

Abstract
Circular clamps are utilised by replicative polymerases to enhance processivity. The topological problem of loading a toroidal clamp onto DNA is overcome by ATP-dependent clamp loader complexes. Different organisms use related protein machines to load clamps, but the mechanisms by which they utilise ATP are surprisingly different. Using mutant clamp loaders that are deficient in either ATP binding or hydrolysis in different subunits, we show how the different subunits of an archaeal clamp loader use ATP binding and hydrolysis in distinct ways at different steps in the loading process. Binding of nucleotide by the large subunit and three of the four small subunits is sufficient for clamp loading. However, ATP hydrolysis by the small subunits is required for release of PCNA to allow formation of the complex between PCNA and the polymerase, while hydrolysis by the large subunit is required for catalytic clamp loading.
Keywords: archaea, ATP hydrolysis, clamp loader, nucleotide binding, PCNA, replication
Introduction
Generally, replicative polymerases obtain processivity using ring-shaped DNA sliding clamps that are loaded onto DNA by clamp loader proteins in ATP-dependent reactions. Although many aspects of clamp-loading systems in different organisms show some similarities, such as the heteropentameric composition and overall structure of the subunits, there are significant differences in the manner by which ATP is used by the various clamp-loading motors.
By far the best-characterised clamp-loading system to date is that from Escherichia coli. The clamp itself is the dimeric β subunit of DNA polymerase III. Although dimeric, each subunit contains three domains of conserved structure to give a pseudo-hexameric ring structure for the β-subunit dimer (Kong et al, 1992). Although the holopolymerase contains several additional subunits, the minimal E. coli clamp loader consists of a heteropentamer of three types of subunits with a stoichiometry of γ3δδ′ in the complex, but only the γ subunits are able to bind ATP (Xiao et al, 1995). Even in the absence of the clamp and DNA, all three binding sites are filled and bind ATP with similar affinity (Johnson and O'Donnell, 2003). The structure of the γ3δδ′ complex revealed that the ATP-binding sites are located at subunit interfaces (Jeruzalmi et al, 2001a). By comparison of the γ3δδ′ structure with two nucleotide-bound structures of other AAA+ ATPases, the authors proposed a mechanism by which one ATP molecule binds to a constitutively open ATP-binding site located at the δ′–γ1 interface. Nucleotide binding at this site is proposed to lead to a structural change of γ3δδ′ involving repositioning of the γ1 sensor-1 motif that allows the binding of two further ATP molecules at the γ1–γ2 and γ2–γ3 interfaces, although a structure of this proposed form of the complex has yet to be determined. Earlier, a biochemical study had shown that the γ3δδ′ complex hydrolyses two to three ATP molecules per clamp-loading cycle (Turner et al, 1999). Different studies suggest this hydrolysis to be either sequential (Hingorani et al, 1999; Johnson and O'Donnell, 2003) or simultaneous (Bertram et al, 2000). However, biochemical studies, together with the crystal structure of the δ subunit bound to a β-subunit monomer (Jeruzalmi et al, 2001b), show that upon filling of the ATP-binding sites in the γ subunits, conformational changes are propagated around the clamp loader complex to enable the δ subunit to contact the β clamp and initiate clamp loading.
Eukaryotes utilise a similar but different clamp-loading system. The clamp is the trimeric PCNA protein. Each monomer contains a tandem repeat of domain structure so that the trimer forms a pseudo-hexameric ring that ends up being similar to the β-subunit clamp of E. coli (Krishna et al, 1994). All five subunits of the heteropentameric eukaryotic clamp loader, replication factor C (RFC), are members of the AAA+ family. Four of the subunits are of similar size (denoted the small subunits) and are very closely related in sequence, with the remaining subunit being somewhat larger (the large subunit). However, both in the human and Saccharomyces cerevisiae clamp loader, one of the small subunits shows deviations of the consensus Walker A and B box sequences making ATP binding and/or hydrolysis by this subunit unlikely. Accordingly, substitutions of the conserved lysine in the Walker A motif in hRFC38, which would normally inactivate the ATPase activity of AAA+ proteins, had only a small effect on the activity of the human clamp loader in vitro (Cai et al, 1998; Podust et al, 1998a). The large subunit is highly conserved (36% identity/55% similarity between human (hRFC140) and yeast (ScRFC1) proteins; Cullmann et al, 1995), but conflicting results have been obtained for the role of ATP binding to this clamp loader subunit. Substitutions of the Walker A lysine in hRFC140 resulted in a marked reduction of the ATPase, clamp loading, and polymerase stimulation activities (Cai et al, 1998; Podust et al, 1998a). In contrast, substitutions at this residue in ScRFC1 had little effect on the yeast clamp loader either in vitro or in vivo (Schmidt et al, 2001a, 2001b). However, in the same series of studies, the authors also showed a maximum binding of four molecules of ATP per RFC complex and therefore implicated ATP binding to the yeast clamp loader large subunit (Gomes et al, 2001). Interestingly, unlike the E. coli clamp loader/β-clamp, filling of the ATP-binding sites in the yeast complex is regulated by both the trimeric clamp (PCNA) and DNA (Gomes et al, 2001). Initially only two sites are filled, followed by binding of one additional molecule of ATP upon interaction with DNA or PCNA, and filling of all four possible ATP-binding sites in the ternary RFC–PCNA–DNA complex. However, which ATP-binding sites of RFC are filled at each stage in the reaction cycle remains unknown.
Another well-studied clamp loader is from bacteriophage T4. The clamp (gp45 protein) is trimeric and similar in structure to PCNA (Moarefi et al, 2000), but the clamp loader complex is different yet again from eukaryotes and E. coli. The T4 clamp loader comprises just two types of subunits gp44 and gp62, with a stoichiometry of 4:1, to give a heteropentameric complex. The gp44 subunits have conserved ATPase motifs, but the smaller gp62 does not. Accordingly, four molecules of ATP are bound by the identical gp44 subunits in each T4 clamp loader complex (Jarvis et al, 1989; Young et al, 1996). However, in contrast to eukaryotic RFC, all four ATP-binding sites are filled in the absence of the clamp and DNA in gp44/gp62. Furthermore, unlike the eukaryotic and E. coli clamp loaders where ATP binding is sufficient for clamp opening (Hingorani and O'Donnell, 1998; Gomes and Burgers, 2001; Gomes et al, 2001), the gp44/gp62 complex requires hydrolysis of two ATP molecules to open the clamp (Trakselis et al, 2003). In eukaryotic and E. coli clamp loaders, ATP turnover only takes place after the ternary clamp loader–clamp–DNA complex has formed and seems to be required to release the clamp loader from the clamp–DNA complex (Turner et al, 1999; Gomes and Burgers, 2001).
Archaea, the third domain of life, utilise yet another version of a clamp-loading system that is the least-studied system to date. Sequence comparisons suggest that archaeal clamp loaders are a closely related, yet simplified, version of their eukaryotic counterparts in that the clamp is similar to PCNA but they only encode single copies of a small and large subunit of RFC. A series of conserved sequence motifs, RFC boxes II–VIII (Cullmann et al, 1995), can be found in both the large and the small subunits (Cann and Ishino, 1999). RFC box I of the eukaryotic large subunit is absent in archaeal clamp loaders, but this motif has been shown to be nonessential for clamp loading (Uhlmann et al, 1997; Podust et al, 1998b) and, therefore, the reason for its conservation remains unclear. With the apparent exception of the euryarchaeotic Methanobacterium thermoautotrophicum clamp loader, for which a heterohexameric composition has been proposed (Kelman and Hurwitz, 2000), archaeal clamp loaders are heteropentameric proteins consisting of one large and four small subunits (Pisani et al, 2000; Cann et al, 2001; Seybert et al, 2002). Sequence alignments show that both the large and small subunits of archaeal clamp loaders contain the conserved ATP-binding motifs of AAA+ proteins (Cann and Ishino, 1999). Notably, the Walker A and B boxes do not show major deviations from the conserved sequences raising the possibility that, unlike the situation in eukaryotes, five ATP-binding sites might be present in archaeal RFC.
