent upon TNFR-1 expression and is a primary determinant of T cell–triggered inflammatory infiltration.
We have demonstrated previously that TNF-α plays an important role in CD8+ T cell–mediated lung injury in vivo (12), and our data indicate that TNF-α is the central mediator of T cell–triggered alveolar epithelial-cell chemokine expression in vitro. To determine the significance of TNF-α on T cell–triggered chemokine expression in vivo, adoptive transfer of CD8+ T cells was performed into HA-transgenic mice that were deficient in TNFR-1 expression (p55–/–). Absence of the p55 receptor significantly reduces lung injury triggered by T cell recognition (R.I. Enelow, et al., unpublished observations). Lungs were harvested 24 hours after transfer, and RNase protection assays were performed, then compared with HA-transgenic recipients with wild-type p55 expression. As shown in Figure 9, deficiency in p55-receptor expression ablates MCP-1 expression at 24 hours after cell transfer in whole-lung homogenates, confirming the central role of TNF-α in T cell–triggered epithelial cell MCP-1 expression in vivo. IP-10 expression was unaffected by the absence of the p55 TNF receptor, consistent with data implicating IFN-γ as the primary inducer of its expression (38).
Figure 9.

TNF-R1 (p55) is required for in vivo expression of MCP-1 triggered by CD8+ T cell recognition. RNase protection assays were performed on RNA extracted from whole-lung homogenates after adoptive transfer of 5 × 106 CD8+ T cell clone 40-2. SPC-HA–transgenic animals were bred into the TNF-R1–deficient (p55–/–) background and compared with wild-type HA+ littermates. Lungs were harvested 24 hours after adoptive transfer and assayed for chemokine expression. MCP-1 expression was absent in the HA+ p55–/– recipients but present in the HA+ p55+/+ animals. IP-10 expression was similar in both.
To isolate the specific contribution of alveolar epithelial-cell activation to the inflammatory infiltration that ensues after T cell transfer, SPC-HA–transgenic animals were simultaneously injected with Ab to MCP-1 (or isotype control) at the time of adoptive transfer, and lungs were harvested at 96 hours for histologic analysis. As shown in Figure 10, lungs harvested from recipients of Ab to MCP-1 demonstrated reduced parenchymal cellular infiltration compared with recipients of control Ab, which was statistically significant by semiquantitative analysis (Figure 11). Immunohistochemical staining of sections from anti–MCP-1 recipients demonstrated that the residual cellular infiltrates were nearly devoid of macrophages (data not shown). These data indicate that a gene product of the alveolar epithelial target cell plays a major role in the lung infiltration that ensues after CD8+ T cell recognition of an antigen presented by the alveolar cell.
Figure 10.

In vivo neutralization of MCP-1 significantly reduces parenchymal infiltration after T cell transfer. Histologic sections from lungs harvested 4 days after adoptive transfer of 2.5 × 106 CD8+ T cell clone 40-2 with coadministration of Ab in the tail vein. (a) The infiltration of lung parenchyma harvested from an SPC-HA–transgenic mouse 96 hours after transfer of T cells with control Ab. (b) The typical pattern of infiltration in the SPC-HA–transgenic animals that received Ab to MCP-1 at the time of T cell transfer (50 μg/animal). The cellular infiltrates are significantly reduced, and the residual infiltrates are nearly devoid of macrophages by immunohistochemistry (not shown).
Figure 11.

Semiquantitative analysis of cellular infiltration after in vivo neutralization of MCP-1 at the time of T cell transfer in HA+ transgenic animals. The parenchymal cellular infiltration is significantly reduced, and the residual infiltrates are nearly devoid of macrophages by immunohistochemistry (not shown). AP < 0.01. The data shown are representative of two separate experiments.
