

Abstract
Transgenic mice broadly expressing JunD (Ubi-junDm) appear phenotypically normal, but have strongly reduced numbers of peripheral lymphocytes. JunD overexpression in lymphocytes does not protect from numerous apoptotic insults; however, transgenic T cells proliferate poorly and exhibit impaired activation due to reduced levels of IL-4, CD25 and CD69. Consistently, in the absence of JunD (junD−/−) T cells hyperproliferate following mitogen induction. Moreover, transgenic T helper (Th) 2 cells have decreased IL-4 and IL-10 expression, whereas junD−/− Th2 cells secrete higher amounts of both Th2 cytokines. Th1-polarized junD−/− CD4+ T cells display enhanced IFN-γ cytokine production associated with upregulated T-bet expression and downregulated expression of suppressor of cytokine signaling-1. These novel findings demonstrate a regulatory role of JunD in T lymphocyte proliferation and Th cell differentiation.
Keywords: IL-4, IL-10, JunD, lymphocytes, SOCS1, T-bet, Th cells
Introduction
Multiple stimuli, such as growth factors, proinflammatory cytokines and physical stress, induce and activate a group of DNA binding proteins that form the dimeric transcription factor complex AP-1. Composed of Fos and Jun dimers, AP-1 binds to specific recognition elements (TRE) found in many enhancer and promoter regions. Hence, being at the end of the signaling cascade, the differential expression of AP-1 proteins converts extracellular signals into cytoplasmic signaling cascades, resulting in the expression of specific target genes (Shaulian and Karin, 2002; Eferl and Wagner, 2003). Three members of the Jun family, c-Jun, JunB and JunD, share a high degree of sequence homology; however, their functions in cellular processes such as growth regulation, transformation, apoptosis or survival are different (Jochum et al, 2001; Mechta-Grigoriou et al, 2001). Mice lacking c-jun die between day 12.5 and 13.5 of embryonic development (Hilberg et al, 1993; Johnson et al, 1993). These mutant embryos exhibit increased apoptosis in the liver and display malformation in the heart outflow tract (Eferl et al, 1999). On the other hand, junB-deficient embryos die due to multiple defects in the extra-embryonic tissues (Schorpp-Kistner et al, 1999). In contrast to c-Jun and JunB, mice lacking junD are viable, suggesting that JunD is dispensable for embryonic development (Thepot et al, 2000). However, mutant male mice display impaired growth, and a lack of JunD results in defective spermatogenesis. In contrast to inactivation, broad overexpression of c-Jun and JunB in transgenic mice does not result in an overt phenotype (Jochum et al, 2001).
JunD was reported to function primarily as a negative regulator of cell proliferation and a protective protein against apoptosis. Overexpression of JunD in immortalized fibroblasts decreases proliferation (Pfarr et al, 1994). In addition, immortalized junD−/− fibroblasts show increased proliferation due to higher levels of cyclin D1 (Weitzman et al, 2000). However, JunD can also positively influence the cell cycle. Primary mouse fibroblasts lacking JunD proliferate less and display premature senescence via a p53-dependent pathway (Weitzman et al, 2000). JunD also plays a major role in cellular transformation in vitro, as JunD expression is decreased in Ras-transformed fibroblasts and JunD overexpression partially suppresses Ras-induced transformation (Pfarr et al, 1994). On the other hand, the tumor suppressor Menin interacts with JunD and represses its transcriptional activity, suggesting that downregulation of JunD is involved in the suppression of neoplastic growth (Agarwal et al, 1999). JunD was also identified as an antiapoptotic gene, as JunD-deficient fibroblasts display increased sensitivity to UV irradiation or TNF-α treatment (Weitzman et al, 2000). Accumulation of p53 in the nuclei of junD−/− fibroblasts could be the reason for the increased sensitivity to proapoptotic signals. In addition, mice lacking JunD were sensitive to the cytotoxic effects of TNF-α-mediated hepatitis in vivo (Weitzman et al, 2000). Recently, increased antiapoptotic gene expression via the JNK/JunD pathway has been linked to NF-κB (Lamb et al, 2003).