We have recently analysed the ATPase, DNA binding, and clamp interacting activities of the heteropentameric clamp loader from the hyperthermophilic euryarchaeon Archaeoglobus fulgidus (Seybert et al, 2002). Here we present an analysis of ATP binding to individual subunits of the A. fulgidus clamp loader and the functional roles of bound nucleotide and nucleotide hydrolysis. We show that A. fulgidus RFC (afRFC) binds two molecules of ATP in the absence of other factors but a maximum of four molecules of ATP in the presence of its clamp, PCNA. These bound nucleotides are located on the large subunit and three of the four, identical, small subunits. Binding of ATP to both the large and the small subunits proved to be essential for the functionality of the A. fulgidus clamp loader. Hydrolysis of ATP bound to the small subunits was required to release the clamp onto the DNA after loading and to allow association with DNA polymerase. However, hydrolysis of the ATP bound to the large subunit played no role in the ability of afRFC to load and release the clamp onto DNA, nor the release of afRFC from its clamp after loading. Instead, a deficiency in ATP hydrolysis at the afRFC large subunit appears to be important for catalytic recycling of afRFC.
Results and discussion
The aim of this study was to analyse nucleotide binding to the A. fulgidus clamp loader and the functional role of bound nucleotide. Sequence alignments show that both the large and small afRFC subunits belong to the AAA+ family (Neuwald et al, 1999) and have a number of conserved sequence motifs. Notably, two motifs critical for nucleotide hydrolysis and binding, namely the P-loop NTP-binding motif (also known as the Walker A box) and the DEAD motif (also known as the Walker B box), do not show major deviations from the consensus sequences. We set out to answer several questions: (1) to investigate whether both the large and the small afRFC subunits bind nucleotide, (2) what contribution each of the subunits makes to the overall ATPase rate of the complex, and (3) to dissect what separate functional roles there might be for nucleotide binding and hydrolysis at each subunit.
In order to address these questions, we designed mutant proteins that were deficient in either nucleotide binding or hydrolysis. In the E. coli clamp loader δ′ subunit, which has been shown to be devoid of nucleotide binding (Guenther et al, 1997), the conserved GKT of the Walker A motif is replaced by GDD. Based on various AAA+ protein structures, the lysine in this motif is expected to interact with the nucleoside triphosphate tail, while the threonine provides a ligand for the bound magnesium ion (reviewed in Ogura and Wilkinson, 2001). The replacement of these residues by aspartate is the reason as to why the δ′ subunit is unable to bind ATP. Consequently, Lys-49 and Thr-50 of the afRFC large subunit were simultaneously substituted by aspartate. Likewise, Lys-51 and Thr-52 of the afRFC small subunit were substituted by aspartate. AfRFC complexes were expressed and purified from E. coli that contained either wild-type or mutant subunits in all possible pairwise combinations. Wild-type large subunits will be designated Largewt and Walker A mutant large subunits as LargeAmut, with a similar notation for the small subunits (Smallwt and SmallAmut).
A different mutation was required to generate proteins that retained the ability to bind ATP but were deficient in hydrolysis. Based again on other AAA+ protein structures, the conserved glutamate residue of the Walker B motif is likely to have a role in polarising the water molecule for in-line nucleophilic substitution of the γ-phosphate of the nucleotide (reviewed in Ogura and Wilkinson, 2001). Therefore, replacement of this residue should affect nucleotide hydrolysis without upsetting nucleotide binding. Consequently, Glu-112 of the large subunit and Glu-110 of the small subunit were substituted by alanine. Again, afRFC complexes were expressed and purified from E. coli. Mutant large and small subunits are designated LargeBmut and SmallBmut, respectively.
All mutant complexes showed the same properties as wild type during purification in terms of yields and elution profiles on gel filtration columns, indicating that complex formation was not affected by any of the mutations.
Nucleotide binding to the afRFC large and small subunits is regulated by both DNA and PCNA
We analysed the binding of nucleotides to afRFC complexes both by equilibrium dialysis and by utilising the rapid separation of free nucleotides from bound nucleotides on spin columns. We have shown previously (Seybert et al, 2002) that ATPγS can substitute for ATP to promote both afRFC–DNA and afRFC–PCNA interactions. Therefore, to avoid any complications due to nucleotide hydrolysis, we used ATPγS in the nucleotide-binding assays. The calculation of protein concentrations in the assays (expressed as molarity of complex) was based on the 1:4 (large:small) stoichiometry of afRFC (Seybert et al, 2002). The amount of ATPγS bound per complex was plotted against the amount of free ATPγS in the reactions (Figure 1). Subsequently, the data were fitted using nonlinear regression analysis (assuming equivalent binding sites), to determine the number of nucleotides bound (Figure 1A and Table IA). Dissociation constants were also modelled to each data set and resulted in values between 1 and 6 μM (data not shown), suggesting that sites that were able to bind nucleotide were not drastically affected by mutations elsewhere in the complex. Initially we analysed nucleotide binding by equilibrium dialysis (Figure 1A), but later found that a spin column assay gave comparable results (compare Figures 1A and B) and was much more convenient for our large reaction numbers.
Figure 1.
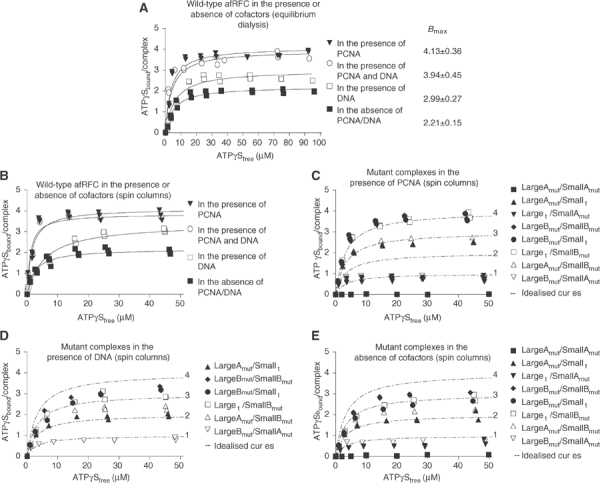
Nucleotide binding by wild-type and mutant afRFC complexes in the absence or presence of DNA and/or PCNA. The amount of bound [35S]ATPγS was measured using equilibrium dialysis (A) or spin column assays (B–E) as described in the Experimental procedures. (A, B) Wild-type afRFC was incubated with [35S]ATPγS in the presence and absence of DNA and PCNA. The data were fitted by nonlinear regression assuming equivalent binding sites to the equation shown in Experimental procedures. Similar studies were carried out with the mutant complexes, (C) in the presence of PCNA, (D) in the presence of DNA, and (E) in the absence of cofactors. Theoretical curves for 1, 2, 3, and 4 bound nucleotides are overlaid for reference in panels C, D, and E.
Table 1.
