

Abstract
Through a multiprotein complex, glycogen synthase kinase-3β (GSK-3β) phosphorylates and destabilizes β-catenin, an important signaling event for neuronal growth and proper synaptic function. δ-Catenin, or NPRAP (CTNND2), is a neural specific member of the β-catenin superfamily, and is also known to modulate neurite outgrowth and synaptic activity. In this study, we investigated the possibility that δ-catenin expression is also affected by GSK-3β signaling, and it participates in the molecular complex regulating β-catenin turnover in neurons. Immunofluorescent light microscopy revealed co-localization of δ-catenin with members of the molecular destruction complex: GSK-3β, β-catenin, and APC in rat primary neurons. GSK-3β formed a complex with δ-catenin, and its inhibition resulted in increased δ-catenin and β-catenin expression levels. LY294002 and amyloid peptide, known activators of GSK-3β signaling, reduced δ-catenin expression levels. Furthermore, δ-catenin immunoreactivity increased and protein turnover decreased when neurons were treated with proteasome inhibitors, suggesting that the stability of δ-catenin, like that of β-catenin, is regulated by proteasome-mediated degradation. Co-immunoprecipitation experiments showed that δ-catenin overexpression promoted GSK-3β and β-catenin interactions. Primary cortical neurons and PC12 cells expressing δ-catenin treated with proteasome inhibitors showed increased ubiquitinated β-catenin forms. Consistent with the hypothesis that δ-catenin promotes the interaction of the destruction complex molecules, cycloheximide treatment of cells overexpressing δ-catenin showed enhanced β-catenin turnover. These studies identify δ-catenin as a new member of the GSK-3β signaling pathway and further suggest that δ-catenin is potentially involved in facilitating the interaction, ubiquitination, and subsequent turnover of β-catenin in neuronal cells.
Keywords: Glycogen synthase kinase-3β, δ-catenin/NPRAP, β-catenin, proteasome, ubiquitination
Introduction
δ-Catenin (CTNND2), or neural plakophilin-related armadillo protein (NPRAP), is a member of the β-catenin superfamily of proteins, which contain a highly homologous repeating central armadillo (ARM) domain (Paffenholz et al., 1997; Zhou et al., 1997; Peifer et al., 1994a). Through this domain these family members interact with cadherin and are linked to the actin cytoskeleton where they modulate cell adhesion and process elaboration (Hatzfeld and Nachtsheim, 1996; Peifer et al., 1994a; Lu et al., 1999; Martinez et al., 2003; Grosheva et al., 2001). In adult neural tissues, δ-catenin is expressed in the dendrites, is enriched in the postsynaptic density, and participates in modulating dendritic arborization (Kim et al., 2002; Lu et al., 2002; Jones et al., 2002; Martinez et al., 2003; Arikkath et al., 2008; Abu-Elneel et al., 2008). In addition to its localization and abundant expression in the brain, there are several lines of evidence indicating that proper expression of δ-catenin is critical for normal brain function. First, hemizygous loss of chromosome 5p15.2 which encodes for δ-catenin, is associated with a severe form of mental retardation in Cri-du-Chat syndrome (Medina et al., 2000). Second, targeted disruption of the δ-catenin gene in mice results in severe impairments in cognitive function and abnormalities in short- and long-term synaptic plasticity which is important in memory and learning (Israely et al., 2004). Although previous studies demonstrated that δ-catenin-induced branching and turnover are modulated by presenilin-1 (PS-1) expression and that PS-1 bearing Alzheimer disease mutations enhances δ-catenin processing, the mechanisms regulating δ-catenin expression and stability are poorly understood (Kim et al., 2006a). Furthermore, little is known about how changes in δ-catenin expression levels affect intracellular signaling pathways that are involved in neuronal morphology and function.
GSK-3β is a serine/threonine protein kinase highly expressed in the central nervous system. While the enzymatic activity of GSK-3β is associated with a diverse number of intracellular signaling pathways, one well-characterized substrate of GSK-3β is β-catenin. Evidence from many studies indicates that GSK-3β has a primary role in down-regulation of β-catenin levels (Rubinfeld et al., 1996; Yost et al., 1996; Sakanaka et al., 1998). GSK-3β is a component of a multiprotein destruction complex that phosphorylates β-catenin thus signaling it for proteasome-mediated degradation, an event which is critical for normal neural development (Peifer et al., 1994b; Peifer et al., 1994c; Aberle et al., 1997; Woodgett, 2001). In the presence of extracellular cues, such as neurotrophins and Wnts, intracellular signal transduction targets the inactivation of GSK-3β resulting in stabilization and accumulation of β-catenin, thereby increasing β-catenin nuclear translocation and binding to transcription factors (Behrens et al., 1996; Huber et al., 1996; Molenaar et al., 1996). Inhibition of GSK-3β has been shown to enhance and modulate accumulation of the destruction complex molecules in growth cones, stabilize β-catenin, and change neuronal morphology (Zhou et al., 2004; Rubinfeld et al., 1995; Zumbrunn et al., 2001).
Shared binding partners, sequence homology, and similarities in the effect of δ-catenin and β-catenin on cellular morphology suggest that δ-catenin is potentially a new member of the GSK-3β signaling complex in neuronal cells. In this study we identify that the GSK-3β destruction complex regulates δ-catenin expression and stability and thereby participates in the molecular complex that regulates β-catenin turnover. We demonstrate that GSK-3β forms a stable complex with δ-catenin and phosphorylates δ-catenin in neurons, an event that mediates ubiquitination and subsequent proteasome degradation of δ-catenin. These findings provide evidence that GSK-3β modulates δ-catenin and β-catenin stability through a similar regulatory pathway and that altering δ-catenin expression levels in neurons effects β-catenin/GSK-3β interactions and β-catenin ubiquitination and turnover.
Materials and Methods
Antibodies
Antibodies used for the detection of δ-catenin were obtained from BD Bioscience and Upstate Biotechnology. All other antibodies were used as follows: anti-β-catenin, anti-pSer33, 37Thr41-β-catenin and anti-GSK-3β (BD Bioscience); anti-ubiquitin (BD Pharmigen); anti-APC and anti-actin (Santa Cruz Biotechnology); anti-pSer9-GSK-3β; anti-Tau (Tau-1) (Sigma); anti-pSer/Thr (Upstate Biotechnology).
