

Abstract
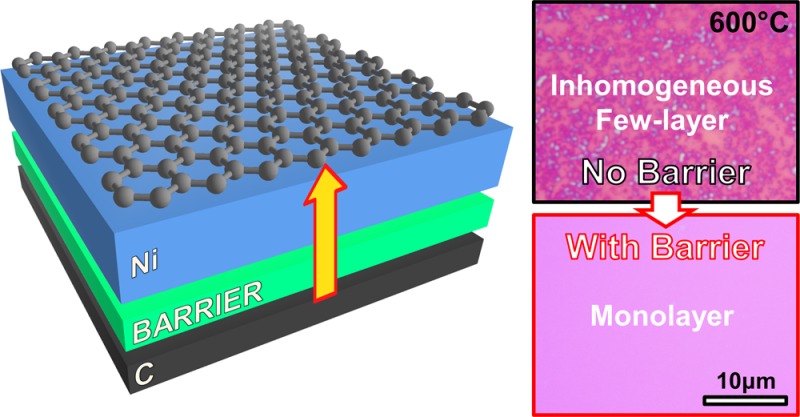
Carbon diffusion barriers are introduced as a general and simple method to prevent premature carbon dissolution and thereby to significantly improve graphene formation from the catalytic transformation of solid carbon sources. A thin Al2O3 barrier inserted into an amorphous-C/Ni bilayer stack is demonstrated to enable growth of uniform monolayer graphene at 600 °C with domain sizes exceeding 50 μm, and an average Raman D/G ratio of <0.07. A detailed growth rationale is established via in situ measurements, relevant to solid-state growth of a wide range of layered materials, as well as layer-by-layer control in these systems.
Keywords: Graphene, solid carbon, low temperature, diffusion barrier, in situ, XPS, XRD
The application potential of graphene depends entirely on the development of growth and integration techniques that are scalable and allow an adequate level of structural control and material quality.1 While chemical vapor deposition (CVD) is widely seen as the most promising approach for this, a potentially equally versatile but much simpler, cheaper and less hazardous technique is the catalytic graphitization of solid carbon sources. Carbon is thereby not supplied from the gas phase as in CVD, but rather as solid carbon film of finite thickness deposited below or atop a catalyst film. Global or local thermal annealing of this stack then yields mono- or few-layer graphene (M-/FLG) at the catalyst surface or interface. This principle has been demonstrated already in a number of variations across the literature, whereby it is commonly assumed that solid-state graphene growth occurs by dissolution of carbon into the catalyst at the annealing temperature followed by precipitation upon cooling.2−7 A particular motivation is thereby layer-by-layer control for FLG growth via the fixed and finite solid carbon supply, which in contrast to CVD is not self-limited by the increasing graphene coverage. However, to date the M-/FLG formed via solid carbon sources remains inferior in terms of uniformity and quality to that achieved by CVD and typically high annealing temperatures (>900 °C) are required to obtain reasonably graphitic films.6−9
Here we report a general and simple method to control the growth process and to significantly improve the quality and homogeneity of graphene formed by the catalytic transformation of solid carbon sources. We focus on technologically relevant low-temperature (≤600 °C) processing and show via complementary in situ X-ray photoelectron spectroscopy (XPS), X-ray reflectivity (XRR), and X-ray diffraction (XRD) measurements of Ni/tetrahedral amorphous carbon (ta-C) stacks that for such temperatures, graphene growth occurs predominantly during ramping up and annealing by carbon dissolution and diffusion through the catalyst, and that the contribution of carbon precipitation on subsequent cooling is minor. We thus show that a key problem with solid-state graphene growth, relevant to all previous literature, is the lack of an “on-switch” for the carbon supply. Carbon is uncontrollably fed during temperature ramping into a catalyst film,10 whose grain size distribution is still rapidly changing, leading to defective and inhomogeneous graphene nucleation at temperatures well below the maximum process temperature, that degrades the overall growth result. Carbon diffusion short-circuits through the evolving grain boundaries of the polycrystalline catalyst that can thereby further lower the graphene growth homogeneity. On the basis of the understanding of the growth process developed herein, we show that all these shortcomings can be effectively addressed by introducing a diffusion barrier between the solid carbon source and the catalyst as a means of controlling the carbon supply during the initial heating ramp. We demonstrate that a thin (1–3 nm) Al2O3 layer inserted into a ta-C/Ni bilayer stack acts as a diffusion barrier, enabling uniform MLG growth at 600 °C with graphene domain sizes exceeding 50 μm, and a quality (based on Raman D/G, 2D/G ratios) that equals that of CVD grown films.11,12 Our method of controlling the growth solute by introducing diffusion barriers is relevant to a large range of carbon solid-state precursors and similar layered materials, as well as to related layer-by-layer control in these systems.