Summary of nucleotide binding and hydrolysis by wild-type or mutant afRFC complexes in the presence or absence of cofactors
| (A) Nucleotide binding | ||||
|---|---|---|---|---|
| afRFC complex | ATPγS binding in the absence of cofactors | ATPγS binding in the presence of DNA | ATPγS binding in the presence of PCNA | |
| Bmax | Bmax | Bmax | ||
| Wild-type afRFC | 2.18 (±0.13) | 3.40 (±0.20) | 4.10 (±0.30) | |
| LargeAmut/SmallAmut | 0.08 (±0.10) | ND | 0.01 (±0.01) | |
| LargeAmut/Smallwt | 1.98 (±0.13) | 2.08 (±0.14) | 2.64 (±0.05) | |
| Largewt/SmallAmut | 0.60 (±0.10) | ND | 0.76 (±0.05) | |
| LargeBmut/SmallBmut | 3.61 (±0.28) | 3.62 (±0.23) | 4.12 (±0.24) | |
| LargeBmut/Smallwt | 2.87 (±0.10) | 3.63 (±0.32) | 4.12 (±0.27) | |
| Largewt/SmallBmut | 3.16 (±0.14) | 3.25 (±0.29) | 4.10 (±0.23) | |
| LargeAmut/SmallBmut | 2.47 (±0.20) | 2.56 (±0.20) | 2.98 (±0.11) | |
| LargeBmut/SmallAmut | 0.75 (±0.08) | 0.82 (±0.06) | 0.89 (±0.05) | |
| (B) Nucleotide hydrolysis | ||||
| afRFC complex | ATP hydrolysis in the absence of cofactors | ATP hydrolysis in the presence of DNA | ATP hydrolysis in the presence of PCNA | ATP hydrolysis in the presence of DNA and PCNA |
| kcat (× 10−3 s−1) | kcat (× 10−3 s−1) | kcat (× 10−3 s−1) | kcat (× 10−3 s−1) | |
| Wild-type afRFC | 18.2 (±1.9) | 369.0 (±33.0) | 12.8 (±0.4) | 1708.4 (±19.1) |
| LargeAmut/SmallAmut | 1.3 (±0.2) | 0.4 (±0.1) | 1.0 (±0.5) | 1.1 (±0.4) |
| LargeAmut/Smallwt | 12.7 (±1.1) | 127.2 (±8.5) | 9.8 (±2.9) | 1098.1 (±12.6) |
| Largewt/SmallAmut | 9.4 (±0.1) | 4.7 (±0.2) | 10.0 (±5.0) | 64.2 (±4.1) |
| LargeBmut/SmallBmut | 8.2 (±0.9) | 16.5 (±0.8) | 6.6 (±0.1) | 35.2 (±0.8) |
| LargeBmut/Smallwt | 14.7 (±0.7) | 381.4 (±23.5) | 12.5 (±2.0) | 1991.6 (±89.9) |
| Largewt/SmallBmut | 11.7 (±0.7) | 29.5 (±1.4) | 10.7 (±4.1) | 39.5 (±1.1) |
| LargeAmut/SmallBmut | 3.2 (±0.1) | 1.7 (±0.1) | 3.2 (±2.3) | 17.9 (±1.8) |
| LargeBmut/SmallAmut | 6.1 (±0.2) | 3.7 (±1.0) | 5.4 (±0.1) | 6.9 (±0.4) |
| The numbers of maximum binding sites per afRFC complex (Bmax) were obtained by fitting the data points shown in Figure 1 by nonlinear regression assuming equivalent binding sites to the equation shown in Experimental procedures. Not determined values are abbreviated as ‘ND'. The values given in parentheses are the 95% confidence interval variations. Bmax in the simultaneous presence of DNA and PCNA was only determined for wild-type afRFC (Bmax, 3.85±0.32). The kcat was determined by dividing the rate of ADP generation by afRFC concentration. | ||||
As predicted, the double Walker A mutant (LargeAmut/SmallAmut), did not bind ATPγS under any conditions tested, confirming that the GDD substitution blocks nucleotide binding. In the absence of DNA and PCNA, wild-type afRFC and mutant LargeAmut/Smallwt complexes both bound two molecules of ATPγS, indicating that the binding of the first two nucleotides to each molecule of afRFC does not involve the large subunit. In the presence of DNA, wild-type afRFC bound an additional molecule of ATPγS, increasing the stoichiometry of binding to three molecules of ATPγS per molecule of afRFC. By contrast, DNA did not increase the number of nucleotides bound to a complex containing LargeAmut/Smallwt. We therefore conclude that the additional nucleotide that binds to the wild-type afRFC in response to DNA binding is bound to the large subunit. The DNA substrate used in these experiments consisted of a 30 base oligonucleotide annealed to an 80 base template, to give a 30 base pair duplex with 5′ (dT)30 and 3′ (dT)20 tails, but we also obtained comparable results with a single-stranded 30-mer (data not shown). In the presence of PCNA, but in the absence of DNA, the stoichiometry of binding increased to four molecules of ATPγS per afRFC complex. When both PCNA and DNA were present in the same reaction, the stoichiometry did not increase beyond four molecules of ATPγS bound per complex. In contrast to wild-type afRFC, the mutant complex LargeAmut/Smallwt bound only one additional molecule of ATPγS in the presence of PCNA, in agreement with the proposal that one of the additional nucleotides normally binds to the large subunit in the wild type. Curiously, the mutant complex Largewt/SmallAmut bound one molecule of ATPγS per complex, irrespective of the presence or absence of either DNA or PCNA cofactors, indicating that the allosteric control of ATP binding to the large subunit is lost in this mutant for some reason.
The Walker B substitutions did not influence the maximum number of ATPγS bound per afRFC complex, confirming that the LargeBmut and SmallBmut subunits were able to bind nucleotide. The LargeAmut/SmallBmut and LargeBmut/SmallAmut complexes had similar binding characteristics as the LargeAmut/Smallwt and Largewt/SmallAmut complexes, respectively, although the Walker B substitutions resulted in increased nucleotide binding in the absence of cofactors compared to wild type. Unlike wild-type afRFC, the number of bound nucleotides was not affected by the presence of DNA. Thus, whereas the exchange of the conserved glutamate in the Walker B box of the afRFC large and small subunits did not affect the overall nucleotide binding to the A. fulgidus clamp loader, the substitutions resulted in a loss of DNA cofactor dependence of nucleotide binding. Thus, there seems to be allostery between sites in the complex that provides crosstalk between subunits.
Taken together, these data confirm that the mutations behaved as predicted, with Walker A mutations blocking nucleotide binding but Walker B mutations allowing binding. Furthermore, the Walker A mutant complexes allowed us to determine which subunits of afRFC are binding nucleotides in the various complexes with DNA and/or PCNA. AfRFC alone binds two ATP molecules and these are located in two of the four small subunits. Upon binding DNA, the large subunit of the complex now picks up an ATP molecule, but once PCNA is bound then a further ATP molecule binds to another of the small subunits to give a total of only four ATP molecules bound to a potential five sites in the complex.
The afRFC large subunit contributes very little to the ATPase activity of the afRFC complex
Since we detected nucleotide binding to both the afRFC large and small subunits, we next analysed the contribution of each subunit to the overall ATPase rate of the complex. The ATPase activity of afRFC is stimulated by a variety of different substrates to a similar degree (Seybert et al, 2002). We chose to use a primed template DNA as a substrate, which was closest to that used in the subsequent assays described in this study (see below).
For the wild-type afRFC alone and in the presence of PCNA, the ATPase activity is very low (Table IB). However, the addition of DNA to the reaction resulted in a stimulation of around 20-fold. The addition of both DNA and PCNA resulted in the largest stimulation with a kcat of 1.7 s−1, a stimulation of almost 100-fold.
We next analysed the ATPase activities of afRFC complexes with subunits carrying Walker A box substitutions. As expected, since the protein is unable to bind nucleotide, the ATPase activity of the complex with Walker A substitutions in both the large and the small subunits (LargeAmut/SmallAmut) was at the detection limits of the assay (less than 0.1% of the wild-type rate). In the presence of DNA and PCNA, the afRFC with Walker A mutant large subunits, LargeAmut/Smallwt, showed an ATPase rate that was comparable to that of the wild type, albeit slightly reduced. By contrast, the kcat of the Largewt/SmallAmut complex was reduced to only 4% of that of the wild-type complex, but retained the stimulation when both DNA and PCNA were present. Taken together, these data suggest that the small subunits are responsible for the major component of the DNA-dependent ATPase activity, with a lesser contribution from the large subunit.
In accordance with the predictions drawn from the sequence comparisons, substitutions in the Walker B motifs drastically reduced the ATPase activity (Table IB), even though these mutant subunits are able to bind nucleotide (Figure 1). As for the Walker A mutations, the data from the mixed complexes (LargeBmut/Smallwt and Largewt/SmallBmut) attribute the ATPase activity of afRFC to the small subunits.
Nucleotide binding to both the large and small afRFC subunits is required for afRFC–PCNA interactions
Since the ATP bound to the afRFC large subunit contributes very little to the overall ATPase rate of the complex, we wondered about the functional importance of this bound nucleotide. Therefore, we compared the interaction of wild-type afRFC and PCNA with that of complexes containing ATP-binding-deficient Walker A mutant subunits, using pull-down assays that we have described previously (Seybert et al, 2002) (Figure 2). The amount of PCNA bound to afRFC in the presence of 1 mM ATP was taken to be 100% and all other values were normalised to this value.
Figure 2.