Primary neuronal cultures and immunofluorescence microscopy
Primary rat cortical and hippocampal neurons were cultured as previously described (Goslin et al. 1998) with the exception of the following modifications. Embryonic day 18 rats (Charles River, MA) were euthanized and the embryos removed according to the approved NIH and ECU IACCU guidelines. Tissues were washed with HBSS (Hank’s balanced salt solution with 10 mM HEPES, pH 7.4). Cortices and hippocampi were dissected and dissociated with 0.25% trypsin at 37°C. The tissues were washed with HBSS and mechanically dissociated with gentle passage through flamed Pasteur pipettes. Cells were plated onto poly-L-lysine coated plates and coverslips in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum overnight. Media was then removed and replaced with Neurobasal media supplemented with B-27 (Invitrogen). Cultures were treated with 5 μM 5′deoxyuridine overnight to limit the growth of glial cells. Primary cortical and hippocampal neurons were grown in culture for 10–14 days unless otherwise indicated. For immunofluorescence light microscopy, cells grown on coverslips were fixed in 4% paraformaldehyde, permeablilized in 0.2% Triton X-100, and blocked with 10% BSA in PBS. Coverslips were incubated with primary antibodies for 1 hour at room temperature followed by the incubation with appropriate secondary antibodies: anti-mouse and anti-rabbit Cy3 (1:400), anti-mouse and anti-rabbit fluoresceine isothyocynate (FITC) (1:100) (Sigma-Aldrich, St. Louis, MO). Immunofluorescence was observed using 40x and 63x objectives on a Zeiss Axiovert S100 and a Zeiss LSM 510 laser scanning confocal light microscope (Carl Zeiss, Thornwood, NY). Control cells, nonspecific staining was determined by incubating coverslips with only secondary antibody. No observable pattern or intensity of immunofluorescence was observed for cells treated in this manner.
Cell lines and transfections
PC12 and PC12-δ-catenin (PC12-δ-cat) cells stably expressing δ-catenin under a Tet-off system were cultured as previously described (Lu et al., 2002). Mouse NIH 3T3 cells were transfected with Lipofectamine 2000 reagent as described by the manufacturer (Invitrogen). Briefly, cells were incubated for 4 hours with the complex containing plasmid DNA and transfection reagent in serum free media. Media was then removed and replaced with serum containing media until further experiments. Cells were then treated with the indicated drugs and lysed 24 to 48 hours after transfection.
Prostate cancer epithelial cells, CWR-RV1 cells, were grown in RPMI 1640 containing 10% FBS. Cells were transfected using the Lipofectamine 2000 method with the following plasmids; HA-GSK-3β-WT (He et al., 1995), HA-GSK-3β-S9A, constituitively active mutant (CA) (Stambolic et al., 1994), and HA-GSK3β-K85A, kinase dead mutant (KD) were kindly provided by Dr. Jim Woodget through Addgene. Primary cortical neurons were transfected with a set of small-hairpin (shRNA) corresponding to rat δ-catenin mRNA. shRNA constructs were designed and synthesized by (Origene) as follows: 5′-CTGGTCACAGGAGTCCTTTGGAACCTCTC-3′ and control shRNA pRS plasmid was used as a negative control. The Amaxa electroporation method was used for transfections of control and shRNA plasmids. Briefly, dissociated neurons were suspended in 100 μl of Amaxa electroporation buffer with 4 μg of plasmid DNA. The cell mixture was then transferred to Amaxa electroporation cuvette and electroporated with Amaxa Nucleofector apparatus set on O-O5 program. After transfection, cells were plated onto poly-L-lysine coated plates and incubated at 37°C with CO2 controlled atmosphere. Cells were collected 3 days after transfection, lysed and immunoblotted.
Tissue homogenization, lysate preparation, immunoprecipitation, and immunoblotting
We prepared tissue culture cell lysates and whole brain homogenates of adult mice in RadioImmuno Precipitation Assay (RIPA) buffer (150mM NaCl, 10mM HEPES pH 7.3, 2mM EDTA, 0.2% SDS, 0.5% Sodium deoxycholate, 1% Triton X-100) supplemented with protease inhibitors (Roche Complete Protease Inhibitor cocktail), followed by 30 minute incubation on ice. Cell debris was removed by 14,000g centrifugation. Supernatant was collected and protein concentration was determined using Pierce BCA assay. For immunoprecipitation experiments one milligram of cell lysate or brain protein extracts were incubated with primary antibody for 4 hours. For δ-catenin immunoprecipitation both monoclonal and polyclonal antibodies were used. Protein A beads (with rabbit primary antibody) and protein G beads (with mouse and goat primary antibody) were added for an additional overnight incubation followed by a 500g centrifugation for 5 minutes. The immuno-complex was washed three times with the extraction buffer, and eluted with 2X SDS sample buffer. Samples were boiled for 5 minutes and loaded on SDS-PAGE gels, transferred to nitrocellulose membrane and subject to Western blot analysis.
Drug treatments
GSK-3β inhibitor-TDZD-8 (Calbiochem) at a final concentration of 10 μM or lithium chloride (LiCl) at a final concentration of 1.0 mM, unless otherwise noted, were added to the cell culture media for 4 hours to inhibit GSK-3β. LY294002 (Sigma), a PI3K inhibitor that activates GSK-3β was added to the media for 1 hour at a final concentration of 50 μM. Synthetic β-amyloid (1-42) Aβ42 were prepared as previously described (Alvarez et al., 1999). Briefly, Aβ42 peptide solutions (> 70% HPLC) from stock solutions were prepared by dissolving lyophilized aliquots of Aβ42 peptide (AnaSpec) in dimethylsulfoxide (DMSO), and the solution was incubated for 1 day. Aβ42 peptide treatments at a final concentration of 10 μM were added to cell culture media with overnight incubation. Proteasome inhibitor treatments were performed as follows: ALLN 10 μM (Calbiochem), MG-132 10 μM (Calbiochem), Lactacystin 10 μM (A.G Scientific, Inc.), and ALLM 25 μM (Sigma). Chemicals were added to cell culture media for 4–8 hours unless otherwise noted.
Determination of protein half-life
δ-Catenin and β-catenin protein half-life were determined by incubating cells with 50 μg/ml cycloheximide for the indicated times. Equal amounts of protein were loaded for corresponding time points and subjected to immunoblot analysis. Western blotted protein bands were semi-quantified using BioRad ChemiDoc Documentation system (BioRad).
Results
δ-Catenin associates with GSK-3β
Previous studies have demonstrated that δ-catenin and GSK-3β are highly regulated during neuronal development and are involved in neurite outgrowth (Ho et al., 2000; Lu et al., 2002; Meijer et al., 2004; Zhou et al., 2004). In order to determine the association between δ-catenin and GSK-3β, we examined the localization of δ-catenin and GSK-3β in primary hippocampal neurons cultured in vitro for 9 days. Endogenous δ-catenin and GSK-3β localized to the neuronal processes and soma (Fig. 1A, B). The observed immunolocalization of δ-catenin and GSK-3β to the dendrites is consistent with previous reports (Leroy and Brion, 1999, Ho et al., 2000; Jones et al., 2002). High magnifciantion imaging and laser confocal immunofluorescent light microscopy using the Z-line function confirmed the partial co-localization of δ-catenin and GSK-3β in dendrites (Fig. 1B, C).
Figure 1. Co-localization of δ-catenin with GSK-3β in primary rat hippocampal neurons.