Discussion
Cytolytic CD8+ T cells represent an important arm of the adaptive immune response to virus infection, although T cell–mediated virus clearance may be associated with significant tissue injury (39–42). The immune response to respiratory virus infection leads to a complex inflammatory cascade in the lung, which may result in virus clearance, lung injury, or both. Virus infection of the respiratory epithelium triggers production of inflammatory mediators by the infected epithelial cells, such as IL-8 (43) and type 1 IFNs (44). This leads to recruitment of cells and mediators of the innate immune system and subsequently to those of the adaptive immune response. Epithelial cells that continue to present viral antigens during this phase become targets of antigen-specific cytolytic T cell recognition. Specific CD8+ T cell responses may play an even greater role in virus clearance (and tissue injury) in immune individuals, since the memory CD8+ T cell response emerges earlier and more vigorously than does the CD8+ T cell response in primary virus infection (45, 46). Although specific CD8+ T cell recognition of a virus-infected target cell may result in apoptotic death of the infected cell, the complexity of factors that influence susceptibility to cytolysis are being increasingly appreciated (29–31, 40). In addition, ample evidence exists to indicate that numerous soluble factors may trigger the expression of a variety of inflammatory mediators by respiratory epithelial cells. However, the specific contribution of lung epithelial cells to the inflammatory milieu that evolves in the context of an immune response to a respiratory virus infection has been difficult to assess because of the complicating effects of virus infection on epithelial cell activities. In this study, we have shown that specific CD8+ T cell recognition of alveolar epithelial cells, in the absence of virus infection, may induce alveolar cell expression of a variety of inflammatory mediators, in vitro and in vivo. The T cell may also produce soluble inflammatory mediators upon specific antigen recognition, and it is possible that both sources of chemokines contribute to the milieu that leads to parenchymal infiltration of host inflammatory cells and the resultant respiratory physiologic impairment. Alveolar epithelial target cells may be important participants in lung injury resulting from CD8+ T cell recognition, since significant macrophage accumulation occurs approximately 72–96 hours after transfer, whereas the transferred CD8+ T cells become essentially undetectable in the lungs after approximately 48 hours (R.I. Enelow, et al., unpublished). Furthermore, CD8+ T cell transfer appears to induce a similar degree of respiratory dysfunction in severe combined immune-deficiency (SCID) mice that express the HA transgene, an argument against a critical role of the host lymphocyte component of the parenchymal infiltration (R.I. Enelow, et al., unpublished observations).
Chemokines are a large family of chemoattractant cytokines that play a major role in the orchestration and amplification of acute and chronic inflammatory processes, particularly in the lung (47–51). The induction of chemokine expression by alveolar epithelial cells appears to be mediated primarily by TNF-α expressed by the CD8+ T cell, a mediator not commonly associated with T cell effector activity. (The results presented in this study have been corroborated with several distinct CD8+ T cell clones, indicating that the phenomena described are not unique to one clone). The activation of alveolar epithelium that occurs as a result of specific CD8+ T cell recognition may reflect either a population effect or an effect of epithelial cells as individual responders to TCR engagement (or both). There are two potential mechanisms that might account for MCP-1 production by alveolar epithelial cells upon specific T cell recognition that are not mutually exclusive. The first involves the direct cytolysis of those target cells whose MHC/peptide complexes engage TCR (by either perforin or membrane-bound TNF-α) with associated antigen-dependent T cell production of soluble TNF-α. The soluble TNF-α produced might therefore lead to the induction of bystander MCP-1 expression by other target cells, which stochastically did not engage the TCR of a lymphocyte and did not undergo apoptosis. The second possibility involves the direct ligation of TCR and TNF receptor(s) on an individual target cell, the result of which may be activation of the engaged target and induction of chemokine expression in the target cell. A critical corollary to this hypothesis is that there may be interactions between cytolytic T cells and target cells that may be below threshold for induction of target-cell death, but above threshold for induction of transcriptional activation. The fact that induction of chemokine expression could occur in a target cell expressing MHC/peptide complexes that have been engaged by a TCR of a cytolytic CD8+ T cell suggests that a perforin/granzyme system may sometimes be quantitatively or qualitatively insufficient to induce cytolysis or that other factors may influence target-cell susceptibility to cytotoxicity. Similarly, the threshold for induction of chemokine expression by transmembrane TNF-α may be lower than the threshold for induction of apoptosis. The identification of cellular regulators of protection against perforin/granzyme–mediated cytotoxicity (29–31) and TNF-mediated apoptosis (32) suggest potential mechanisms by which regulation of in vivo susceptibility of target cells to T cell–mediated cytolysis might occur.