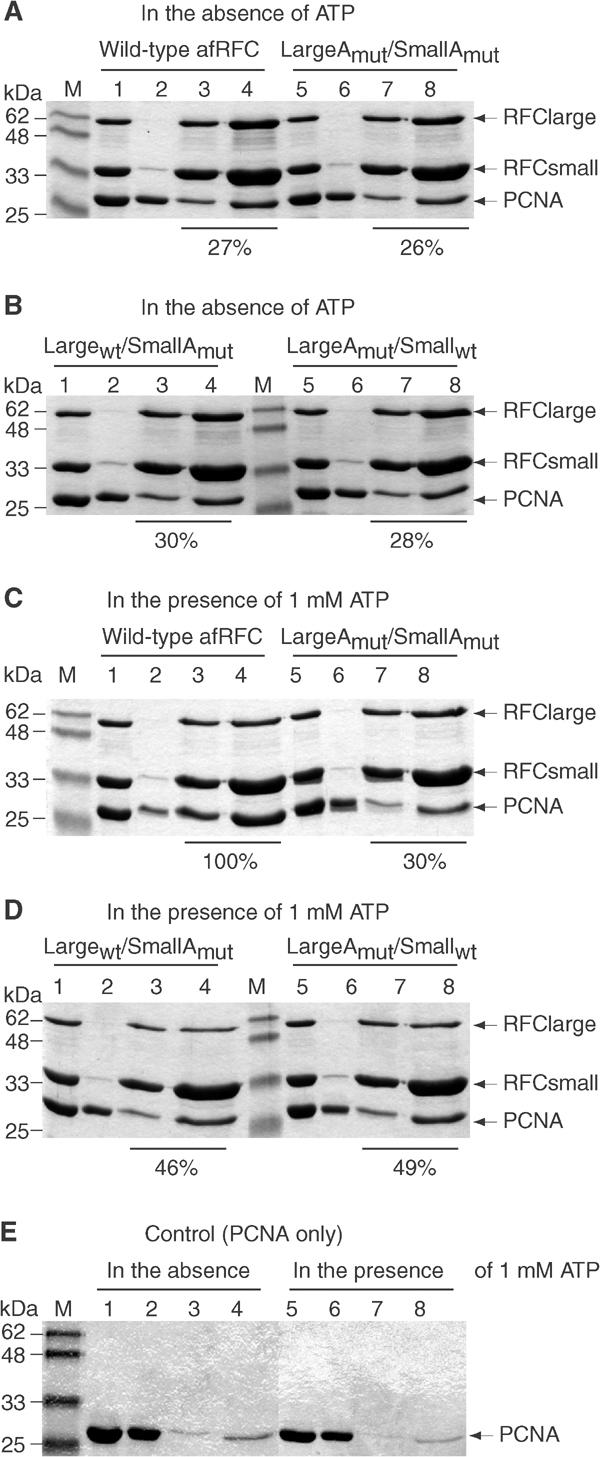
ATP binding to both the large and small subunits is required to stabilise afRFC–PCNA complexes. In all, 150 pmol of the afRFC proteins was incubated with 400 pmol of PCNA in the absence (A, B) or presence of 1 mM ATP (C, D). AfRFC–PCNA complexes were captured by incubation with Ni-agarose beads (via the His-tag located at the N-terminus of the large subunit), eluted by boiling in SDS sample buffer and analysed by SDS–PAGE in 15% gels stained with Coomassie brilliant blue dye. To improve quantification, 30% as well as 70% of the eluates were run on the gels. In the presence of 1 mM ATP, after subtraction of nonspecific PCNA binding (E), the total amount of PCNA in the eluates bound to the 60 pmol of afRFC recovered from the beads was estimated to be 80 pmol. This ratio of bound PCNA per afRFC was taken to be 100% and all other values were normalised to this value. The results of these calculations are shown below the respective lanes. Lane M, molecular mass markers; lanes 1 and 5, 10% of reaction mixtures; lanes 2 and 6, 10% of supernatants after incubation with Ni-agarose beads; lanes 3 and 7, 30% of eluates; lanes 4 and 8, 70% of eluates.
The amount of PCNA that is pulled down by wild-type afRFC is reduced to 27% when ATP is omitted from the reaction. The binding of PCNA to the LargeAmut/SmallAmut, Largewt/SmallAmut, and LargeAmut/Smallwt complexes was indistinguishable, either from each other or from wild type, under these conditions. In the presence of ATP, the mutant complexes showed differing properties. For the ATP-binding-deficient LargeAmut/SmallAmut complex, the amount of PCNA binding did not increase significantly in the presence of ATP and even though both the Largewt/SmallAmut and LargeAmut/Smallwt complexes did show stimulated binding of PCNA, in both cases this was less than half of that observed for the wild type.
These data show that ATP binding to both the large and small subunits is required to stabilise the afRFC–PCNA complex.
ATP binding to both the large and the small afRFC subunits is both necessary and sufficient to stabilise PCNA–DNA complexes
Earlier studies have shown that nonhydrolysable ATP analogues support the loading of sliding clamps by their clamp loaders (Turner et al, 1999). Therefore, we were interested in investigating whether ATP binding to the large and small subunits of afRFC is both necessary and sufficient to stabilise PCNA–DNA complexes.
As shown in Figure 3A, we achieved consistent recovery of the nicked DNA substrate after the gel filtration. For wild-type afRFC, in the presence of either ATPγS or ATP, we detected levels of PCNA–DNA complex that were consistent with single loading events on our nicked DNA substrate. The ATP-binding-competent, but ATPase-deficient, Walker B mutants LargeBmut/SmallBmut and Largewt/SmallBmut (Figure 3) were also able to load PCNA efficiently. These findings demonstrate that ATP binding to the afRFC large and small subunits is sufficient to stabilise PCNA–DNA complexes, and that ATP hydrolysis is not required at either the large or small subunits. Furthermore, ATP binding to both the large and small subunits is strictly necessary to promote PCNA loading, as shown by the severe defects of those mutant complexes with impaired ATP binding (LargeAmut/SmallAmut, Largewt/SmallAmut, and LargeAmut/Smallwt). Negligible loading of PCNA was detected on either supercoiled or linearised DNA substrates in the presence of ATP (Figure 3C). The lack of loading on supercoiled plasmids indicates a specificity for a nick site on the DNA, although it should be noted that we observed single loading events on this substrate. In the case of nicked, linear DNA, PCNA can slide off the ends of the DNA and is not stably loaded. This has been demonstrated for other sliding clamps (Stukenberg et al, 1991; Yao et al, 2000).
Figure 3.
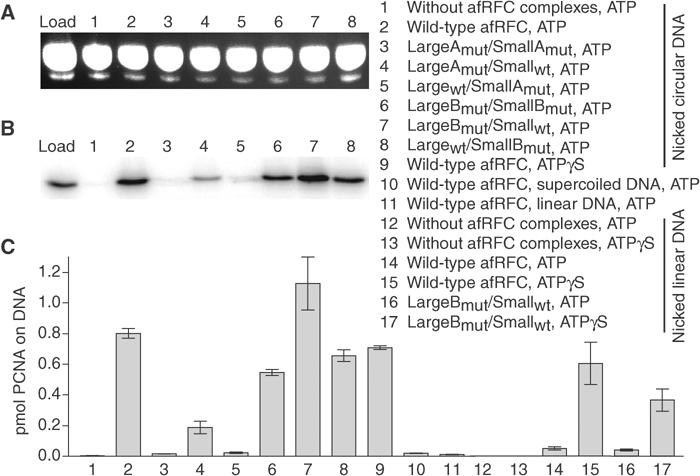
ATP binding at both the afRFC large and small subunits is necessary and sufficient to promote stable PCNA–DNA complexes, whereas ATP hydrolysis at only the afRFC small subunits is necessary for PCNA release. Loading assays were performed using a spin column assay. Reactions contained 8 pmol 32P-PCNA, 4 pmol nicked pUC19 DNA (reactions 1–9), 4 pmol supercoiled pUC19 DNA (reaction 10), 4 pmol linearised pUC19 DNA (reaction 11), or 4 pmol nicked, linearised pUC19 DNA (reactions 12–17). Reactions 1–8, 10–12, 14, and 16 contained 5 mM ATP. Reactions 9, 13, 15, and 17 contained 5 mM ATPγS. The mixtures were substituted with 4 pmol wild-type (reactions 2, 9–11, 14, and 15) or mutant (reactions 3–8 and 16–17) afRFC complexes. The spin columns were pre-equilibrated in 200 mM NaCl. (A) Control of DNA retention on the spin columns. In all, 10% of the input DNA (load) and 10% each of the eluates of reactions 1–8 were loaded onto a 1% agarose–TBE gel stained with ethidium bromide. (B) Analysis of 32P-afPCNA in the eluates by SDS–PAGE. In all, 5% of the input 32P-PCNA (load) or 90% of the eluates of reactions 1–8 were separated by SDS–PAGE in a 15% gel and visualised on a phosphorimager. (C) Quantitative depiction of results obtained by phosphorimager analysis.