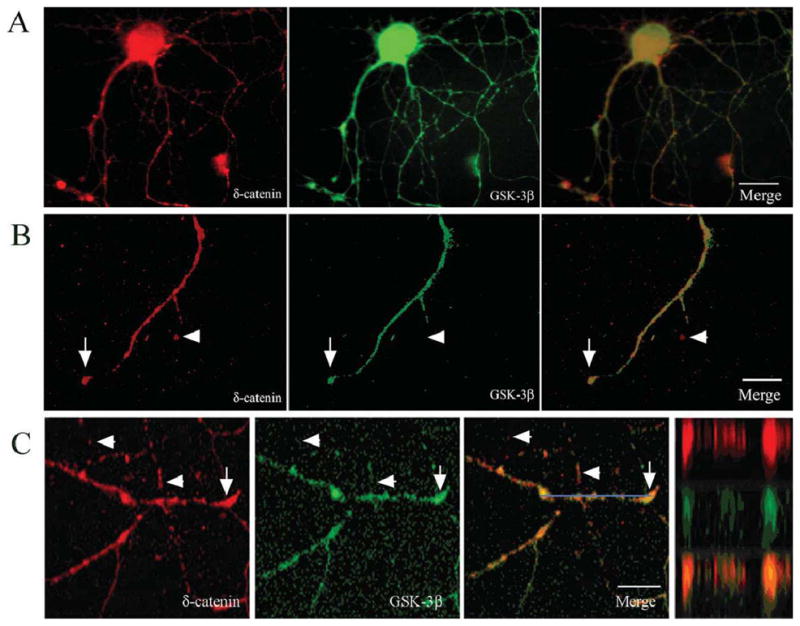
A. Fluorescent light microscopic imaging using a Zeiss Axiovert microscope shows intracellular distribution of δ-catenin and GSK-3β in a DIV 9 neuron. Merged images show co-localization in soma and dendrites. B. Higher magnification of a dendrite shows partial overlapping (arrows) and non-overlapping (arrowheads) localization of δ-catenin with GSK-3β. C. Confocal imaging of a primary hippocampal neuron (DIV 9) along a dendrite shows partial overlapping (arrows) and non-overlapping (arrowheads) localization of δ-catenin and GSK-3β. Blue bar: Z-line image. Far right panel shows Z-line and merge. Bars: 15 μm (A) and 10 μm (B and C).
Next, we performed co-immunoprecipitation experiments to investigate the association of δ-catenin with GSK-3β in neuronal tissues. Endogenous δ-catenin was immunoprecipitated from adult mouse brain homogenates. Western blot analysis demonstrated that GSK-3β was co-immunoprecipitated with δ-catenin (Fig 2A, left panel). Reverse co-immunoprecipitation using anti-GSK-3β antibody followed by δ-catenin immunoblotting confirmed this interaction (Fig. 2A, right panel). Similar findings on primary cortical neurons further corroborated this observation (Fig. 2B). In addition, when cultured neurons were treated with LiCl, a selective GSK-3β inhibitor, anti-δ-catenin immunoprecipitated lysates showed a decrease in δ-catenin phospho-serine-threonine immunoreactivity compared to untreated controls (Fig. 2B, left panel and 2C). These results suggest that δ-catenin serine-threonine phosphorylation may be controlled by GSK-3β activity in primary cortical neurons.
Figure 2. δ-Catenin co-immunoprecipitates with GSK-3β in neuronal tissues.

A. (Left panel) Western blot analysis of δ-catenin immunoprecipitated (IP) from mouse brain homogenates and probed with indicated antibodies; (Right panel) GSK-3β IP from mouse brain homogenates probed with indicated antibody. B. Primary cortical neurons were treated with GSK-3β inhibitor (LiCl) as indicated and immunoprecipitated. (Left panel) δ-catenin IP probed with GSK-3β and phospho-serine threonine antibody (S/TP); (Right panel) GSK-3β IP probed with indicated antibodies. C. Bar graph shows mean values of relative densitometric phospho-δ-catenin immunoreactivity. Error bars represent ± SEM with n =3.
GSK-3β activity affects δ-catenin expression and stability
To further explore the effects of GSK-3β activity on δ-catenin expression levels we treated primary cortical neurons with increasing doses of LiCl. Western blot analysis showed that cells treated within therapeutic relevant doses of LiCl corresponded with an increase in δ-catenin immunoreactivity in a dose dependent manner (Fig. 3A). Treatments above the 1.0 mM concentration corresponded with a tapering decrease in δ-catenin expression, emphasizing the importance of dosage effects on the regulation GSK-3β and its substrates (Kim et al., 2006b). Tau-1 immunoreactivity (recognizing dephosphorylated Ser 195/198/199/202) has been shown to increase in the presence of LiCl in a dose-dependent manner referring to the involvement of GSK-3β activity in phosphorylation of tau at the Tau-1 site (Takahashi et al. 1999; Tatebayashi et al. 2004). Both anti-dephospho-Tau (Tau-1) and anti-phospho-GSK-3β (GSK-3βP) immunreactivities increased with increasing LiCl concentrations demonstrating the effectiveness of LiCl in the inhibition of GSK-3β activity (Fig. 3A). Treatment with TDZD-8, another GSK-3β specific inhibitor, had a similar effect on increasing δ-catenin immunoreactivity (Fig. 3E).
Figure 3. Regulation of δ-catenin by the GSK-3β pathway in primary cortical neurons.

A. Western blot analysis showed that inhibition of GSK-3β with LiCl (up to 1.0 mM) increased δ-catenin levels in primary cortical neurons. Tau1 (dephospho-Tau) was used to verify effectiveness of pharmacological treatment. GSK-3β activity was verified by probing with GSK-3βP-Ser9 (numbers represent mM concentrations). B. Western blot analysis of primary cortical neurons treated with 1 mM of LiCl, shows decreased δ-catenin expression compared with untreated controls (C). C. Inhibition of PI3K activity with LY294002 (LY) decreased δ-catenin protein expression in primary cortical neurons. D. Treatment of neurons with Aβ42 (Aβ) decreased the amount of phosphorylated GSK-3β and showed decreased δ-catenin expression. E. Neurons treated with LiCl, as well as a different GSK-3β specific inhibitor TDZD-8 (GSK-3βi), showed increased δ-catenin and β-catenin immunoreactivity. Four-hour pretreatment of neurons with GSK-3β inhibitors shows rescue of δ-catenin and β-catenin expression in the presence of LY294002. F. CWR22-Rv-1 cells transfected with either GSK-3β-HA WT (wild type), GSK-3β KD (kinase dead), and GSK-3β CA (constitutively active). Transfection with GSK-3β KD and GSK-3β CA demonstrate corresponding increases and decreases in endogenous δ-catenin immunoreactivity. Bar graphs to the right of representative Western blots in panel B, C, D, E, and F show mean values of relative densitometric of δ-catenin from Western blots. Analysis was performed with SigmaStat Version 3.11 (Systat Software, Inc., San Jose, CA) using the t-test module. Error bars represent ± SEM with n = 5 (LiCl); 4 = (LY); 4 = (Aβ); 3 = (E and F).
While these results showed that GSK-3β inhibition increased δ-catenin levels in cultured cells, we sought to determine if activation of GSK-3β negatively affects δ-catenin expression levels. To activate GSK-3β we treated neurons with either LY294002 or Aβ42 (Zhou et al., 2004; Takashima et al., 1996) and observed a decrease in δ-catenin immunoreactivity under these conditions (Fig. 3C, D, and E). We then pretreated neurons with LiCl followed by incubation with LY294002. Western blot analysis showed a recovered δ- and β-catenin expression levels (Fig. 3E). These results are consistent with previous reports which demonstrate that β-catenin levels can be modulated by GSK-3β activity (Grimes and Jope, 2001b; Zhou et al.,2004; Fuentealba et al., 2004) but provide new information with regard to the effects of GSK-3β activity on δ-catenin expression. Together, these results are in line with the notion that modulation of GSK-3β activity affects δ- and β-catenin levels in primary cortical neurons, which are potentially important downstream signaling events in the neuroprotective effects of GSK-3β inhibition.