Figure 1 highlights the major advantages that can be achieved with the introduction of a diffusion barrier. As a reference system, we use SiO2(300 nm)/Si substrates covered by filtered cathodic vacuum arc (FCVA) deposited ta-C (∼10 nm) and a top layer of physical vapor deposited Ni (∼550 nm, see Methods). We adopt a simple one-step annealing procedure in which samples are held at the chosen temperature for 5 min (see Methods). The plan-view, postgrowth scanning electron microscope (SEM) images in Figure 1A–C show that for the given conditions the onset of graphitic film formation at the top Ni surface is ∼300 °C, at which small (<200 nm in lateral dimensions) multilayered graphene islands are formed close to Ni grain boundaries (Figure 1B). This is indicative of short-circuit diffusion of carbon through the grain boundaries to the catalyst surface. For higher annealing temperatures, the graphene coverage expands and increases in thickness (Figure 1C). The as-grown graphene, however, is very inhomogeneous and no significant increase in the size of regions of constant contrast with (maximum) annealing temperature is apparent. This indicates that the as-formed layer quality is largely defined by the temperature at which formation begins during the heating ramp, rather than the maximum temperature reached during the process.
Figure 1.

SEM micrographs of Ni(550 nm)/ta-C(10 nm) (A–C) and Ni(550 nm)/Al2O3(2 nm)/ta-C(10 nm) (D–F) annealed at 200 °C (A,D), 300 °C (B), 500 °C (E), 600 °C (C,F) for 5 min (heated and cooled at a fixed rate of 100 °C min–1). The inset of D shows a higher-magnification micrograph of the sample showing the Ni grain structure (scale bar is 200 nm). The insets of E,F show lower-magnification micrographs of the same samples (scale bars are 100 μm). Sketches indicating the effect of annealing for each of the samples are also shown.
Figure 1D–F shows the results of identical thermal annealing but for a Ni/Al2O3/ta-C stack with the Al2O3 (∼2 nm) layer deposited by atomic layer deposition (ALD) on the ta-C prior to the Ni top layer deposition. In this case, no graphene formation or other carbon deposit is observed on the Ni surface for temperatures up to 500 °C (Figure 1 E). Instead the SEM image clearly shows the increase in average lateral Ni grain dimensions to typically ∼1 μm, significantly larger than those of ∼20 nm observed following annealing at 200 °C (Figure 1D). We note here the close analogy to the catalyst film pretreatment step used in graphene CVD to achieve a fully reduced catalyst layer with larger grain sizes.13,14 Increasing the annealing temperature to 600 °C results in the formation of large MLG domains of constant contrast (∼100 μm in lateral dimensions) that appear to grow out from a single nucleation point and extend across numerous Ni grains (Figure 1F). The secondary electron (SE) contrast variation within each island can be attributed to electron channelling contrast arising from the different grain orientations of the underlying polycrystalline Ni.15 The onset of growth, triggered by the diffusion of carbon through the Al2O3 diffusion barrier, is thereby clearly dependent on the Al2O3 thickness. We find the onset of graphene formation to occur at annealing temperatures of 400–500 °C for an Al2O3 (1 nm) layer, at 500–600 °C for an Al2O3 (2 nm) layer, while for an Al2O3 (3 nm) layer no graphene formation was observed even after annealing for 5 min at 600 °C.
Figure 2A compares typical Raman spectra for catalytically graphitized ta-C with and without an Al2O3 diffusion barrier, following transfer to clean SiO2(300 nm)/Si substrates (see Methods). The Ni/ta-C sample gives a D/G ratio of ∼2 and a very weak 2D peak, indicative of highly defective nanocrystalline, graphitic deposits.16 The introduction of the Al2O3 layer between the ta-C and Ni leads to a reduction in the D/G ratio to ∼0.5 indicating a significant improvement in quality. The 2D peak is also well fitted by a single Lorentzian peak with a full width at half-maximum (fwhm) of ∼32 cm–1 and 2D/G ratio of ∼3.1 confirming the presence of MLG,17 as previously suggested based on the SE contrast of the islands in Figure 1E. Analogous to our previously reported CVD results,11,18 a further improvement in graphitic quality can be achieved if the Ni film is decorated with a 5 nm evaporated Au film. For such a Au(5 nm)/Ni(550 nm)/Al2O3(2 nm)/ta-C(10 nm) stack heated to 600 °C, a Raman D/G ratio of ∼0.07 is observed, confirming high graphitic quality. The 2D peak is well fitted by a single Lorentzian with a fwhm of ∼34 cm–1, and a 2D/G ratio of >2 which is again consistent with the presence of MLG (Figure 2A). Figure 2B–D shows corresponding optical micrographs of the transferred graphene and 75 × 100 μm maps of the Raman 2D/G and D/G intensity ratios for this sample. The optical micrograph shows uniform contrast across the imaged area, again indicative of MLG coverage. The mapped region has an average 2D/G ratio of ∼3.5 with 100% of the area having a 2D/G ratio >2 (Figure 2C) suggesting a corresponding areal coverage of MLG. Figure 2D shows the D/G ratio is uniformly low with an average value of <0.07 demonstrating a similar quality to the graphene produced by CVD at similar temperatures.11 Careful inspection of the map of the D/G intensity ratio (Figure 2D) reveals an interconnected pattern with slightly higher D/G ratio, which reflects the lateral polycrystallinity of the continuous as-grown graphene. The Raman pattern indicates domains of ∼50 μm in lateral dimensions that bond together to form a continuous graphene film. This domain size is comparable to those deduced from SEM data shown in Figure 1F for elemental Ni. Six contact Hall-geometry devices fabricated on the as-transferred MLG give sheet resistance (RS) values of ∼1 kΩ/□, consistent with values measured for CVD graphene for the given support (SiO2) and transfer process.11,12 We emphasize that the graphene grown by annealing at 600 °C shows significantly better uniformity than other reports of growth from solid carbon across the literature, while the graphitic quality is comparable to the best reported values, even when notably higher growth temperatures (≥900 °C) were used.6−8
Figure 2.