ATP hydrolysis at the afRFC large subunit is not required for PCNA to move away from the site of loading
The observation that PCNA can be deposited stably onto a circular, but not linearised, nicked DNA substrate suggests that PCNA can slide freely on DNA after loading by afRFC in the presence of ATP. Therefore, we decided to analyse the role of ATP hydrolysis in this process by testing whether PCNA could be loaded stably on linearised nicked DNA in the absence of ATP hydrolysis and in the presence of ATP hydrolysis only at the afRFC small subunits. In the presence of ATPγS, the amount of PCNA deposited on DNA by wild-type afRFC complexes on nicked, linearised DNA was comparable to that on nicked DNA circles. However, in the presence of ATP, 10-fold less PCNA was observed on the nicked, linearised DNA substrate, indicating that PCNA was released from RFC and slid off the linear DNA. Likewise, in the presence of ATP, but not ATPγS, PCNA loaded onto DNA by the LargeBmut/Smallwt complex, was efficiently released. Consequently, ATP hydrolysis by the afRFC small subunits permits sliding of PCNA along DNA after loading. ATP hydrolysis by the large subunit is not required for this process.
ATP hydrolysis at the afRFC small subunits permits release of afRFC from PCNA after loading to enable afPolB1 to form a complex with PCNA
Since ATP hydrolysis by the small subunits permits movement of PCNA along DNA, we wished to investigate whether this involved release of PCNA from afRFC to allow access to the polymerase. To address this question, we analysed the eluates of spin columns from PCNA-loading reactions containing singly nicked, circular DNA (Figure 4A). A large amount of afRFC co-eluted with PCNA and DNA in the presence of ATPγS. Under these conditions, afRFC was bound to DNA in a stable complex with PCNA since we could not detect significant amounts of afRFC in the eluates in the absence of PCNA. By contrast, in the presence of ATP rather than ATPγS, wild-type afRFC was efficiently released from PCNA. Furthermore, release of PCNA from afRFC was essential to allow formation of the PCNA–afPolB1–DNA complex. Thus, in the presence of ATPγS, we detected significantly less afPolB1 in the eluates compared to reactions that were carried out in the presence of ATP.
Figure 4.
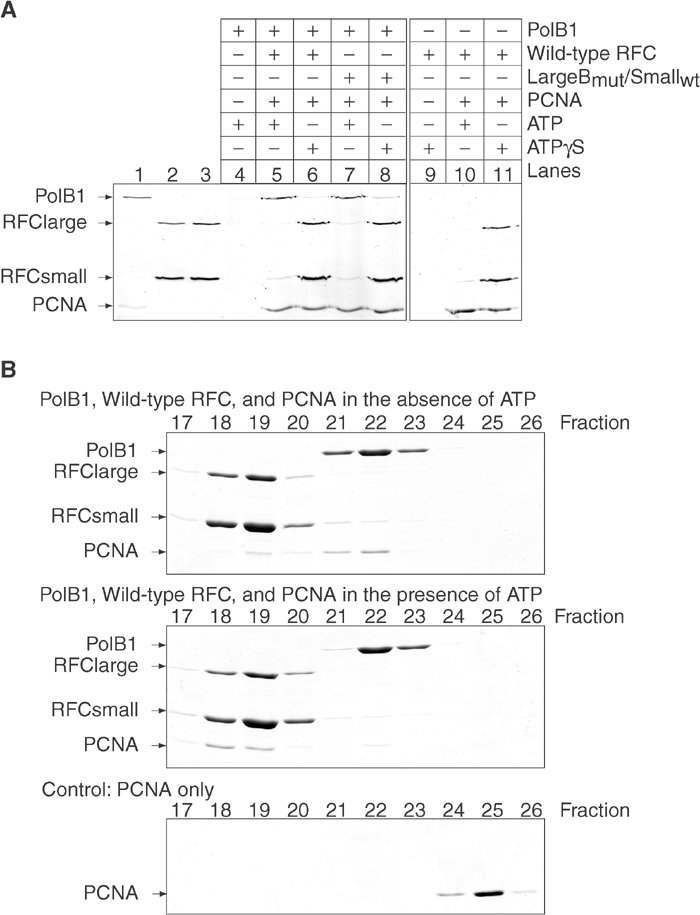
Release of PCNA from afRFC is necessary for interaction with afPolB1. (A) ATP bound to the afRFC small subunits inhibits release of afRFC and blocks access of afPolB1 to PCNA. Proteins in the eluates of the spin columns were visualised after electrophoresis in a 12% SDS–PAGE gel by silver staining. Reactions contained 5 pmol nicked pUC19 DNA, 8 pmol afPolB1 (lanes 4–8), 16 pmol afRFC (lanes 5, 6, and 9–11), 16 pmol LargeBmut/Smallwt (lanes 7 and 8), 16 pmol pk-PCNA (lanes 5–8, 10, and 11), 1 mM ATP (lanes 4, 5, 7, and 10), or 1 mM ATPγS (lanes 6, 8, 9, and 11). In addition, 5% of the input afPolB1 and PCNA (lane 1), afRFC (lane 2), or LargeBmut/Smallwt (lane 3) proteins were loaded onto the gel. (B) In all, 1 nmol PCNA and, where indicated, 3 nmol afPolB1 and 3 nmol afRFC were separated at room temperature by gel filtration in the presence or absence of 5 mM ATP. A measure of 15 μl of the 750 μl fractions 17–26 were separated in a 12% SDS–PAGE gel stained with Coomassie brilliant blue.
In the presence of ATP, PCNA was efficiently released from the mutant LargeBmut/Smallwt complex after loading and was able to bind afPolB1. By contrast, in the presence of ATPγS, the mutant complex formed a stable complex with PCNA and access of afPolB1 was blocked. We therefore conclude that hydrolysis at only the afRFC small subunits is sufficient for release of PCNA after loading and for the subsequent interaction with afPolB1.
We also analysed the competition of afRFC and afPolB1 for PCNA when PCNA was not loaded onto DNA. Figure 4B shows that afRFC, PCNA, and afPolB1 are unable to form a complex that is stable to gel filtration at room temperature. In the absence of ATP, PCNA interacts preferentially with afPolB1, but in the presence of ATP, PCNA binds more tightly to afRFC. Since the ATPase activity of the afRFC–PCNA complex is very low in the absence of DNA (kcat=0.013 s−1 at 55°C, and decreases even further at lower temperatures; Seybert et al, 2002), ATP hydrolysis should be minimal during the gel filtration.
In summary, the interactions of afRFC and afPolB1 with PCNA are mutually exclusive in both the presence and absence of DNA. ATP hydrolysis by the afRFC small subunits is required to release afRFC from PCNA and allow formation of the PCNA–afPolB1 complex. Once again, ATP hydrolysis by the large subunit is not required for this process.
ATP hydrolysis by afRFC is necessary for catalytic loading of PCNA
Since we had been unable to demonstrate any aspect of afRFC-dependent PCNA loading that was dependent upon ATP hydrolysis by the afRFC large subunits, we analysed PCNA-dependent DNA synthesis by afPolB1 in the presence of wild-type afRFC and our afRFC mutant complexes to test whether ATP hydrolysis by the large subunits was required at any stage in the reaction. Although the simple assay system we set up did not include factors required for processive DNA replication in vitro in other systems including archaea (such as single-stranded DNA binding proteins, for example, RPA), we were able to observe significant primer extension with the wild-type complex (Figure 5). Our singly primed ssM13 template can accommodate the loading of multiple clamps, so we established conditions for the assay under which the rate of PCNA loading was rate limiting to maximise the effects of any defects in clamp loading (Figure 5). Under these conditions, as expected, the mutants with nucleotide-binding defects (namely LargeAmut/SmallAmut, LargeAmut/Smallwt, and Largewt/SmallAmut) did not stimulate DNA synthesis by afPolB1 since we have shown that these are unable to load PCNA. Furthermore, complexes with ATPase-deficient small subunits (LargeBmut/SmallBmut and Largewt/SmallBmut) also showed strong defects in stimulation. These data are consistent with the notion that hydrolysis by the afRFC small subunits is essential to allow release of PCNA from afRFC to enable a complex to be formed between afPolB1 and PCNA. By raising the amount of PCNA and afRFC (each to 10 pmol) to compensate for the reduced ATPase activity (and hence reduced efficiency of PCNA loading), we were able to observe some stimulation with the Largewt/SmallBmut complex but this was still reduced compared to the wild type. However, this stimulation was lost when the amount of polymerase was reduced, even at these higher afRFC and PCNA concentrations, presumably due to decreased competition from polymerase for the RFC-bound PCNA.