It is likely that multiple mechanisms account for the activation of epithelial cells as a population, in response to specific CD8+ T cell recognition, the net result of which is the production of chemokines by the alveolar cells, which may in turn amplify inflammatory responses in the lung. In our adoptive transfer model, this may contribute to the recruitment of host inflammatory cells, particularly macrophages, into the lung parenchyma, the infiltration of which strongly correlates with the physiologic dysfunction associated with interstitial pneumonitis, i.e., restrictive mechanics and diminished diffusing capacity (11). The data in this study suggest that alveolar epithelial cells may be active participants in the inflammation and injury associated with CD8+ T cell recognition of alveolar antigens in the lung, irrespective of the influence of virus infection. Furthermore, apoptotic target-cell death is not a necessary outcome of CD8+ cytolytic T cell antigen recognition, which may instead (or in addition) lead to inflammatory activation of the cell being recognized.
Acknowledgments
The authors would like to gratefully acknowledge the technical assistance of Alyssa Stell, Jennifer Liebermann, and Gwen Baber. This work was supported by US Public Health Service grant HL-58660, as well as the Johnie Walker Murphy Career Investigator Award from the American Lung Association. Support of the Beirne B. Carter Foundation, the American Lung Association of Virginia, and the Cardiovascular Research Center at the University of Virginia is gratefully acknowledged. We thank Thomas J. Braciale for critical review of the manuscript.
References
- 1.Braciale TJ, Ada GL, Yap KL. Functional and structural considerations in the recognition of virus-infected cells by cytotoxic T lymphocytes. Contemp Top Mol Immunol. 1978;7:319–371. doi: 10.1007/978-1-4757-0779-3_10. [DOI] [PubMed] [Google Scholar]
- 2.Ada G, Jones P. The immune responses to influenza infection. Curr Top Microbiol Immunol. 1986;128:1–54. doi: 10.1007/978-3-642-71272-2_1. [DOI] [PubMed] [Google Scholar]
- 3.Askonas B. Our immune defense against influenza. Biochem Soc Trans. 1980;8:257–260. doi: 10.1042/bst0080257. [DOI] [PubMed] [Google Scholar]
- 4.Leatherman J, Michael A, Schwartz B, Hoidal J. Lung T cells in hypersensitivity pneumonitis. Ann Intern Med. 1984;100:390–396. doi: 10.7326/0003-4819-100-3-390. [DOI] [PubMed] [Google Scholar]
- 5.Mukae H, Kadota J, Kohno S, Matsukura S, Hara K. Increase of activated T-cells in BAL fluid of Japanese patients with bronchiolitis obliterans organizing pneumonia and chronic eosinophilic pneumonia. Chest. 1995;108:123–128. doi: 10.1378/chest.108.1.123. [DOI] [PubMed] [Google Scholar]
- 6.Spurzem, J., and Rennard, S. 1996. Immunology of idiopathic pulmonary fibrosis. In Immunopathology of lung disease. R. Kradin and B. Robinson, editors. Butterworth-Heinemann, Boston, Massachusetts, USA. 119–131.