(A) Raman spectra of the M-/FLG grown from Ni(550 nm)/ta-C(10 nm) (corresponding to Figure 1C), Ni(550 nm)/Al2O3(2 nm)/ta-C(10 nm) (corresponding to Figure 1F), and Au(5 nm)/Ni(550 nm)/Al2O3(2 nm)/ta-C(10 nm) samples annealed for 5 min at ∼600 °C and subsequently transferred to Si/SiO2(300 nm) using the bubbling transfer method. (B) Optical micrograph of the as-transferred MLG grown from a Au(5 nm)/Ni(550 nm)/Al2O3(2 nm)/ta-C(10 nm) sample under the annealing conditions used in (A). The sheet resistance (RS) of the as-transferred graphene is ∼1 kΩ/□, measured using six contact Hall-geometry devices (see Methods). (C,D) Raman maps of 2D/G peak intensity (average 2D/G ratio of ∼3.5 with 100% of the area >2 ) (C) and D/G peak intensity (average D/G ratio of <0.07) (D) for the region of graphene corresponding to the optical micrograph in (B).
As the basis of our process rationale, we use a combination of in situ XRR, XRD, and XPS to directly reveal the underlying growth mechanisms and kinetics. Figure 3 shows XRR and XRD data of a Ni(70 nm)/ta-C(10 nm) stack for which the temperature was increased in a stepwise manner. The sample was held at each temperature for ∼60 min, during which the respective measurements were performed. The XRR curves (Figure 3A,B) show two important changes during the heating process. First, between ∼250 and ∼350 °C the total reflective angle shifts to a lower value (Figure 3A), indicative of a decrease in the electron density of the topmost layer (as-deposited Ni). Concurrently the oscillations seen at higher angles, arising from the ta-C layer, vanish (Figure 3B). These two observations represent a direct signature of the dissolution of the ta-C layer and subsequent diffusion of this carbon into the Ni layer.
Figure 3.

(A,B) In situ XRR curves of a Ni(70 nm)/ta-C(10 nm)/SiO2(300 nm)/Si sample taken during vacuum heating (base pressure ∼10–6 mbar) for reflecting angles (2θ) of 0.5–0.9° (A) and 2–4.0° (B). The dashed horizontal arrow in A indicates the shift in total reflective angle on heating, while the vertical dashed lines in A and B indicate the oscillations associated with the Ni and ta-C layers respectively. (C) In situ grazing incidence XRD of a Ni(70 nm)/ta-C(10 nm)/SiO2(300 nm)/Si sample taken during the same stepwise annealing process with a fixed incident angle of αi = 0.75° (information depth of ∼80 nm). Note that the temperature-dependent shift in the reflection angles is due to thermal expansion. A monochromatic X-ray beam of 11.5 keV and a wavelength of 1.07812 Å (selected by a Si(111) double crystal monochromator) is used, and the reflected/diffracted X-rays are measured using a Mythen detector system. (D) Sketch showing the Ni/ta-C stacks that were probed and indicating the diffusion of carbon to the exposed catalyst surface which leads to M-/FLG formation.
X-ray diffractograms, taken in grazing incident geometry at each temperature step (Figure 3C), show sharpening of the Ni(111) and Ni(200) reflections19 on heating of the film above ∼250 °C, indicating significant grain growth and that the catalyst is metallic and of face-centered-cubic (fcc) structure. The Ni remains metallic and fcc throughout the annealing process and notably we find no evidence of a bulk, crystalline Ni-carbide20,21 using XRD. Between ∼450 and ∼575 °C, the graphite (002) peak22 at the sample surface reaches measurable intensity confirming M-/FLG formation during heating of the Ni/ta-C stack. The further increase in peak intensity at the next temperature step (∼692 °C), corroborates the increase in graphene thickness observed in our ex situ annealing experiments and shows that carbon diffusion to the catalyst surface and the resulting graphene formation continues as the sample is ramped to higher temperatures (Figure 3D). Upon cooling to room temperature, the graphite (002) peak intensity is not significantly altered, suggesting that the contribution to growth by precipitation on cooling is small. The observed behavior is consistent with previous in situ XRD observations of the solid-state formation of multilayer graphitic films,10 and has strong similarities to the metal-induced crystallization of amorphous silicon (a-Si).23 Here, we are able to further reveal the breakdown of the ta-C layer (using XRR) and generalize the observed growth mode to other metastable carbon sources (see ex situ observations below).