Figure 5.
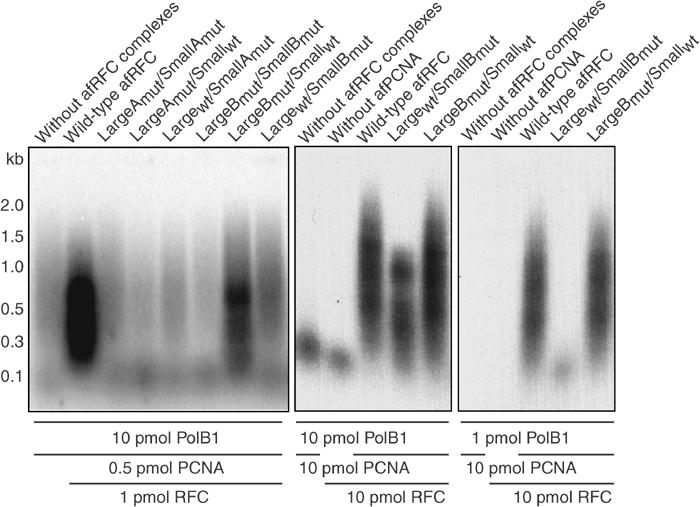
Ability of afRFC wild-type or mutant complexes to stimulate DNA synthesis catalysed by afPolB1. Reaction mixtures (20 μl) were supplemented with 200 mM NaCl and singly primed, closed circular M13mp18 DNA (25 fmol). After incubation for 30 min at 65°C, the DNA was precipitated and analysed by alkaline gel electrophoresis. Autoradiograms of dried gels are shown.
Surprisingly, a significant defect was also observed for the LargeBmut/Smallwt mutant complex, albeit less drastic than for the other mutants. However, this defect with respect to wild type could be overcome by using a higher concentration of the mutant afRFC and PCNA, even when the amount of polymerase in the reaction is reduced 10-fold. The simplest explanation for these observations is that the rate of catalytic PCNA loading is reduced in the mutant complex such that the effect is only apparent in the reaction when the rate of PCNA loading is the rate-limiting step in the assay. When the amount of afRFC and PCNA is increased, this rate is no longer limiting and the reaction proceeds to the same extent as for the wild type.
These data finally reveal a role for ATP hydrolysis by the large subunit. We have shown that in a mutant afRFC with an ATPase-deficient large subunit, all steps of the clamp-loading process up to, and including, the release of PCNA are unaffected. Furthermore, a defect in clamp loading is only revealed in this mutant afRFC when the rate of catalytic clamp loading is limiting. Consequently, the only role remaining for ATP hydrolysis by the large subunit is one of catalytic recycling of afRFC, perhaps either controlling release of the complex from the DNA template or a conformational change that is required prior to PCNA binding.
A model for archaeal clamp loaders
The experiments we describe above allow us to deduce a detailed model for the distinct roles of ATP binding and hydrolysis by afRFC (Figure 6). Although these experiments have been undertaken with an archaeal RFC, we believe that this may prove a useful model for eukaryotic RFC. RFC in eukaryotes is a multisubunit complex that contains one large subunit and four, closely related, small subunits. However, it has been inferred by sequence analysis (Cullmann et al, 1995) that one of these small subunits is not able to bind ATP. Consequently, eukaryotic RFC is only able to bind a total of four nucleotides because it has only four sites that are competent for nucleotide binding in the complex and this has been demonstrated directly for the yeast complex (Gomes et al, 2001). As we show here for afRFC, the loading of these sites in yeast RFC was shown to be sequential, dependent upon the nature of cofactors associated with RFC (i.e. DNA and PCNA), and presumed to involve the large subunit and three of the small subunits. However, the requirement for ATP hydrolysis remained unclear, particularly any difference between hydrolysis at the different sites. Using the archaeal RFC, we can now suggest how and why this might take place in the eukaryotic system. Note, however, that the E.coli clamp loader utilises ATP in a distinctly different way (Johnson and O'Donnell, 2003).
Figure 6.
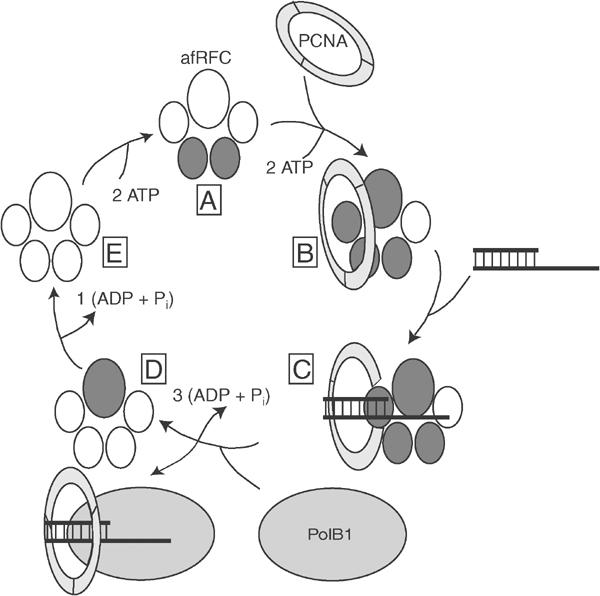
Model for clamp loading by afRFC. A proposed mechanism for ATP utilisation by the archaeal clamp loader includes the following (clockwise, starting from the top): (A) initial binding of two molecules of ATP by the afRFC small subunits, (B) upon interaction with PCNA, binding of two additional molecules of ATP (one of which binds to the large subunit), (C) loading of PCNA onto DNA, (D) hydrolysis of ATP at the afRFC small subunits, which results in both release of PCNA onto DNA and release of the clamp loader from the PCNA–DNA complex, thereby enabling the polymerase to associate with PCNA, and (E) hydrolysis of ATP at the afRFC large subunit, which results in recycling of afRFC and in initiation of a new cycle of clamp loading.
Bacteriophage T4 and archaea have simpler clamp loaders as eukaryotes, with four identical small subunits and an additional subunit to constitute a heteropentamer. This additional subunit in archaea has ATPase motifs and is homologous with the large subunit in eukaryotic RFC. Curiously, however, the additional subunit in the T4 clamp loader (gp62) is smaller than the other subunits (themselves homologues of the small subunits of RFC) and has no ATPase motifs. It has been demonstrated that the T4 clamp loader complex can bind four ATP molecules, presumably to each of the four identical gp44 subunits. Despite having four identical small subunits in archaeal RFC, we show that only three of these are competent to bind ATP. How these complexes are able to prevent ATP binding to one of their potential binding sites despite having identical small subunits is very interesting but remains a mystery in the absence of structural information. However, there are precedents for this kind of behaviour in a related system, namely the hexameric helicase from bacteriophage T7 (gp4), which has six identical sites but only binds four ATP molecules (Singleton et al, 2000). The reason for this relates to a rotation of subunits around the ring such that the ATP-binding sites, which are located between the subunits, are not identical. It is tempting to speculate that a similar mechanism might operate in RFC.
Experimental procedures
Materials
Unless stated otherwise, all chemicals were supplied by Sigma. The radioactive stock solution used in the nucleotide-binding assays was prepared as follows: 2.5 μl [35S]ATPγS (10 μCi/μl, >1000 Ci/mmol, Amersham Biosciences) and 25 μl cold 10 mM ATPγS were mixed in a final volume of 1 ml in 2 mM DTT and 10 mM Tris–HCl (pH 7.5). This stock solution was stored in small aliquots at −80°C and only thawed once.