- 7.Karpel JP, Norin AJ. Association of activated cytolytic lung lymphocytes with response to prednisone therapy in patients with idiopathic pulmonary fibrosis. Chest. 1989;96:794–798. doi: 10.1378/chest.96.4.794. [DOI] [PubMed] [Google Scholar]
- 8.Costabel U, Teschler H, Guzman J. Bronchiolitis obliterans organizing pneumonia (BOOP): the cytological and immunocytological profile of bronchoalveolar lavage. Eur Respir J. 1992;5:791–797. [PubMed] [Google Scholar]
- 9.Kradin RL, et al. Usual interstitial pneumonitis is a T-cell alveolitis. Clin Immunol Immunopathol. 1986;40:224–235. doi: 10.1016/0090-1229(86)90025-5. [DOI] [PubMed] [Google Scholar]
- 10.Enelow R, et al. A lung-specific neo-antigen elicits specific CD8+ T cell tolerance with preserved CD4+T cell reactivity. J Clin Invest. 1996;98:914–922. doi: 10.1172/JCI118874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enelow RI, et al. Experimental T cell-mediated lung disease: structural and functional consequences of alveolar cell recognition by CD8+T lymphocytes. J Clin Invest. 1998;102:1652–1661. doi: 10.1172/JCI4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu AN, et al. Perforin-independent cytotoxicity in T cell-mediated cytotoxicity of alveolar epithelial cells is preferentially mediated by tumor necrosis factor-α: relative insensitivity to Fas ligand. Am J Respir Cell Mol Biol. 1999;20:849–858. doi: 10.1165/ajrcmb.20.5.3585. [DOI] [PubMed] [Google Scholar]
- 13.Kusters S, et al. In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur J Immunol. 1997;11:2870–2875. doi: 10.1002/eji.1830271119. [DOI] [PubMed] [Google Scholar]
- 14.Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53:45–53. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- 15.Kinkhabwala M, et al. A novel addition to the T cell repertory. Cell surface expression of tumor necrosis factor/cachectin by activated normal human T cells. J Exp Med. 1990;171:941–946. doi: 10.1084/jem.171.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Standiford TJ, et al. Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J Clin Invest. 1990;86:1945–1953. doi: 10.1172/JCI114928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauty A, et al. The T cell-specific CXC chemokines IP-10, Mig, and I-TAC are expressed by activated human bronchial epithelial cells. J Immunol. 1999;162:3549–3558. [PubMed] [Google Scholar]
- 18.Lilly CM, et al. Expression of eotaxin by human lung epithelial cells: induction by cytokines and inhibition by glucocorticoids. J Clin Invest. 1997;99:1767–1773. doi: 10.1172/JCI119341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu B, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luster A. Chemokines: chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 21.Gerszten RE, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–723. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 22.Graham M, Braciale T, Braciale V. Use of antiviral T-lymphocyte clones to characterize antigen presentation and T-lymphocyte subsets. Methods. 1996;9:439–444. doi: 10.1006/meth.1996.0050. [DOI] [PubMed] [Google Scholar]
- 23.McKall-Faienza, et al. Absence of TNFRp55 influences virus-induced autoimmunity despite efficient lymphocytic infiltration. Int Immunol. 1998;10:405–412. doi: 10.1093/intimm/10.4.405. [DOI] [PubMed] [Google Scholar]
- 24.Wikenheiser K, et al. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci USA. 1993;90:11029–11033. doi: 10.1073/pnas.90.23.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fine A, Anderson NL, Rothstein TL, Williams MC, Gochuico BR. Fas expression in pulmonary alveolar type II cells. Am J Physiol. 1997;273:L64–L71. doi: 10.1152/ajplung.1997.273.1.L64. [DOI] [PubMed] [Google Scholar]
- 26.Kagi D, et al. Fas and perforin as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- 27.Kojima H, et al. Two distinct pathways of specific killing revealed by perforin mutant cytototoxic T lymphocytes. Immunity. 1994;1:357–364. doi: 10.1016/1074-7613(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 28.Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 1994;370:650–652. doi: 10.1038/370650a0. [DOI] [PubMed] [Google Scholar]