To establish if these changes in δ-catenin expression were specific to GSK-3β activity, we transfected CWR22-Rv1 cells, endogenously expressing δ-catenin, with GSK-3β WT, GSK-3β kinase dead mutant (KD), or GSK-3β-constitutively active mutant (CA) constructs. Consistent with our pharmacological data, we found that transfecting cells with GSK-3β-CA resulted in decreased δ-catenin expression and GSK-3β-KD resulted in decreased δ-catenin expression further linking GSK-3β activity to δ-catenin expression (Fig. 3F).
δ-Catenin is ubiquitinated and undergoes proteosome-mediated degradation
Next we tested the possibility that GSK-3β affects δ-catenin degradation. To determine if δ-catenin is degraded by the proteasome pathway, we incubated NIH 3T3 cells transfected with δ-catenin and treated them with ALLN (N-acetyl-Leu-Leu-norleucinal, LLnL, or calpain I inhibitor), a widely used inhibitor of proteosome-mediated degradation. Western blot analysis showed that ALLN treatment resulted in increased δ-catenin immunoreactivity (Fig. 4A). To demonstrate this effect on endogenous δ-catenin, time course experiments using primary cortical neurons showed that ALLN treatment resulted in increased δ-catenin and β-catenin immunoreactivity within 2 hour of incubation with ALLN (Fig. 4B, C). Here, the increased expression of β-catenin was used as positive control for the effectiveness of the drug treatments and was consistent with the report by Aberle et al (1997). To verify that the change in δ-catenin immunoreactivity was due to proteasome specific inhibition, we treated primary cortical neurons with ALLM, Pepstatin A, Leupeptin, or sodium orthovanadate, and we observed no increase in δ-catenin expression compared to treatments with either the ALLN, MG132, or lactacystin (data not shown). Furthermore, protein turnover experiments demonstrated increased δ-catenin stability in primary cortical neurons treated with ALLN, suggesting that δ-catenin turnover involves proteasome-mediated degradation (Fig. 4D).
Figure 4. Proteasome inhibitor treatment reveals increased δ-catenin levels, ubiquitinated δ-catenin, and increased δ-catenin stability.

A. Western blot of NIH 3T3 cells transfected with δ-catenin showing increased δ-catenin immunoreactivity following treatment with the proteasome inhibitor ALLN. B. Neurons treated with ALLN for indicated times and immunoblotted with δ-catenin and β-catenin antibodies show increased immunoreactivity. Accompanying bar graph represents ubiquitinated densitometric measurements from untreated control and ALLN treated neurons. C. Bar graph representing relative densitometric values of δ-catenin in primary cortical neurons treated with ALLN or DMSO controls. Error bars represents the mean ± SEM from 5 independent experiments. D. δ-Catenin turnover was decreased in neurons treated with ALLN. Quantitative measurements of δ-catenin band intensities of cells treated with cycloheximide at indicated time points from three independent experiments. Data are reported as mean ± SEM. All data was normalized for each condition (Control and ALLN) at the 0 hour time point. E. Lysates from treated neurons immunoprecipitated with δ-catenin antibody show slower migrating and ubiquitated δ-catenin forms. F. Western blot of lysates from cultured neurons treated with LY294002 or LiCl immunoprecipitated with anti-δ-catenin. LY294002 enhanced phospho-serine threonine immunoreactivity whereas LiCl decreased phospho-serine threonine immunoreactivity and ubiquitination compared to untreated controls. Western blots (WB) of non-immunoprecipitated lysates are shown in bottom panel of D and E. Bar graph represents densitometric measurements from control, LY294002, and LiCl neuronal cells.
To determine if δ-catenin is ubiquitinated, we treated primary cortical neurons with ALLN and immunoprecipitated δ-catenin from the cell lysates. Western blot analysis using an anti-ubiquitin antibody detected ubiquitin suggesting association of this molecule with δ-catenin (Fig. 4E). To assess if GSK-3β activity is involved in δ-catenin ubiquitination, cell lysates from primary neurons treated with or without LY294002 or LiCl were immunoprecipitated with a δ-catenin antibody and immunoblotted using an anti-ubiquitin or anti-phospho-serine/threonine antibody (Fig. 4F). Western blot analysis showed that inhibition of GSK-3β activity with LiCl treatment decreased δ-catenin ubiquitination and serine-theonine phosphorylation compared to untreated controls (Fig. 4F). Conversely, GSK-3β activation by LY294002 treatment showed an increase in δ-catenin phosphorylation and decrease in its ubiquitination. A decreased δ-catenin ubiquitination under LY294002 conditions is possibly due to its enhanced degradation and turnover. Taken together, these results suggest that δ-catenin stability is regulated by GSK-3β thereby tagging it for proteasome-mediated degradation.
δ-Catenin expression promotes GSK-3β association with β-catenin
It is well established that β-catenin stability is regulated by a multiprotein complex that includes GSK-3β and APC (Adenomatous Polyposis Coli) proteins (Hart et al., 1998; Peifer and Polakis, 2000; Woodgett, 2001; Ha et al., 2004). Since our data supports that δ-catenin stability is also regulated by the GSK-3β-proteasome signaling pathway, we tested the hypothesis that δ-catenin may participate in the multiprotein complex by promoting the interaction of these molecules to influence β-catenin degradation. To explore this possibility, we first sought to determine if δ-catenin co-localizes with the destruction complex molecules in primary neurons. Double immunofluorescent labeling showed partial co-localization of δ-catenin with β-catenin, GSK-3β, and APC in the dendrites of mature hippocampal neurons (Fig. 5, arrows). Immunoprecipitation of δ-catenin from primary cortical neurons confirmed the association of δ-catenin with APC, β-catenin and GSK-3β (Fig. 6A). Next we performed co-immunoprecipitation experiments to determine if δ-catenin expression enhanced the interactions among β-catenin, GSK-3β and APC. We used PC12 cells stably overexpressing δ-catenin under the control of a tetracycline-regulated transactivator system (Lu et al., 2002). Western blot analysis of proteins immunoprecipitated from cell lysates by anti-β-catenin indicated higher levels of GSK-3β and APC in PC12 cells overexpressing δ-catenin (PC12-δ-cat) compared to PC12 controls (Fig. 6B). Reverse co-immunoprecipitation by anti-GSK-3β confirmed the increased association of GSK-3β with β-catenin in PC12-δ-cat compared to controls PC12 cells (Fig. 6C); however, APC levels were not different between PC12-δ-cat and PC12 cells (Fig. 6C), suggesting that δ-catenin expression does not affect GSK-3β and APC interactions.
Figure 5. Localization of δ-catenin with GSK-3β destruction complex members in the neurites of primary hippocampal neurons.