Figure 4 summarizes in situ, time- and depth-resolved XPS data that provides complementary surface-sensitive information on the graphene formation process. Figure 4A shows the time-resolved evolution of XP C1s core level spectra for a Ni(550 nm)/ta-C(10 nm) stack during heating to ∼600 °C in vacuum. We have previously identified four key components in the C1s spectra at ∼283.2 eV (CA), ∼283.8 eV (CDis), ∼284.4 eV (CGr), and ∼284.8 eV (CB).18 The peak evolution at the catalyst surface is consistent with that previously observed for graphene CVD on Ni catalysts during isothermal hydrocarbon exposures.13,18 The CA and CDis components emerge together first and are assigned to carbon bound to Ni surface sites (including a surface carbide reconstruction), and interstitial carbon dissolved in the Ni catalyst, respectively.13,18 The CGr and CB peaks then appear simultaneously some time later, reflecting the formation of graphitic carbon at the catalyst surface. Importantly, this formation of graphitic carbon is observed during the heating ramp and subsequent annealing of the Ni/ta-C stacks, again confirming that here the predominant M-/FLG growth mode is not via carbon precipitation upon cooling.
Figure 4.
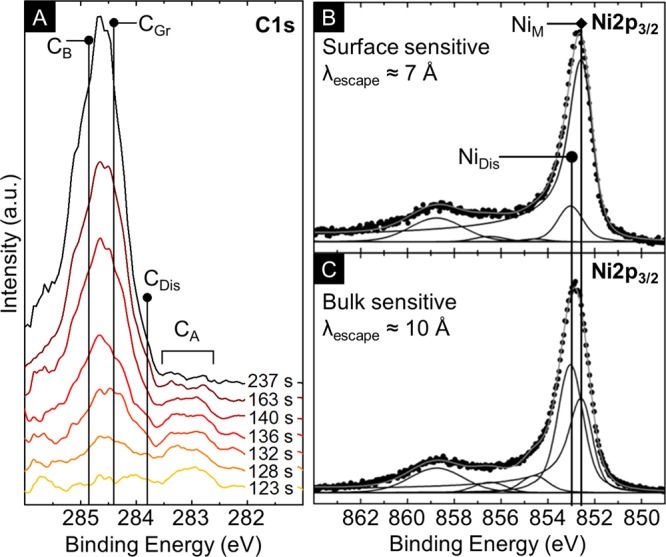
(A) Time-resolved in situ XPS C1s core level lines for Ni(550 nm)/ta-C(10 nm) stacks during vacuum heating to ∼600 °C at ∼100 °C/min. Acquisition times are relative to the start of the heating ramp from room temperature. Spectra are collected in normal emission geometry at photon energies of 435 eV (surface sensitive; λescape ≈ 7 Å) with a spectral resolution of ∼0.3 eV. (B) Depth-resolved in situ XPS Ni2p3/2 core level lines for Ni(550 nm)/ta-C(10 nm) stacks at the end of vacuum annealing at ∼600 °C. Spectra are background corrected (Shirley) and collected in normal emission geometry at photon energies of 1010 eV (surface sensitive; λescape ≈ 7 Å) and 1300 eV (bulk sensitive; λescape ≈ 10 Å) with a spectral resolution of ∼0.3 eV. Increased information depth is achieved using higher-incident X-ray energies and hence increased electron mean free path lengths. The spectra are fitted using Doniach-Šùnjić functions convoluted with Gaussian profiles with an accuracy of ∼0.05 eV. All binding energies are referenced to the Fermi edge.
Figure 4B,C shows depth resolved Ni2p3/2 core level spectra taken at the end of the vacuum annealing, prior to cooling. The information depth is varied by changing the incident X-ray energies (hv) and hence the inelastic mean free path lengths of the photoelectrons (λescape) to obtain a more surface sensitive spectrum (hv = 1010 eV, λescape ≈ 7 Å) and a more bulk sensitive spectrum (hv = 1300 eV, λescape ≈ 10 Å). We assign two main components NiM and NiDis which are related to metallic Ni and an interstitial solid solution of C in Ni, respectively.13,18 Comparison of the relative intensities of these components shows that the dissolved carbon species (NiDis) is stronger in the more bulk sensitive spectra. This indicates that although we observe effects of short-circuit diffusion at Ni grain boundaries, dissolved carbon is incorporated into the catalyst subsurface during the growth process, further highlighting the similarity between the solid-state M-/FLG formation observed here and the isothermal growth seen in CVD.
We have so far considered graphene growth from ta-C, however a wide variety of solid carbon sources may be used for solid-state growth with different bonding ratios and clustering as well as additional elements beyond carbon. Criteria for selection typically include the level of control over the quantity of carbon deposited,8 the cost associated with this deposition, and the potential for graphene doping,6,9 whether intended or not. We therefore further investigate HOPG and nanocrystalline diamond, two forms of elemental carbon that represent the two extremes of carbon bonding with almost exclusively sp2 and sp3 bonding respectively. Ni catalyst films are deposited on these samples by evaporation to minimize any alteration of the carbon bonding by the impingement of the Ni atoms, which is found to be more severe for the higher energy atoms commonly produced by sputtering (see Supporting Information).