Site-directed mutagenesis
A recombination PCR method (Yao et al, 1992) was used to introduce point mutations into the afRFC large and small subunits. The plasmids pET28-afRFClarge or pET22-afRFCsmall (Seybert et al, 2002) were used to introduce substitutions into the coding sequences of the afRFC large and small subunits, respectively. The primers for the substitutions, with the exchanged codon(s) underlined in each case, were as follows: for Lys-49 to aspartate and Thr-50 to aspartate of the afRFC large subunit, 5′-CGGCGTGGGAGATGATTCCCTCGCTCTGGCCCTTGC-3′ and 5′-GCCAGAGCGAGGGAATCATCTCCCACGCCGGGAGGACCG-3′; for Lys-51 to aspartate and Thr-52 to aspartate of the afRFC small subunit, 5′-CTGGAACTGGTGATGATGCAACAGCAATAGCTCTGG-3′ and 5′-GCTATTGCTGTTGCATCATCACCAGTTCCAGGAGGTCCG 3′; for Glu-112 to alanine of the afRFC large subunit, 5′-CTAATTATTCTTGACGCTGTTGACAACATTCAc-3′ and 5′- GAATGTTGTCAACAGCGTCAAGAATAATTAGC-3′; for Glu-110 to alanine of the afRFC small subunit, 5′ CTTCCTCGACGCTGCTGATGCACTAACTGC-3′ and 5′-GTGCATCAGCAGCGTCGAGGAAGATTATC-3′.
Expression and purification of recombinant proteins
AfRFC, afPCNA, and afPolB1 were purified as described (Seybert et al, 2002). The afRFC mutant complexes were expressed and purified using the same procedure as described for wild-type afRFC.
Nucleotide-binding assays
Nucleotide binding to afRFC complexes was determined by equilibrium dialysis or in a spin column assay (Tamura et al, 1992). For the equilibrium dialysis, disposable dialysers with a cutoff of 10 000 Da were used (DispoEquilibrium dialyser, Harvard Apparatus). One dialyser chamber contained, in a final volume of 50 μl, 2 μM wild-type afRFC complexes in reaction buffer (20 mM Tris–HCl (pH 7.5), 20 mM MgCl2, 2 mM DTT, 0.1 mM EDTA, and 5% (v/v) glycerol) and where indicated, 3 μM afPCNA and/or 3 μM DNA (5′-AGGGAAGGGAGAGGGAGGAGAAGAAGGGAG-3′ annealed to 5′-(dT)30-CTCCCTTCTTCTCCTCCCTCTCCCTTCCC-(dT)21-3′). In a final volume of 50 μl, the second dialyser chamber contained 4–200 μM [35S]ATPγS, reaction buffer, where indicated, PCNA and/or DNA, but no afRFC. As determined by reactions analysed in the absence of afRFC, equilibrium was reached after the dialysers had been gently shaken for 48 h at room temperature. Note that, as verified by ATPase assays, the stability of the RFC complex was not affected by this prolonged incubation at room temperature. After 48 h, aliquots of the dialysates were counted along with standards of known [35S]ATPγS concentration in a liquid scintillation counter.
For the spin column assay, in a final volume of 50 μl, 2 μM wild-type or mutant afRFC complexes were incubated at 50°C in reaction buffer (see above) that contained, where indicated, 3 μM afPCNA and/or DNA (see above for sequence). After 2 min incubation, [35S]ATPγS was added to final concentrations of 0.1–100 μM and the reactions were incubated for a further 15 min at 50°C. In the meantime, Micro Bio-Spin P30 Tris chromatography columns (Biorad) were pre-equilibrated in reaction buffer according to the supplier's instructions. The reaction mixtures were applied to the gel filtration columns and within 10 s the centrifugation was started (4 min at 1000 g). The eluates were counted along with standards of known [35S]ATPγS concentration in a liquid scintillation counter. Using GraphPad Prism 3.0 software, the data were fitted by nonlinear regression assuming equivalent binding sites according to the equation: Y=Bmax:X/(KD+X) where Y is the amount of ATPγS bound per complex, X is the amount of free ATPγS (ATPγSfree=ATPγStotal − ATPγSbound), Bmax is the maximum number of binding sites, and KD is the dissociation constant.
ATPase assays
ATPase rates were determined by analysing the turnover of radioactively labelled ATP. The 20 μl reaction mixtures contained 50 mM HEPES–KOH (pH 7.5), 20 mM magnesium chloride, 25 mM potassium chloride, 250 μM [γ32P]ATP, and 25 nM to 2 μM afRFC complexes. Where indicated, reactions contained 3 μM afPCNA and/or 300 μM (nucleotide concentration) singly primed M13mp18 DNA prepared as described previously (Seybert et al, 2002). After various times of incubation at 55°C, 0.5 μl aliquots were spotted onto polyethyleneimine–cellulose thin-layer plates that were developed in 0.15 M formic acid–0.15 M LiCl (pH 3.0) as the liquid phase and analysed by phosphorimaging.
Determination of afRFC–PCNA interactions
Pull-down assays (in which complexes were captured via the His-tag located at the N-terminus of the afRFC large subunit) were used to determine interactions between wild-type versus mutant afRFC complexes and afPCNA as described in Seybert et al (2002).
PCNA-loading assays
For the quantitative analysis of afRFC-dependent afPCNA loading onto DNA, radioactively labelled afPCNA had to be generated. To this end, afPCNA carrying a protein kinase recognition site at its carboxy terminus (designated pk-afPCNA) was constructed. Two oligonucleotides (5′-TAATACGACTCACTATAGGG-3′ and 5′- TTTACGGCCGTTACTAACCAACAGAAGCTCTTCGAAGCT CCGACTCTATG-3′, the sequence of the noncoding strand of the protein kinase recognition site is underlined) were used to amplify the afPCNA coding sequence from pET22-afPCNA (Seybert et al, 2002) and the PCR product was inserted into the NdeI/HindIII sites of pET22b (Novagen). Pk-afPCNA was expressed and purified as described for afPCNA (Seybert et al, 2002). For the 32P labelling, reaction mixtures (50 μl) contained 20 mM Tris–HCl (pH 7.5), 2 mM DTT, 12 mM MgCl2, 100 mM NaCl, 100 μCi [γ32P]ATP (3000 Ci/mmol), 40 μg pk-afPCNA, and 9 U protein kinase A (catalytic subunit from bovine heart). After 60 min incubation at 30°C, the reactions were purified over Micro Bio-Spin P6 Tris chromatography columns (Biorad), which had been pre-equilibrated in 50 mM Tris–HCl (pH 7.5), 1 mM DTT, 1 mM EDTA, and 150 mM NaCl. An afPCNA derivative carrying the protein kinase recognition site at its amino terminus resulted in at least 200-fold lower phosphorylation under these reaction conditions. For the loading assays, reaction mixtures (20 μl) contained 20 mM Tris–HCl (pH 7.5), 5 mM DTT, 8 mM MgCl2, 100 μM EDTA, 4% (v/v) glycerol, and DNA, proteins, and nucleotides as indicated in the figure legends. Reactions were incubated for 5 min at 65°C and applied to Microspin S-400 HR columns (Amersham Biosciences) that had been pre-equilibrated in 50 mM Tris–HCl (pH 7.5), 200 mM NaCl, 1 mM DTT, 20 mM MgCl2, and 5% (v/v) glycerol. Within 10 s, centrifugation was started (2 min at 735 g) and aliquots of the eluates were analysed on ethidium bromide-stained native agarose gels as well as by SDS–PAGE followed by phosphorimager analysis or silver staining. Nicked DNA was generated by incubation of pUC19 DNA with a limited amount of DNaseI in the presence of ethidium bromide and purified by phenol–CHCl3/isoamyl alcohol (24:1, v/v) extraction followed by ethanol precipitation (Greenfield et al, 1975). Nicked linearised DNA was generated by cutting nicked pUC19 DNA with HindIII.
Analysis of afRFC–PCNA–PolB1 interactions by gel filtration
A Superdex 200 HR 10/30 gel filtration column (Amersham Biosciences) was equilibrated in column buffer (25 mM CHES 8.5, 1 mM DTT, 40 mM MgCl2, and 150 mM NaCl) that contained, where indicated, 5 mM ATP. In all, 3 nmol afPolB1, 3 nmol afRFC, and 1 nmol afPCNA were mixed in a final volume of 200 μl and applied to the column. The flow rate was set to 0.5 ml/min and 750 μl fractions were collected.
Elongation of a singly primed M13 DNA template by PolB1
AfPolB1 catalysed elongation of singly primed M13mp18 single-stranded circular DNA was carried out essentially as described in Seybert et al (2002).
Acknowledgments
This work was supported by Cancer Research UK and a postdoctoral fellowship from the European Molecular Biology Organization (to AS).