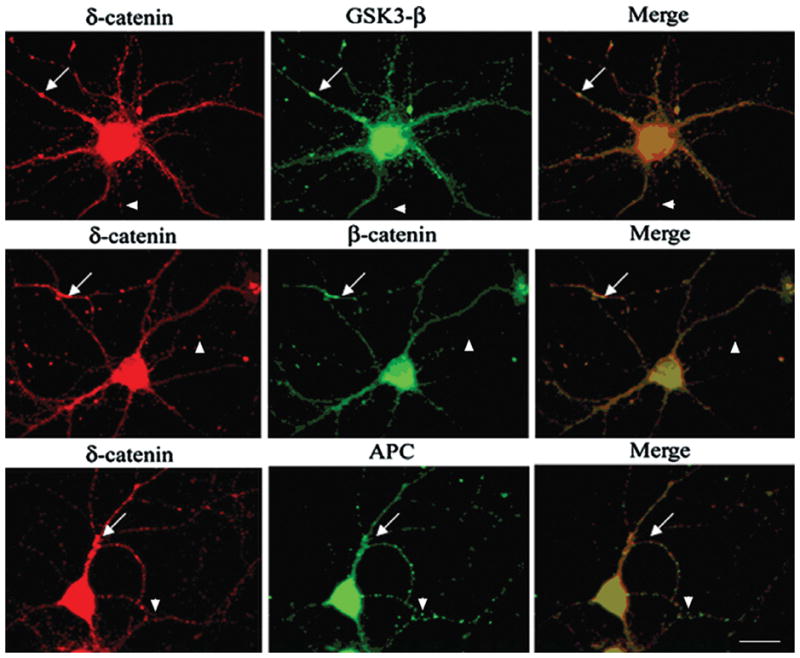
Double immunofluorescence staining shows intracellular distribution of δ-catenin, β-catenin, GSK-3β, and APC. Arrows show co-localization of GSK-3β destruction complex molecules with δ-catenin whereas arrowheads show non-overlapping localization of δ-catenin with β-catenin, GSK-3β, and APC. Bar: 15 μm.
Figure 6. δ-Catenin expression facilitates β-catenin, GSK-3β, and APC interactions.

A. δ-Catenin co-immunoprecipitated with APC, β-catenin, and GSK-3β from cultured neurons. ALLN treatment showed an enhanced co-immunoprecipitation of δ-catenin with β-catenin and GSK-3β. Bar graph to the right represents mean densitometric values from δ-catenin immunoprecipitated cells treated with ALLN. Densitometric values for APC, β-catenin, and GSK-3β were compared to δ-catenin. B. and C. Lysates from tetracycline-inducible PC12 cells overexpressing δ-catenin (PC12-δ-cat) were immunoprecipitated with either anti-β-catenin or anti-GSK-3β antibodies as indicated. Immunoprecipitated samples were then immunoblotted with indicated antibodies. Greater amounts of GSK-3β (B) and β-catenin (C) were co-precipitated from PC12-δ-cat cells compared to PC12. Non-immunoprecipitated samples were subject to Western (WB) analysis (bottom panel) using anti-δ-catenin antibody. Bar graphs to the right of B and C demonstrate Western blot densitometric values of β-catenin, GSK-3β and APC from PC-12-δ-cat and PC12 cells. Data shown in panels A, B, and C are representative of gels from three separate experiments.
We then examined if δ-catenin expression affected β-catenin ubiquitination by transfecting NIH 3T3 cells with δ-catenin (δ-cat) or vector alone (control). Transfected cells were treated with ALLN as indicated, and Western blot analysis showed additional slower migrating β-catenin forms when δ-catenin was overexpressed (Fig. 7A, arrows). These higher molecular weight forms of β-catenin are indicative of ubiquitination (Aberle et al., 1997). Similar results of increased ubiquitination of β-catenin were observed using PC12 cells overexpressing δ-catenin (Fig. 7B, β-catenin, arrows). In addition, immunoblot analysis using a phospho-β-catenin (Ser33,37/Thr41) antibody, corresponding to phosphorylation sites by GSK-3β, showed an enhanced signal in PC12-δ-cat compared to PC12 controls (Fig. 7B, β-cateninp, arrows). To verify this effect using cells endogenously expressing δ-catenin, we altered δ-catenin expression in primary cortical neurons by transfecting them with either vector alone (control) or siRNA targeted against δ-catenin. Anti-β-catenin immunoblotting showed decreased levels of the slower migrating β-catenin forms (decreased ubiquitination) when δ-catenin expression was reduced (Fig. 7C, arrows). Anti-actin was used as a loading control for Western blot analysis and was used as reference to determine δ-catenin knock-down efficiency (Fig. 7C and D). Although individual experiments did not show robust knock-down for endogenous δ-catenin, overall trends were clear and β-catenin showed decreased ubiquitination (Fig 7D, right panel), Therefore, the results from all these three cell model systems indicated that δ-catenin expression effected β-catenin ubiquitination.
Figure 7. δ-Catenin expression facilitates the ubiquitination of β-catenin.

A. NIH 3T3 cells transfected with δ-catenin cDNA (δ-cat) or vector alone as control (Con) and treated with ALLN. Western blot analysis illustrates that δ-catenin transfected cells show additional slower migrating β-catenin forms compared to controls (indicated by the arrows). Similar results were obtained from three independent experiments. B. ALLN treatment increased levels of slower migrating β-catenin forms (arrows) in PC12-δ-cat compared to control cells. The levels of phospho-β-catenin antibody were greater in PC12-δ-cat versus control cells. Similar results were obtained from three independent experiments. Graphs to the right of A and B represent desitometric values of ubiquitinated β-catenin under ALLN conditions. Measurements were normalized to control (Con) or PC12-δ-cat. C. Primary cortical neurons transfected with either vector alone as control (Con) or siRNA targeted against δ-catenin (siRNA-δ-cat) were treated with ALLN. Western blot analysis demonstrated decreased β-catenin ubiquitination as δ-catenin expression decreased. Bar graph to the right represents relative densitometric values of β-catenin in primary cortical neurons treated with or without ALLN; all values were normalized to actin as a loading control. Similar results were obtained from 2 independent experiments, data shown from representative Western blot.
δ-Catenin expression promotes β-catenin turnover
Since δ-catenin expression increased β-catenin-GSK-3β interactions as well as β-catenin ubiquitination, we sought to determine what effect δ-catenin expression had on β-catenin turnover. Cells were treated with cycloheximide to block protein synthesis, and then cell lysates taken at different time points were analyzed by Western blot analysis with antibodies to β-catenin according to a previously established procedure (Kim et al., 2006a). β-Catenin turnover was increased in PC12-δ-cat cells when compared to PC12 controls (Fig. 8A and B). δ-Catenin expression reduced the half-life of β-catenin from 12 hours to less than 8 hours (Fig. 8B).
Figure 8. Effects of δ-catenin expression on β-catenin stability.