The differentiation of naive T cells into Th1 and Th2 helper cells is a hallmark of T cell-dependent immune responses (Mosmann and Coffman, 1989; Paul and Seder, 1994). Th1 cells promote the inflammatory and cellular immune response by producing IFN-γ, lymphotoxin α (TNF-β) and IL-2. In contrast, Th2 cells induce humoral immunity by secreting IL-4, IL-5, IL-6 and IL-10 cytokines. The most potent factors that influence Th1 and Th2 differentiation are IL-12 and IL-4, respectively. IL-12 activates the Stat-4 signaling pathway (Glimcher and Murphy, 2000; Ho and Glimcher, 2002; O'Shea et al, 2002), and IL-4 mediates its effects through the Stat-6 signaling pathway (Wurster et al, 2000; O'Shea et al, 2002). Several transcription factors have been identified that promote the proliferation and differentiation of T cells into Th effector cells. Among the AP-1 members, JunB was found to regulate IL-4 expression and to bias Th differentiation toward the Th2 lineage (Rincon et al, 1997; Li et al, 1999; Hartenstein et al, 2002). Other transcription factors associated in the regulation of IL-4 expression and commitment of Th2 cells include GATA-3 (Zhang et al, 1997, 1998, 1999; Zheng and Flavell, 1997), c-maf (Ho et al, 1996; Kim et al, 1999), NFAT (Rooney et al, 1995a; Macian et al, 2001) and NF-κB (Ferreira et al, 1999; Das et al, 2001), most of them being able to dimerize or to interact functionally with AP-1.
To investigate the role of JunD in cellular processes, such as proliferation, differentiation and apoptosis in vivo, we generated mice expressing a JunD transgene in many tissues. Ubi-junDm transgenic mice are viable, but exhibit profound defects in lymphoid cells. Overexpression of JunD suppresses B and T lymphocyte proliferation and Th differentiation, whereas lymphocyte apoptosis is not affected. JunD appears to mediate these effects by modulating the expression of key molecules involved in T cell activation (CD25, CD69) and differentiation (IL-4, IL-10). IFN-γ was also found to be increased in JunD-deficient Th1 or Th2 polarized CD4+ T cells, which is probably due to increased expression of Th1-specific transcription factor T-bet and reduced expression of suppressor of cytokine signaling-1 (SOCS1). Thus, these data identify a novel function for JunD in T lymphocyte proliferation and Th differentiation.
Results
Characterization of Ubi-junDm transgenic mice
Transgenic mice were generated expressing the murine junD gene under the control of the human ubiquitin C promoter (Ubi-junDm), which directs ubiquitous expression to most mouse tissues (Figure 1A; Schorpp et al, 1996). The junD cDNA was linked to the SV40 polyadenylation signal and was tagged at the C-terminus with a single myc epitope to be able to distinguish the exogenous from the endogenous JunD protein (Togel et al, 1998). Two transgenic lines with different copy numbers, line Tg5 (two to three copies), and Tg8 (five copies) were established. Tg5 was mainly used in subsequent analyses, as initial experiments gave identical results. Western blot analysis showed that transgenic JunD (JunDm) protein was widely expressed in many organs (Figure 1A), including B and T cells and thymocytes (Figure 1B). Western blot analysis performed on purified T cells displayed similar JunD expression in transgenic cells compared to wild-type cells. The expression of myc-tagged full-length JunD yielded a slightly larger protein that was detected with antibodies to JunD. This slight increase in size was expected from the addition of the 14-amino acid long myc-tag (Figure 1C; Togel et al, 1998).
Figure 1.