Figure 5 compares scanning electron micrographs of Ni(550 nm)/HOPG and Ni(550 nm)/diamond samples annealed at 600 °C for 5 min (heated at a constant rate of 100 °C min–1, and cooled at ∼300 °C min–1). The annealed Ni/diamond samples show the formation of thick carbon layers at the exposed surface of the Ni catalyst (Figure 5A). Removal of the Ni catalyst by etching in FeCl3 reveals a corresponding depletion of the diamond layer at the SiO2/catalyst interface (observed by optical microscopy). The Ni covered HOPG substrates, conversely show no graphene or graphitic growth at the exposed catalyst surface (Figure 5B), and the HOPG appears to remain largely intact. Raman spectra measured on the Ni catalyst surfaces further confirms this (see insets) with the presence of D, G, and 2D peaks on the Ni/diamond sample indicating the formation of a graphitic carbon layer, while no peaks related to carbon are observed on the Ni/HOPG sample. The 2D peak of the Ni/diamond sample can be well fitted with a single Lorentzian peak of ∼79 cm–1 width, upshifted in position by ∼20 cm–1 compared to that of MLG, indicating that the graphitic layers formed are turbostratic (i.e., non-AB stacked).17,24 We note that in these experiments, the rapid quenching of the samples means that growth by precipitation on cooling is largely suppressed, as we have previously observed for CVD growth at similar temperatures.13 Ar+ plasma treatments were also performed on cleaned HOPG substrates prior to Ni evaporation in order to controllably induce defects/sp3 bonding at the HOPG surface with harsher plasma treatments leading to an increasing D/G ratio (see Supporting Information). Following Ni evaporation and the standard annealing process at 600 °C, M-/FLG formation is observed at the Ni surface, with the HOPG samples that underwent harsher Ar+ plasma treatments yielding increased M-/FLG nucleation density and coverage (see Supporting Information Figure S1). The comparison of carbon sources indicates that the driving force for the observed growth behavior is the thermodynamic stability of sp2 bonded, crystalline graphite relative to the solid carbon source used (e.g., ta-C, nanocrystalline diamond, and plasma treated HOPG). This has been highlighted already in comparison to similar layer exchange mechanisms such as the metal-induced crystallization of a-Si films.10 One of the key roles of the metal catalyst, here Ni, is thereby to lower the activation barriers typically associated with solid state transformations.
Figure 5.

Scanning electron micrographs of Ni(550 nm)/diamond(∼100 nm) (A) and Ni(550 nm)/HOPG (B) annealed at ∼600 °C for 5 min in vacuum (heated at a fixed rate of 100 °C min–1, cooled at ∼300 °C min–1). All scalebars are 2 μm. Insets show the corresponding Raman spectra measured on the as-grown samples. Sketches indicating the effect of annealing on each of the samples are also shown.
Our data reveals the following coherent model for solid-state graphene growth from Ni/metastable carbon stacks. The carbon source is broken down during heating and the liberated carbon diffuses through the catalyst layer toward the exposed catalyst surface, appearing as carbon bound to Ni surface sites and forming a subsurface Ni–C solid solution. Once sufficient carbon has diffused to the Ni catalyst surface, M-/FLG nucleation occurs and film growth continues while at elevated temperatures. This indicates that the solid-state formation of M-/FLG is not limited to a mechanism based on carbon precipitation on cooling, as has been suggested in other literature.2−7 Instead we observe a mechanism based on direct Ni-catalyzed transformation of the metastable carbon source into M-/FLG, which resembles the isothermal growth that occurs during hydrocarbon exposure for graphene CVD.18 The introduction of a diffusion barrier between the metastable carbon source and the catalyst prevents premature dissolution of the carbon source and delays graphene formation until a higher temperature has been reached. We note that for higher-temperature annealing (>900 °C) with no diffusion barrier present,6,7 M-/FLG is likely to form (as discussed here) during the initial stages of the heating ramp, but then can redissolve into the catalyst film as the temperature (and thus carbon solubility of Ni) is further increased. Upon subsequent cooling, M-/FLG may then be formed again by precipitation,10 which is consistent with the high-quality, yet inhomogeneous M-/FLG that has so far been reported in the literature for growth at these high temperatures.
Using the understanding developed from our in situ measurements in the context of existing literature, we are thus able to rationalize the improvement in quality and uniformity achieved by introducing Al2O3 barrier layers between the catalyst and carbon source. Al2O3 has been shown to be stable in the presence of carbon for the temperature range considered here,25 and the formation of bulk nickel-aluminate is also not expected under our vacuum annealing conditions.26 Our results show that the Al2O3 films effectively retard the diffusion of carbon into the Ni catalyst. With increasing Al2O3 film thickness, a higher temperature is reached before adequate carbon diffuses to the catalyst surface for graphene nucleation to occur. Previous literature reports on the onset of measurable carbon diffusion into single crystal sapphire at temperatures >500 °C.100 Further, Al2O3 is known to be stable during carbon nanotube (CNT) growth by CVD and CNT forest growth beneath a thin (∼10 nm) physical vapor deposited alumina layer has been reported,27 all of which is consistent with the diffusion of carbon through ALD deposited alumina as suggested here. In the context of CNT CVD, it is also known that transition metal catalysts such as Ni and Fe can diffuse in to Al2O3 layers28 which may also affect the dissociation of the solid carbon layer and subsequent carbon permeation. We note that other materials besides Al2O3 may also make suitable barrier layers and selection requires careful consideration of the material’s deposition and stability in the presence of both the catalyst and solid carbon source for the chosen processing conditions.