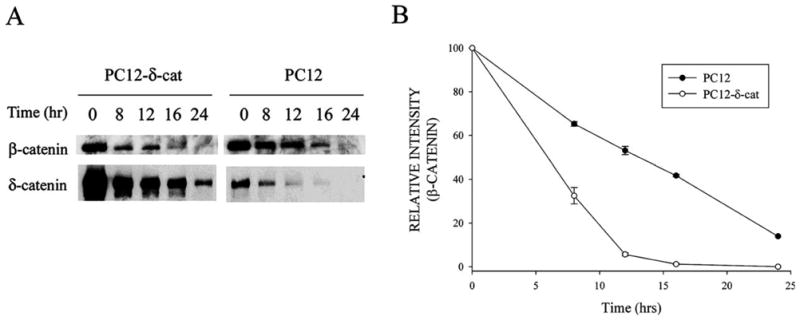
A. Immunoblotting with anti-δ-catenin and anti-β-catenin antibodies in PC12-δ-cat and PC12 cells treated with cycloheximide for indicated times. B. Semi-quantitative measurements of β-catenin immunoreactivity in PC12-δ-cat and PC12 cells shows increased β-catenin turnover in cells overexpressing δ-catenin. Data are from three independent experiments and are reported as mean ± SEM. All data was normalized for each condition (PC12 and PC12-δcat) at the 0 hour time point.
Discussion
In this study we examined the hypothesis that GSK-3β regulates δ-catenin expression by changing its stability in neuronal cells. We demonstrated that δ-catenin is associated with GSK-3β, is ubiquitinated, and undergoes proteosome-mediated degradation. Our studies also provided evidence that δ-catenin expression affects β-catenin-GSK-3β interactions as well as β-catenin ubiquitination and turnover. Our data in corroboration with others support a model where the GSK-3β molecular destruction complex is associated with the regulation of δ-catenin and β-catenin in neuronal cells (Votin et al., 2005).
We found that δ-catenin expression levels were correlated with GSK-3β activity when neurons were treated with various pharmacological agents to modulate GSK-3β activity. Interestingly, we found that only low doses of LiCl (up to 3mM) resulted in increased δ-catenin expression, whereas higher doses of LiCl reduced δ-catenin levels. It is known that the degree of GSK-3β inhibition is associated with dramatically different morphological presentations in neurons. Previous reports show that while partial inhibition of GSK-3β favors process extension of neurites from developing neurons, complete inactivation of GSK-3β leads to impaired neurite growth (Munoz-Montano et al., 1999; Kim et al., 2006b). It is also well established that δ-catenin expression is capable of inducing process elongation and branching in cells (Kim et al., 2002; Martinez et al., 2003; Kim et al., 2008). Therefore, our data which supports that partial inhibition of GSK-3β increases δ-catenin expression, raises an interesting possibility that δ-catenin may be an important downstream target of GSK-3β signaling that participates in modulating neuronal morphology.
There is growing evidence that the regulated degradation of proteins by localized control of GSK-3β activity and activation of the ubiquitin-proteosome pathway may be an important modulator of synaptic plasticity and neuronal morphology (Zhou et al., 2004; Bingol and Schuman, 2005). Several studies show that synaptic activity drives the ubiquitin machinery and β-catenin into neuronal spines which promotes changes in neurite morphology (Bozdagi et al., 2000; Murase et al., 2002). Here we demonstrated that the ubiquitin-proteosome pathway is one such element involved in regulating the turnover of δ-catenin. As a newly identified component of this signaling, adequate changes in δ-catenin expression have been associated with normal neuronal development, function, and dendritic arborization (Ho et al., 2000; Israely et al., 2004; Kim et al., 2002). Given that δ-catenin is localized to the post-synaptic compartment and undergoes rapid redistribution with synaptic activity (Jones et al., 2002), it suggests that signaling control of δ-catenin turnover may be an important localized event influencing synaptic structures and dendritic morphogenesis. Future experiments to address the localization and time-dependent nature of this signaling, through the identification of ubiquitin ligases and other kinases that contribute to promoting catenin turnover will help in elucidating the roles of δ-catenin in neuronal morphogenesis and function.
Our data supports that δ-catenin may be a new member of the GSK-3β molecular destruction complex involved in promoting β-catenin turnover. In this complex, GSK-3β phosphorylates β-catenin which is facilitated by APC (Munemitsu et al., 1995; Rubinfeld et al., 1996). Previous studies demonstrate that β-catenin and APC are enriched in the growth cones of axons and display a punctuated pattern along the dendritic processes in primary mouse neuronal cultures (Morrison et al., 1997; Zhou et al., 2004; Shimomura et al., 2005). δ-Catenin is preferentially localized to dendrite in mature neurons and also displays a similar punctuate distribution pattern (Lu et al., 1999; Ho et al., 2000). In this study, we showed by immunofluorescent light microscopy that endogenous δ-catenin partially co-localizes with β-catenin, APC, and GSK-3β in the processes of primary hippocampal neurons. We also demonstrated that both endogenous and exogenous δ-catenin co-immunoprecipitates with β-catenin, APC, and GSK-3β in neuronal cells, identifying a new association between δ-catenin and molecular members of the destruction complex.
Regulation of the destruction complex and its central role in ubiquitin-dependent destabilization of β-catenin has largely been studied in the context of oncogenesis and embryogenesis due to its role in transcriptional regulation (Cadigan and Nusse, 1997; Polakis, 2000). There is emerging evidence to suggest that the destruction complex molecules play an important role in neuronal function and that stabilized β-catenin can inhibit neurite outgrowth independent of its transcriptional activity (Pollack et al., 1997; Krylova et al., 2002; Yu and Malenka, 2003; Zhou et al., 2004; Votin et al., 2005). We found that overexpression of δ-catenin led to a greater interaction between β-catenin and GSK-3β and enhanced β-catenin phosphorylation and ubiquitination. In addition, overexpression of δ-catenin promoted the turnover of β-catenin in PC12 cells. Taken together, our data highlight the importance of proper δ-catenin expression for promoting β-catenin turnover and suggest that aberrant δ-catenin expression may be associated with altered efficiency of the destruction complex effecting β-catenin stability which has been shown to negatively regulate neurite outgrowth and function (Votin et al., 2005).
There are several lines of evidence that support our finding that δ-catenin and β-catenin stability is controlled by similar mechanisms. First, δ-catenin and β-catenin share several binding partners, which include PS-1, S-SCAM, and cadherin (Levesque et al.,1999; Ide et al., 1999). Of these partners, PS-1 has been implicated in the turnover of both δ-catenin and β-catenin, and pathogenic mutations in PS-1 have been associated with enhanced δ-catenin and β-catenin destabilization (Kim et al., 2006a; Zhang et al., 1998). PS-1 proteins are located in the plasma membrane and participate in the cleavage of transmembrane proteins (Georgakopoulos et al., 1999; Kaether et al., 2006). Since neither δ-catenin nor β-catenin fulfill the classical criteria of PS-1/γ-secretase substrate for intramembranous proteolysis, an alternative role for PS-1 may need to be defined in the turnover of δ-catenin and β-catenin. Previous reports demonstrate that GSK-3β, β-catenin, and δ-catenin all bind to the hydrophilic loop region of PS-1 (Takashima et al., 1998; Murayama et al., 1997; Yu et al., 1998; Tanahashi and Tabira, 1999; Levesque et al., 1999). Therefore PS-1 may act as a scaffolding molecule for the destruction complex molecules, thereby participating in the turnover of δ-catenin and β-catenin. For example, δ-catenin may compete with β-catenin for the same binding site on PS-1, which could make β-catenin more vulnerable to cytoplasmic proteasome-mediated degradation. In support of this hypothesis, several reports demonstrate that PS-1 is associated with GSK-3β, which modulates the turnover of β-catenin (Kang et al., 1999; Takashima et al., 1998; Twomey and McCarthy, 2006; Prager et al., 2007). Further studies are needed to address the role of PS-1 in the regulation of GSK-3β, β-catenin, and δ-catenin at the plasma membranes as well as its role in signaling events, which contribute to time-dependent association or disassociation of these molecules.