Expression of the Ubi-junDm transgene. (A) Schematic representation of the 3.0 kb Ubi-junDm transgene construct. The complete mouse junD cDNA was ligated to the human Ubi-C promoter and a myc-tag was inserted at the C terminus. Expression of Ubi-junDm transgene in various organs of 3-week-old wild-type (Wt) and transgenic mice (Tg5) analyzed by Western blot using a myc-specific polyclonal antibody. Br, brain; Spl, spleen; Ki, kidney; Te, testis; Thy, thymus. (B) Ubi-junDm protein expressed in splenic B cells, T cells and thymocytes analyzed by Western blot using a myc-specific polyclonal antibody. (C) Ubi-junDm protein (arrow) expressed in splenic CD4+ T cells analyzed by Western blot using a JunD-specific polyclonal antibody. Both wild-type JunD isoforms are recognized by the antibody (double arrow). Actin protein levels were used as loading control. (D) Serum Ig isotype levels obtained from 8-week-old wild-type (open bars; n=9) and Ubi-junDm transgenic mice (filled bars; n=9). The amounts of each IgM, IgA, IgG2a, IgG2b, IgG3, IgE and IgG1 subclass were determined by ELISA. Results are the mean±s.d. of the indicated numbers of mice. aP⩽0.015.
Ubi-junDm transgenic mice in a 129/sv × C57BL/6 mixed genetic background were fertile and did not show any gross phenotypic abnormalities, except for signs of infections localized mainly around the eyelids (data not shown). Peripheral white blood analysis of 12-week-old transgenic mice revealed that the absolute lymphocyte number was reduced to half (wild-type 82%±10; Ubi-junDm 45%±8), while neutrophil counts had increased 2.5-fold (wild-type 14%±6; Ubi-junDm 35%±3). Histological analyses also revealed massive focal inflammation by neutrophils with subsequent ulceration of the colon mucosa in 10% of Ubi-junDm mice (data not shown).
T helper cell-derived cytokines differentially regulate the production of immunoglobulin (Ig) classes by B cells. Th2 cells secrete IL-4, IL-6 and IL-10, which are involved in B lymphocyte class switching from IgM and IgD to IgG1 and IgE; by contrast, the Th1 cytokines IL-2 and IFN-γ induce class switching to type IgG2a and IgG2b isotypes (Mosmann and Coffman, 1989; Paul and Seder, 1994). To assess whether JunD is involved in Th1- or Th2-dependent Ig production, serum Ig levels were measured by enzyme-linked immunoassay (ELISA) with isotype-specific antibodies. The mean concentration of IgM, IgA, IgG2a, IgG2b, IgG3 and IgE was comparable between wild-type and transgenic mice (Figure 1D). However, transgenic mice showed significantly reduced levels of IgG1 compared to wild-type mice, suggesting that this reduction is mediated by a defect in Th2-dependent cytokine expression, which might explain the observed susceptibility to infections in these mice. Thus, overexpression of JunD seems to affect the lymphoid system and therefore the B and T cell lineage was further analyzed.
Analysis of 4–6-week-old transgenic thymi indicated similar numbers of single and double positive CD4 and CD8 T lymphocytes compared to wild-type. These cells also expressed normal levels of TCRα/CD3 complexes (Figure 2A and data not shown). Lymphocyte cell numbers in the thymus of transgenic mice were also within the normal range and without consistent differences compared to nontransgenic littermates. Analysis of B cell lineage development did not reveal any significant differences between wild-type and transgenic mice in either splenic cellularity or expression of B220, CD43, IgM, IgD, Gr-1 and Mac1 (data not shown). While thymocyte development was normal, peripheral CD4+ T cell numbers in spleen and in particular in lymph nodes were reduced compared to controls (Figure 2A).
Figure 2.