The increase in graphene formation temperature achieved by introducing Al2O3 barrier layers is analogous to an increase in exposure temperature for CVD graphene growth. The accompanying increase in graphene domain size and improvement in thickness uniformity may be explained based on considerations of carbon diffusivity. For an increase in growth temperature from 300 to 600 °C, there is a more than 5 orders of magnitude increase in the lattice diffusivity of carbon in Ni.29 Grain boundary and surface diffusion will also be significantly increased,30−32 meaning that carbon reaching the catalyst surface can more easily diffuse and attach to the edge of an existing graphene island, rather than a local supersaturation developing (e.g., at catalyst grain boundaries) and leading to nucleation of additional layers. We note that a higher temperature of graphene nucleation might also be achieved by a faster ramp rate. However, we found that even for a 10-fold increase in the heating rate to 1000 °C min–1, there was little improvement in the thickness homogeneity (data not shown). This can be understood in the context of the Arrhenius relationship between carbon diffusivity and temperature,29 which means that extremely high heating rates are required to significantly increase the M-/FLG formation temperature without a barrier layer.
A key feature of our solid-state growth, is that M-/FLG formation is fed from below the catalyst surface, therefore the formation of additional graphene layers is not limited by the already formed graphene layers (and leakage of carbon precursor through these), in contrast to CVD.11,12 The thickness of FLG films instead may be defined prior to the annealing process by the quantity of solid carbon source deposited. We note that throughout our experiments, thicker FLG regions generally showed lower D/G ratios compared to MLG regions, indicating that the additional layers formed have lower defect densities (compare, for example, the growth from elemental Ni in Figures 2A and 5A). This may relate to these additional layers forming once the catalyst has reached a higher temperature due to continued carbon diffusion to the catalyst surface. These additional layers were also found to be turbostratic with 2D peaks well fitted by single Lorentzian peaks but with fwhm of >40 cm–1.17,24 This is in contrast to the Bernal stacked graphene formed during CVD on Ni-based catalysts11 and may relate to a more rapid feeding of carbon to the catalyst surface from the underlying solid source, or a different distribution of dissolved carbon within the catalyst,33 arising from the way in which carbon is supplied.
In summary, we establish the introduction of carbon diffusion barriers as a general and simple method to control and improve graphene formation from the catalytic transformation of solid carbon sources. We focus on (Au-alloyed) Ni atop ta-C, nanocrystalline diamond, and plasma-treated HOPG as model systems to highlight via complementary in situ XRR, XRD, and XPS measurements that graphene growth for technologically relevant low temperatures (<600 °C) occurs predominantly during ramping up and annealing by carbon dissolution and diffusion through the catalyst, driven by the thermodynamic stability of graphene. This is also relevant for higher temperature annealing, for which an additional redissolution of as-formed graphene might occur. Hence a key problem with solid-state graphene growth, relevant to all previous literature, is the lack of an “on-switch” for the carbon supply. We address this key problem by introducing a diffusion barrier between the solid carbon source and the catalyst, here in the form of a nanometer-thick Al2O3 layer, which effectively prevents premature carbon dissolution and allows us to demonstrate a significantly improved M-/FLG quality and uniformity comparable to that achieved by CVD. We note that M-/FLG growth from solid carbon sources is a scalable technique,34 and importantly offers a route for the direct integration of graphene in device architectures.14 We expect our method of controlling the growth by introducing diffusion barriers to be relevant to a large range of carbon solid-state precursors and similar layered materials, as well as to related layer-by-layer control in these systems.
Methods
We investigate polycrystalline Ni films (550 nm thick unless otherwise stated) thermally evaporated, or sputtered onto various substrates chosen to act as solid carbon sources: highly orientated pyrolytic graphite (HOPG), tetrahedral amorphous carbon (ta-C), and nanocrystalline diamond. HOPG(0001) substrates (Mikromasch, ZYH grade, < 3.5° mosaic spread) are cleaved to ∼0.1 mm thickness and the surfaces cleaned by mechanical exfoliation using the well-established scotch-tape method. Ta–C was deposited on to SiO2(300 nm)/Si substrates using a filtered cathodic vacuum arc (FCVA) system. Nanocrystalline diamond films (∼100 nm thick) were deposited on SiO2(500 nm)/Si substrates using microwave plasma enhanced CVD.35 Al2O3 (1–3 nm) layers are deposited by atomic layer deposition using a Cambridge Nanotech Savannah ALD system with a 200 °C process that uses tri[methyl]aluminium as a precursor and water as an oxidant both carried in a N2(20 sccm) flow for 10–30 cycles.36,37 For the Au decorated Ni catalysts, Au(5 nm) was deposited by thermal evaporation on to the exposed Ni surface. The samples are annealed under vacuum in a custom-built cold-wall reactor at selected temperatures using a standard one step procedure [<10–6 mbar, 200–600 °C, 5 min, heated and cooled at a constant rate of 100 °C min–1] unless otherwise stated.