- 29.Muller C, Tschopp J. Resistance of CTL to perforin-mediated lysis. Evidence for a lymphocyte membrane protein interacting with perforin. J Immunol. 1994;153:2470–2478. [PubMed] [Google Scholar]
- 30.Bird CH, et al. Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol Cell Biol. 1998;18:6387–6398. doi: 10.1128/mcb.18.11.6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun J, et al. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J Biol Chem. 1996;271:27802–27809. doi: 10.1074/jbc.271.44.27802. [DOI] [PubMed] [Google Scholar]
- 32.Pimentel-Muinos FX, Seed B. Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity. 1999;11:783–793. doi: 10.1016/s1074-7613(00)80152-1. [DOI] [PubMed] [Google Scholar]
- 33.Gu L, et al. In vivo properties of monocyte chemoattractant protein-1. J Leukoc Biol. 1997;62:577–580. doi: 10.1002/jlb.62.5.577. [DOI] [PubMed] [Google Scholar]
- 34.Xu N, Chen CY, Shyu AB. Modulation of the fate of cytoplasmic mRNA by AU-rich elements: key sequence features controlling mRNA deadenylation and decay. Mol Cell Biol. 1997;17:4611–4621. doi: 10.1128/mcb.17.8.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Black RA, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor- alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 36.Kayagaki N, et al. Metalloproteinase-mediated release of human Fas ligand. J Exp Med. 1995;182:1777–1783. doi: 10.1084/jem.182.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gearing AJ, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 38.Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10) J Exp Med. 1987;166:1084–1097. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alwan WH, Kozlowska WJ, Openshaw PJ. Distinct types of lung disease caused by functional subsets of antiviral T cells. J Exp Med. 1994;179:81–89. doi: 10.1084/jem.179.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guidotti LG, Chisari FV. To kill or to cure: options in host defense against viral infection. Curr Opin Immunol. 1996;8:478–483. doi: 10.1016/s0952-7915(96)80034-3. [DOI] [PubMed] [Google Scholar]
- 41.Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- 42.Openshaw PJ. Immunopathological mechanisms in respiratory syncytial virus disease. Springer Semin Immunopathol. 1995;17:187–201. doi: 10.1007/BF00196165. [DOI] [PubMed] [Google Scholar]
- 43.Fiedler MA, Wernke-Dollries K, Stark JM. Respiratory syncytial virus increases IL-8 gene expression and protein release in A549 cells. Am J Physiol. 1995;269:L865–L872. doi: 10.1152/ajplung.1995.269.6.L865. [DOI] [PubMed] [Google Scholar]
- 44.Biron CA. Initial and innate responses to viral infections: pattern setting in immunity or disease. Curr Opin Microbiol. 1999;2:374–381. doi: 10.1016/s1369-5274(99)80066-6. [DOI] [PubMed] [Google Scholar]
- 45.Selin LK, Varga SM, Wong IC, Welsh RM. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J Exp Med. 1998;188:1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flynn KJ, et al. Virus-specific CD8+T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683–691. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 47.Chensue S, et al. Monocyte chemotactic protein expression during schistosome egg granuloma formation. Sequence of production, localization, contribution, and regulation. Am J Pathol. 1995;146:130–138. [PMC free article] [PubMed] [Google Scholar]
- 48.Smith R, Streiter R, Phan S, Kunkel S. C-C chemokines: novel mediators of the profibrotic response to bleomycin challenge. Am J Respir Cell Mol Biol. 1996;15:693–702. doi: 10.1165/ajrcmb.15.6.8969262. [DOI] [PubMed] [Google Scholar]
- 49.Agostini C, et al. Involvement of the IP-10 chemokine in sarcoid granulomatous reactions. J Immunol. 1998;161:6413–6420. [PubMed] [Google Scholar]
- 50.Keane MP, et al. The CXC chemokines, IL-8 and IP-10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol. 1997;159:1437–1443. [PubMed] [Google Scholar]
- 51.Hogaboam CM, Lukacs NW, Chensue SW, Strieter RM, Kunkel SL. Monocyte chemoattractant protein-1 synthesis by murine lung fibroblasts modulates CD4+T cell activation. J Immunol. 1998;160:4606–4614. [PubMed] [Google Scholar]