Therefore, our studies are consistent with the literature that proteins of the destruction complex, such as GSK-3β and APC, can regulate levels of β-catenin (Munemitsu et al. 1995). Our studies demonstrate that δ-catenin is a new member in this complex that may participate in modulating signaling on β-catenin. We propose that δ-catenin expression facilitates the GSK-3β-β-catenin interaction, leading to enhanced ubiquitination and degradation of β-catenin.
Acknowledgments
Grant sponsor: NIH/NCI; Grant number: CA111891
Grant sponsor: NIH/NIA; Grant number: AG026630
We would like to thank Dr. Warren Knudson for helpful discussions, and Melissa Clark and Christi Boykin for technical assistance. This study was supported in part by NIH/NCI (CA111891) and NIH/NIA (026630) grants (Q.L.).
Reference List
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Elneel K, Ochiishi T, Medina M, Remedi M, Gastaldi L, Caceres A, Kosik K. A delta-catenin signaling pathway leading to dendritic protrusions. J Biol Chem. 2008;283:32781–91. doi: 10.1074/jbc.M804688200. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Toro R, Caceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999;459:421–426. doi: 10.1016/s0014-5793(99)01279-x. [DOI] [PubMed] [Google Scholar]
- Arikkath J, Israely I, Tao Y, Mei L, Xin L, Reichardt L. Erbin controls dendritic morphogenesis by regulatin localization of δ-catenin. J Neurosci. 28:7047–7056. doi: 10.1523/JNEUROSCI.0451-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC. Signal transduction during amyloid-beta-peptide neurotoxicity: role in Alzheimer disease. Brain Res Brain Res Rev. 2004;47:275–289. doi: 10.1016/j.brainresrev.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos A, Marambaud P, Efthimiopoulos S, Shioi J, Cui W, Li HC, Schutte M, Gordon R, Holstein GR, Martinelli G, Mehta P, Friedrich VL, Jr, Robakis NK. Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. Mol Cell. 1999;4:893–902. doi: 10.1016/s1097-2765(00)80219-1. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low density culture. Boston: MIT; 1998. pp. 339–370. [Google Scholar]
- Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001a;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001b;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Grosheva I, Shtutman M, Elbaum M, Bershadsky AD. p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: a link between cell-cell contact formation and regulation of cell locomotion. J Cell Sci. 2001;114:695–707. doi: 10.1242/jcs.114.4.695. [DOI] [PubMed] [Google Scholar]
- Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol Cell. 2004;15:511–521. doi: 10.1016/j.molcel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Hart MJ, de los SR, Albert IN, Rubinfeld B, Polakis P. Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol. 1998;8:573–581. doi: 10.1016/s0960-9822(98)70226-x. [DOI] [PubMed] [Google Scholar]
- Hatzfeld M, Nachtsheim C. Cloning and characterization of a new armadillo family member, p0071, associated with the junctional plaque: evidence for a subfamily of closely related proteins. J Cell Sci. 1996;109:2767–2778. doi: 10.1242/jcs.109.11.2767. [DOI] [PubMed] [Google Scholar]
- Ho C, Zhou J, Medina M, Goto T, Jacobson M, Bhide PG, Kosik KS. Delta-catenin is a nervous system-specific adherens junction protein which undergoes dynamic relocalization during development. J Comp Neurol. 2000;420:261–276. [PubMed] [Google Scholar]
- Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59:3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- Ide N, Hata Y, Deguchi M, Hirao K, Yao I, Takai Y. Interaction of S-SCAM with neural plakophilin-related Armadillo-repeat protein/delta-catenin. Biochem Biophys Res Commun. 1999;256:456–461. doi: 10.1006/bbrc.1999.0364. [DOI] [PubMed] [Google Scholar]
- Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, Liu X. Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr Biol. 2004;14:1657–1663. doi: 10.1016/j.cub.2004.08.065. [DOI] [PubMed] [Google Scholar]
- Jones SB, Lanford GW, Chen YH, Morabito M, Kim K, Lu Q. Glutamate-induced delta-catenin redistribution and dissociation from postsynaptic receptor complexes. Neuroscience. 2002;115:1009–1021. doi: 10.1016/s0306-4522(02)00532-8. [DOI] [PubMed] [Google Scholar]
- Kaether C, Haass C, Steiner H. Assembly, trafficking and function of gamma-secretase. Neurodegener Dis. 2006;3:275–283. doi: 10.1159/000095267. [DOI] [PubMed] [Google Scholar]
- Kang DE, Soriano S, Frosch MP, Collins T, Naruse S, Sisodia SS, Leibowitz G, Levine F, Koo EH. Presenilin 1 facilitates the constitutive turnover of beta-catenin: differential activity of Alzheimer’s disease-linked PS1 mutants in the beta-catenin-signaling pathway. J Neurosci. 1999;19:4229–4237. doi: 10.1523/JNEUROSCI.19-11-04229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Han JR, Park J, Oh M, James SE, Chang S, Lu Q, Lee KY, Ki H, Song WJ, Kim K. Delta-catenin-induced dendritic morphogenesis. An essential role of p190RhoGEF interaction through Akt1-mediated phosphorylation. J Biol Chem. 2008;283:977–987. doi: 10.1074/jbc.M707158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Bareiss S, Kim KK, Tatum R, Han JR, Jin YH, Kim H, Lu Q, Kim K. Presenilin-1 inhibits delta-catenin-induced cellular branching and promotes delta-catenin processing and turnover. Biochem Biophys Res Commun. 2006a;351:903–908. doi: 10.1016/j.bbrc.2006.10.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Sirota A, Chen YH, Jones SB, Dudek R, Lanford GW Thakore C, Lu Q. Dendrite-like process formation and cytoskeletal remodeling regulated by delta-catenin expression. Exp Cell Res. 2002;275:171–184. doi: 10.1006/excr.2002.5503. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR, Anton ES, Snider WD. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006b;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylova O, Herreros J, Cleverley KE, Ehler E, Henriquez JP, Hughes SM, Salinas PC. WNT-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3-responsive spinal sensory neurons. Neuron. 2002;35:1043–1056. doi: 10.1016/s0896-6273(02)00860-7. [DOI] [PubMed] [Google Scholar]
- Leroy K, Brion JP. Developmental expression and localization of glycogen synthase kinase-3beta in rat brain. J Chem Neuroanat. 1999;16:279–93. doi: 10.1016/s0891-0618(99)00012-5. [DOI] [PubMed] [Google Scholar]
- Levesque G, Yu G, Nishimura M, Zhang DM, Levesque L, Yu H, Xu D, Liang Y, Rogaeva E, Ikeda M, Duthie M, Murgolo N, Wang L, VanderVere P, Bayne ML, Strader CD, Rommens JM, Fraser PE, St George-Hyslop P. Presenilins interact with armadillo proteins including neural-specific plakophilin-related protein and beta-catenin. J Neurochem. 1999;72:999–1008. doi: 10.1046/j.1471-4159.1999.0720999.x. [DOI] [PubMed] [Google Scholar]