In situ high-pressure XPS measurements during vacuum annealing were performed at the BESSY II synchrotron at the ISISS end station of the FHI-MPG. In situ XRR and (grazing incidence) XRD were performed during vacuum annealing at the European Synchrotron Radiation Facility (beamline BM20/ROBL, operated by the Helmholtz-Zentrum Dresden- Rossendorf). Ex situ characterization is performed on as-grown samples using scanning electron microscopy (SEM, Zeiss SigmaVP, 1 kV) or after transfer of the M-/FLG films to SiO2(300 nm)/Si substrates using optical microscopy, and Raman spectroscopy (Renishaw Raman InVia Microscope, 532 nm excitation). Transfer to SiO2(300 nm)/Si substrates is carried out using an electrolysis-based bubbling in a NaOH (1 M) aqueous solution to detach PMMA-supported graphene from the catalyst,38,39 with the PMMA subsequently removed in acetone. Electrical measurements are performed at room temperature using six contact Hall-geometry devices fabricated by e-beam lithography on the as-transferred MLG. Cr/Au contacts are evaporated on top of the MLG which is then patterned by an O2 plasma etch.
Acknowledgments
We are grateful to Stephen Fleming for assistance with e-beam lithography. We also thank Dr. Oliver A. Williams for providing the nanocrystalline diamond films used in this work. R.S.W. acknowledges funding from EPSRC (Doctoral training award) and Sidney Sussex College. S.H. acknowledges funding from ERC Grant InsituNANO (279342). P.R.K. acknowledges funding from the Cambridge Commonwealth Trust. This research was partially supported by the EUFP7 Work Programme under grant GRAFOL (project reference 285275), and EPSRC under Grant GRAPHTED (project reference EP/K016636/1) and Grant EP/H047565/1. We acknowledge the Helmholtz-Zentrum-Berlin Electron storage ring BESSY II for provision of synchrotron radiation at the ISISS beamline and we thank the BESSY staff for continuous support of our experiments. We acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation and we thank the staff for assistance in using beamline BM20/ROBL.
Supporting Information Available
Controlled HOPG plasma damage experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Novoselov K. S.; Fal’ko V. I.; Colombo L.; Gellert P. R.; Schwab M. G.; Kim K. Nature 2012, 490, 192–200. [DOI] [PubMed] [Google Scholar]
- Zheng M.; Takei K.; Hsia B.; Fang H.; Zhang X.; Ferralis N.; Ko H.; Chueh Y.-L.; Zhang Y.; Maboudian R.; Javey A. Appl. Phys. Lett. 2010, 96, 063110. [Google Scholar]
- Hofrichter J.; Szafranek B. N.; Otto M.; Echtermeyer T. J.; Baus M.; Majerus A.; Geringer V.; Ramsteiner M.; Kurz H. Nano Lett. 2010, 10, 36–42. [DOI] [PubMed] [Google Scholar]
- Suzuki S.; Takei Y.; Furukawa K.; Hibino H. Appl. Phys. Express 2011, 4, 065102. [Google Scholar]
- García J. M.; He R.; Jiang M. P.; Kim P.; Pfeiffer L. N.; Pinczuk A. Carbon 2011, 49, 1006–1012. [Google Scholar]
- Sun Z.; Yan Z.; Yao J.; Beitler E.; Zhu Y.; Tour J. M. Nature 2010, 468, 549–552. [DOI] [PubMed] [Google Scholar]
- Peng Z.; Yan Z.; Sun Z.; Tour J. M. ACS Nano 2011, 5, 8241–8247. [DOI] [PubMed] [Google Scholar]
- Shin H.-J.; Choi W. M.; Yoon S.-M.; Han G. H.; Woo Y. S.; Kim E. S.; Chae S. J.; Li X.-S.; Benayad A.; Loc D. D.; Gunes F.; Lee Y. H.; Choi J.-Y. Adv. Mater. 2011, 23, 4392–4397. [DOI] [PubMed] [Google Scholar]
- Xiong W.; Zhou Y. S.; Jiang L. J.; Sarkar A.; Mahjouri-Samani M.; Xie Z. Q.; Gao Y.; Ianno N. J.; Jiang L.; Lu Y. F. Adv. Mater. 2013, 25, 630–634. [DOI] [PubMed] [Google Scholar]
- Saenger K. L.; Tsang J. C.; Bol A. A.; Chu J. O.; Grill A.; Lavoie C. Appl. Phys. Lett. 2010, 96, 153105. [Google Scholar]
- Weatherup R. S.; Dlubak B.; Hofmann S. ACS Nano 2012, 6, 9996–10003. [DOI] [PubMed] [Google Scholar]
- Kidambi P. R.; Ducati C.; Dlubak B.; Gardiner D.; Weatherup R. S.; Martin M.-B.; Seneor P.; Coles H.; Hofmann S. J. Phys. Chem. C 2012, 116, 22492–22501. [Google Scholar]
- Weatherup R. S.; Bayer B. C.; Blume R.; Baehtz C.; Kidambi P. R.; Fouquet M.; Wirth C. T.; Schlögl R.; Hofmann S. ChemPhysChem 2012, 13, 2544–2549. [DOI] [PubMed] [Google Scholar]
- Dlubak B.; Martin M.-B.; Weatherup R. S.; Yang H.; Deranlot C.; Blume R.; Schloegl R.; Fert A.; Anane A.; Hofmann S.; Seneor P.; Robertson J. ACS Nano 2012, 6, 10930–10934. [DOI] [PubMed] [Google Scholar]
- Goldstein J.; Newbury D. E.; Joy D. C.; Lyman C. E.; Echlin P.; Lifshin E.; Sawyer L.; Michael J. R.. Scanning Electron Microscopy and X-Ray Microanalysis, 3rd ed.; Kluwer Academic Publishers: New York, 2003. [Google Scholar]
- Ferrari A.; Robertson J. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar]
- Ferrari A.; Meyer J.; Scardaci V.; Casiraghi C.; Lazzeri M.; Mauri F.; Piscanec S.; Jiang D.; Novoselov K.; Roth S.; Geim A. Phys. Rev. Lett. 2006, 97, 187401. [DOI] [PubMed] [Google Scholar]
- Weatherup R. S.; Bayer B. C.; Blume R.; Ducati C.; Baehtz C.; Schlögl R.; Hofmann S. Nano Lett. 2011, 11, 4154–4160. [DOI] [PubMed] [Google Scholar]
- Powder Diffraction File 04-0850. JCPDS-International Centre for Diffraction Data; Newtown Square, PA.