References
- Bertram JG, Bloom LB, Hingorani MM, Beechem JM, O'Donnell M, Goodman MF (2000) Molecular mechanism and energetics of clamp assembly in Escherichia coli. The role of ATP hydrolysis when gamma complex loads beta on DNA. J Biol Chem 275: 28413–28420 [DOI] [PubMed] [Google Scholar]
- Cai J, Yao N, Gibbs E, Finkelstein J, Phillips B, O'Donnell M, Hurwitz J (1998) ATP hydrolysis catalyzed by human replication factor C requires participation of multiple subunits. Proc Natl Acad Sci USA 95: 11607–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann IK, Ishino Y (1999) Archaeal DNA replication: identifying the pieces to solve a puzzle. Genetics 152: 1249–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann IK, Ishino S, Yuasa M, Daiyasu H, Toh H, Ishino Y (2001) Biochemical analysis of replication factor C from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol 183: 2614–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullmann G, Fien K, Kobayashi R, Stillman B (1995) Characterization of the five replication factor C genes of Saccharomyces cerevisiae. Mol Cell Biol 15: 4661–4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes XV, Burgers PM (2001) ATP utilization by yeast replication factor C. I. ATP-mediated interaction with DNA and with proliferating cell nuclear antigen. J Biol Chem 276: 34768–34775 [DOI] [PubMed] [Google Scholar]
- Gomes XV, Schmidt SL, Burgers PM (2001) ATP utilization by yeast replication factor C. II. Multiple stepwise ATP binding events are required to load proliferating cell nuclear antigen onto primed DNA. J Biol Chem 276: 34776–34783 [DOI] [PubMed] [Google Scholar]
- Greenfield L, Simpson L, Kaplan D (1975) Conversion of closed circular DNA molecules to single-nicked molecules by digestion with DNAase I in the presence of ethidium bromide. Biochim Biophys Acta 407: 365–375 [DOI] [PubMed] [Google Scholar]
- Guenther B, Onrust R, Sali A, O'Donnell M, Kuriyan J (1997) Crystal structure of the delta' subunit of the clamp-loader complex of E. coli DNA polymerase III. Cell 91: 335–345 [DOI] [PubMed] [Google Scholar]
- Hingorani MM, Bloom LB, Goodman MF, O'Donnell M (1999) Division of labor—sequential ATP hydrolysis drives assembly of a DNA polymerase sliding clamp around DNA. EMBO J 18: 5131–5144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani MM, O'Donnell M (1998) ATP binding to the Escherichia coli clamp loader powers opening of the ring-shaped clamp of DNA polymerase III holoenzyme. J Biol Chem 273: 24550–24563 [DOI] [PubMed] [Google Scholar]
- Jarvis TC, Paul LS, von Hippel PH (1989) Structural and enzymatic studies of the T4 DNA replication system. I. Physical characterization of the polymerase accessory protein complex. J Biol Chem 264: 12709–12716 [PubMed] [Google Scholar]
- Jeruzalmi D, O'Donnell M, Kuriyan J (2001a) Crystal structure of the processivity clamp loader gamma (γ) complex of E. coli DNA polymerase III. Cell 106: 429–441 [DOI] [PubMed] [Google Scholar]
- Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O'Donnell M, Kuriyan J (2001b) Mechanism of processivity clamp opening by the δ subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell 106: 417–428 [PubMed] [Google Scholar]
- Johnson A, O'Donnell M (2003) Ordered hydrolysis in the gamma complex clamp loader AAA+ machine. J Biol Chem 278: 14406–14413 [DOI] [PubMed] [Google Scholar]
- Kelman Z, Hurwitz J (2000. A unique organization of the protein subunits of the DNA polymerase clamp loader in the archaeon Methanobacterium thermoautotrophicum H. J Biol Chem 275: 7327–7336 [DOI] [PubMed] [Google Scholar]
- Kong XP, Onrust R, O'Donnell M, Kuriyan J (1992) Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell 69: 425–437 [DOI] [PubMed] [Google Scholar]
- Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J (1994) Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 79: 1233–1234 [DOI] [PubMed] [Google Scholar]
- Moarefi I, Jeruzalmi D, Turner J, O'Donnell M, Kuriyan J (2000) Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J Mol Biol 296: 1215–1223 [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV (1999) AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9: 27–43 [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6: 575–597 [DOI] [PubMed] [Google Scholar]
- Pisani FM, De Felice M, Carpentieri F, Rossi M (2000) Biochemical characterization of a clamp-loader complex homologous to eukaryotic replication factor C from the hyperthermophilic archaeon Sulfolobus solfataricus. J Mol Biol 301: 61–73 [DOI] [PubMed] [Google Scholar]
- Podust VN, Tiwari N, Ott R, Fanning E (1998a) Functional interactions among the subunits of replication factor C potentiate and modulate its ATPase activity. J Biol Chem 273: 12935–12942 [DOI] [PubMed] [Google Scholar]
- Podust VN, Tiwari N, Stephan S, Fanning E (1998b) Replication factor C disengages from proliferating cell nuclear antigen (PCNA) upon sliding clamp formation, and PCNA itself tethers DNA polymerase delta to DNA. J Biol Chem 273: 31992–31999 [DOI] [PubMed] [Google Scholar]
- Schmidt SL, Gomes XV, Burgers PM (2001a) ATP utilization by yeast replication factor C. III. The ATP-binding domains of Rfc2, Rfc3, and Rfc4 are essential for DNA recognition and clamp loading. J Biol Chem 276: 34784–34791 [DOI] [PubMed] [Google Scholar]
- Schmidt SL, Pautz AL, Burgers PM (2001b) ATP utilization by yeast replication factor C. IV. RFC ATP-binding mutants show defects in DNA replication, DNA repair, and checkpoint regulation. J Biol Chem 276: 34792–34800 [DOI] [PubMed] [Google Scholar]
- Seybert A, Scott DJ, Scaife S, Singleton MR, Wigley DB (2002) Biochemical characterisation of the clamp/clamp loader proteins from the euryarchaeon Archaeoglobus fulgidus. Nucleic Acids Res 30: 4329–4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Sawaya MR, Ellenberger T, Wigley DB (2000) Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 101: 589–600 [DOI] [PubMed] [Google Scholar]
- Stukenberg PT, Studwell-Vaughan PS, O'Donnell M (1991) Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J Biol Chem 266: 11328–11334 [PubMed] [Google Scholar]
- Tamura JK, Bates AD, Gellert M (1992) Slow interaction of 5′-adenylyl-beta,gamma-imidodiphosphate with Escherichia coli DNA gyrase. Evidence for cooperativity in nucleotide binding. J Biol Chem 267: 9214–9222 [PubMed] [Google Scholar]
- Trakselis MA, Berdis AJ, Benkovic SJ (2003) Examination of the role of the clamp-loader and ATP hydrolysis in the formation of the bacteriophage T4 polymerase holoenzyme. J Mol Biol 326: 435–451 [DOI] [PubMed] [Google Scholar]
- Turner J, Hingorani MM, Kelman Z, O'Donnell M (1999) The internal workings of a DNA polymerase clamp-loading machine. EMBO J 18: 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F, Cai J, Gibbs E, O'Donnell M, Hurwitz J (1997) Deletion analysis of the large subunit p140 in human replication factor C reveals regions required for complex formation and replication activities. J Biol Chem 272: 10058–10064 [DOI] [PubMed] [Google Scholar]
- Xiao H, Naktinis V, O'Donnell M (1995) Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. IV. ATP-binding site mutants identify the clamp loader. J Biol Chem 270: 13378–13383 [DOI] [PubMed] [Google Scholar]
- Yao N, Leu FP, Anjelkovic J, Turner J, O'Donnell M (2000) DNA structure requirements for the Escherichia coli gamma complex clamp loader and DNA polymerase III holoenzyme. J Biol Chem 275: 11440–11450 [DOI] [PubMed] [Google Scholar]
- Yao Z, Jones DH, Grose C (1992) Site-directed mutagenesis of herpesvirus glycoprotein phosphorylation sites by recombination polymerase chain reaction. PCR Methods Appl 1: 205–207 [DOI] [PubMed] [Google Scholar]
- Young MC, Weitzel SE, von Hippel PH (1996) The kinetic mechanism of formation of the bacteriophage T4 DNA polymerase sliding clamp. J Mol Biol 264: 440–452 [DOI] [PubMed] [Google Scholar]