- Lu Q, Mukhopadhyay NK, Griffin JD, Paredes M, Medina M, Kosik KS. Brain armadillo protein delta-catenin interacts with Abl tyrosine kinase and modulates cellular morphogenesis in response to growth factors. J Neurosci Res. 2002;67:618–624. doi: 10.1002/jnr.10151. [DOI] [PubMed] [Google Scholar]
- Lu Q, Paredes M, Medina M, Zhou J, Cavallo R, Peifer M, Orecchio L, Kosik KS. Delta-catenin, an adhesive junction-associated protein which promotes cell scattering. J Cell Biol. 1999;144:519–532. doi: 10.1083/jcb.144.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MC, Ochiishi T, Majewski M, Kosik KS. Dual regulation of neuronal morphogenesis by a delta-catenin-cortactin complex and Rho. J Cell Biol. 2003;162:99–111. doi: 10.1083/jcb.200211025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina M, Marinescu RC, Overhauser J, Kosik KS. Hemizygosity of delta-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome. Genomics. 2000;63:157–164. doi: 10.1006/geno.1999.6090. [DOI] [PubMed] [Google Scholar]
- Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de WM, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Morrison EE, Askham J, Clissold P, Markham AF, Meredith DM. Expression of beta-catenin and the adenomatous polyposis coli tumour suppressor protein in mouse neocortical cells in vitro. Neurosci Lett. 1997;235:129–132. doi: 10.1016/s0304-3940(97)00739-8. [DOI] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Montano JR, Lim F, Moreno FJ, Avila J, az-Nido J. Glycogen Synthase Kinase-3 Modulates Neurite Outgrowth in Cultured Neurons: Possible Implications for Neurite Pathology in Alzheimer’s Disease. J Alzheimers Dis. 1999;1:361–378. doi: 10.3233/jad-1999-1602. [DOI] [PubMed] [Google Scholar]
- Murase S, Mosser E, Schuman EM. Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron. 2002;35:91–105. doi: 10.1016/s0896-6273(02)00764-x. [DOI] [PubMed] [Google Scholar]
- Murayama O, Honda T, Mercken M, Murayama M, Yasutake K, Nihonmatsu N, Nakazato Y, Michel G, Song S, Sato K, Takahashi H, Takashima A. Different effects of Alzheimer-associated mutations of presenilin 1 on its processing. Neurosci Lett. 1997;229:61–64. doi: 10.1016/s0304-3940(97)00417-5. [DOI] [PubMed] [Google Scholar]
- Paffenholz R, Franke WW. Identification and localization of a neurally expressed member of the plakoglobin/armadillo multigene family. Differentiation. 1997;61:293–304. doi: 10.1046/j.1432-0436.1997.6150293.x. [DOI] [PubMed] [Google Scholar]
- Peifer M, Berg S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell. 1994a;76:789–791. doi: 10.1016/0092-8674(94)90353-0. [DOI] [PubMed] [Google Scholar]
- Peifer M, Pai LM, Casey M. Phosphorylation of the Drosophila adherens junction protein Armadillo: roles for wingless signal and zeste-white 3 kinase. Dev Biol. 1994b;166:543–556. doi: 10.1006/dbio.1994.1336. [DOI] [PubMed] [Google Scholar]
- Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- Peifer M, Sweeton D, Casey M, Wieschaus E. wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development. 1994c;120:369–380. doi: 10.1242/dev.120.2.369. [DOI] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Pollack AL, Barth AI, Altschuler Y, Nelson WJ, Mostov KE. Dynamics of beta-catenin interactions with APC protein regulate epithelial tubulogenesis. J Cell Biol. 1997;137:1651–1662. doi: 10.1083/jcb.137.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager K, Wang-Eckhardt L, Fluhrer R, Killick R, Barth E, Hampel H, Haass C, Walter J. A structural switch of presenilin 1 by glycogen synthase kinase 3beta-mediated phosphorylation regulates the interaction with beta-catenin and its nuclear signaling. J Biol Chem. 2007;282:14083–14093. doi: 10.1074/jbc.M608437200. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Munemitsu S, Polakis P. The APC protein and E-cadherin form similar but independent complexes with alpha-catenin, beta-catenin, and plakoglobin. J Biol Chem. 1995;270:5549–5555. doi: 10.1074/jbc.270.10.5549. [DOI] [PubMed] [Google Scholar]
- Sakanaka C, Weiss JB, Williams LT. Bridging of beta-catenin and glycogen synthase kinase-3beta by axin and inhibition of beta-catenin-mediated transcription. Proc Natl Acad Sci U S A. 1998;95:3020–3023. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura A, Kohu K, Akiyama T, Senda T. Subcellular localization of the tumor suppressor protein APC in developing cultured neurons. Neurosci Lett. 2005;375:81–86. doi: 10.1016/j.neulet.2004.10.074. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Yasutake K, Tomizawa K. Lithium inhibits neurite growth and tau protein kinase I/glycogen synthase kinase-3beta-dependent phosphorylation of juvenile tau in cultured hippocampal neurons. J Neurochem. 1999;73:2073–83. [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci U S A. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, Imahori K. Exposure of rat hippocampal neurons to amyloid beta peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci Lett. 1996;203:33–36. doi: 10.1016/0304-3940(95)12257-5. [DOI] [PubMed] [Google Scholar]
- Tanahashi H, Tabira T. Isolation of human delta-catenin and its binding specificity with presenilin 1. Neuroreport. 1999;10:563–568. doi: 10.1097/00001756-199902250-00022. [DOI] [PubMed] [Google Scholar]
- Tatebayashi Y, Haque N, Tung YC, Iqbal K, Grundke-Iqbal I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J Cell Sci. 2004;117:1653–63. doi: 10.1242/jcs.01018. [DOI] [PubMed] [Google Scholar]
- Twomey C, McCarthy JV. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett. 2006;580:4015–4020. doi: 10.1016/j.febslet.2006.06.035. [DOI] [PubMed] [Google Scholar]
- Votin V, Nelson WJ, Barth AI. Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J Cell Sci. 2005;118:5699–5708. doi: 10.1242/jcs.02679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Judging a protein by more than its name: GSK-3. Sci STKE. 2001;2001:RE12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, St George-Hyslop PH, Fraser PE. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin. J Biol Chem. 1998;273:16470–16475. doi: 10.1074/jbc.273.26.16470. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de WM, Clevers H, Saftig P, De SB, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS. Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport. 1997;8:2085–2090. doi: 10.1097/00001756-199705260-00054. [DOI] [PubMed] [Google Scholar]
- Zumbrunn J, Kinoshita K, Hyman AA, Nathke IS. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol. 2001;11:44–49. doi: 10.1016/s0960-9822(01)00002-1. [DOI] [PubMed] [Google Scholar]