- Powder Diffraction File 72-1467. JCPDS-International Centre for Diffraction Data; Newtown Square, PA.
- Portnoi V. K.; Leonov A. V.; Mudretsova S. N.; Fedotov S. A. Phys. Metals Metallogr. 2010, 109, 153–161. [Google Scholar]
- Powder Diffraction File 89-8487. JCPDS-International Centre for Diffraction Data; Newtown Square: PA.
- Gösele U. In Alloying; Walters J. L., Jackson H. R., Sims C. T., Eds.; ASM International: Metals Park, Ohio, 1988; pp 489–519. [Google Scholar]
- Lespade P.; Marchand A.; Couzi M.; Cruege F. Carbon 1984, 22, 375–385. [Google Scholar]
- Halmann M.; Frei A.; Steinfeld A. Miner. Process. Extr. Metall. Rev. 2011, 32, 247–266. [Google Scholar]
- Trumble K.; Rühle M. Acta Metall. Mater. 1991, 39, 1915–1924. [Google Scholar]
- Guenette M. C.; Tucker M. D.; Ionescu M.; Bilek M. M. M.; McKenzie D. R. J. Appl. Phys. 2011, 109, 083503. [Google Scholar]
- Yan Z.; Ma L.; Zhu Y.; Lahiri I.; Hahm M.; Liu Z.; Yang S.; Xiang C.; Lu W.; Peng Z.; Sun Z.; Kittrell C.; Lou J.; Choi W.; Ajayan P. M.; Tour J. M. ACS Nano 2013, 7, 58–64. [DOI] [PubMed] [Google Scholar]
- Amama P. B.; Pint C. L.; Kim S. M.; McJilton L.; Eyink K. G.; Stach E. A.; Hauge R. H.; Maruyama B. ACS Nano 2010, 4, 895–904. [DOI] [PubMed] [Google Scholar]
- Lander J. J.; Kern H. E.; Beach A. L. J. Appl. Phys. 1952, 23, 1305–1309. [Google Scholar]
- Mishin Y.; Herzig C. Mater. Sci. Eng., A 1999, 260, 55–71. [Google Scholar]
- Parthasarathy T.; Shewmon P. Scr. Metall. 1983, 17, 943–946. [Google Scholar]
- Hofmann S.; Csányi G.; Ferrari A.; Payne M.; Robertson J. Phys. Rev. Lett. 2005, 95, 036101. [DOI] [PubMed] [Google Scholar]
- Rinaldi A.; Tessonnier J.-P.; Schuster M. E.; Blume R.; Girgsdies F.; Zhang Q.; Jacob T.; Abd Hamid S. B.; Su D. S.; Schlögl R. Angew. Chem., Int. Ed. 2011, 50, 3313–3317. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Yan Z.; Yao J.; Beitler E.; Zhu Y.; Tour J. M. Nature 2010, 468, 549–552. [DOI] [PubMed] [Google Scholar]
- Williams O. A.; Kriele A.; Hees J.; Wolfer M.; Müller-Sebert W.; Nebel C. E. Chem. Phys. Lett. 2010, 495, 84–89. [Google Scholar]
- Dlubak B.; Kidambi P. R.; Weatherup R. S.; Hofmann S.; Robertson J. Appl. Phys. Lett. 2012, 100, 173113. [Google Scholar]
- Puurunen R. L. J. Appl. Phys. 2005, 97, 121301. [Google Scholar]
- Wang H.; Taychatanapat T.; Hsu A. IEEE Electron Device Lett. 2011, 32, 1209–1211. [Google Scholar]
- Gao L.; Ren W.; Xu H.; Jin L.; Wang Z.; Ma T.; Ma L.-P.; Zhang Z.; Fu Q.; Peng L.-M.; Bao X.; Cheng H.-M. Nat. Commun. 2012, 3, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.