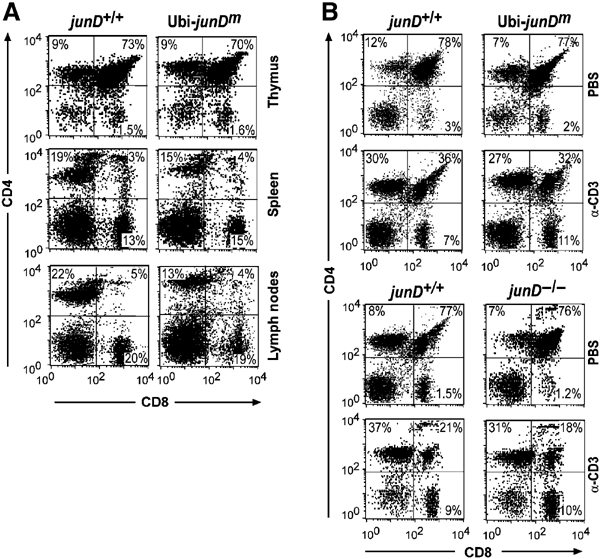
Lymphocyte populations and anti-CD3 mediated depletion of double-positive thymocytes are unaffected in Ubi-junDm transgenic and junD−/− mice. (A) Flow cytometric analysis of thymi, spleens and lymph nodes. Single-cell suspensions were stained with the indicated antibody combination. Percentages of positive cells within a quadrant are indicated. (B) Wild-type, Ubi-junDm mice or junD−/− mice (12 weeks old) were injected with PBS or with 40 μg/ml anti-CD3 antibody. Thymi were removed 48 h after injection and single-cell suspensions were stained with anti-CD4 and anti-CD8 fluorescent antibody conjugates and analyzed by flow cytometry. The results are representative of five mice per group. CD4 and CD8 expression of wild-type and Ubi-junDm mice (upper panel), wild-type and junD−/− mice (lower panel).
JunD does not protect lymphoid cells from apoptosis
To investigate whether overexpression of JunD in the lymphoid lineage influences apoptotic susceptibility, Ubi-junDm peripheral B and T lymphocytes as well as thymocytes were exposed to various apoptotic stimuli. When B and T cells were treated with either UV-irradiation or TNF-α, no appreciable difference in viability was found between wild-type and transgenic cells (Table I). Analysis of apoptosis using Ubi-junDm thymocytes stimulated with Fas (anti-CD95) antibodies, anti-CD3, UV-irradiation or TNF-α did not reveal any significant difference compared to wild type (Table I). These results indicate that under ex vivo conditions, JunD overexpression does not prevent cells from undergoing apoptosis. Administration of anti-CD3 has been shown to result in the rapid deletion of double-positive (DP) thymocytes by apoptosis (Shi et al, 1991). Wild-type and Ubi-junDm mice treated with anti-CD3 showed a similar reduction of DP thymocytes as assessed by flow cytometry (Figure 2B, upper panel). Furthermore, no increased sensitivity of junD−/− thymocytes was observed, as the level of depletion was similar compared to wild-type mice (Figure 2B, lower panel). Thus, neither increased JunD levels nor the absence of JunD can modulate the susceptibility of thymocytes to anti-CD3-induced cell death.
Table 1.
Induction of apoptosis in wild-type and Ubi-junDm B cells, T cells and thymocytes
| Treatment | CD19+ B cells | Thy1.2+ T cells | Thymocytes | |||
|---|---|---|---|---|---|---|
| Wt | Ubi-junDm | Wt | Ubi-junDm | Wt | Ubi-junDm | |
| UV (40 J/m2) | 75%±8 | 73%±5 | 50%±10 | 55%±9 | 62%±6 | 55%±9 |
| TNF-α (10 ng/ml) | 35%±10 | 30%±8 | 52%±5 | 58%±7 | 55%±8 | 60%±6 |
| anti-Fas (0.1 μg/ml) | n.d. | n.d. | n.d. | n.d. | 32%±5 | 30%±7 |
| anti-CD3 (1 μg/ml) | n.d. | n.d. | 48%±7 | 51%±3 | 73%±9 | 74%±8 |
| PMA/Iono | n.d | n.d. | 23%±0.9 | 23%±2 | n.d. | n.d. |
| 8- to 12-week-old wild-type and Ubi-junDm mice were used. Cells were stimulated for 24 h with the indicated death-promoting stimuli. Cell viability was determined in duplicate by AnnexinV and propidium iodide staining. The result of three independent experiments (±s.d.) is shown for each activation; n.d., not done. | ||||||
JunD and its role in mitogen-induced T lymphocyte proliferation
To analyze whether JunD functions as a negative regulator during lymphocyte proliferation, ex vivo proliferation assays were performed with purified splenic T cells. Ubi-junDm Thy-1.2+ or CD4+ T cells showed a marked decrease in proliferation when grown in the presence of anti-CD3 alone or in combination with anti-CD28 (Figure 3A), and responded poorly to phorbol 12-myristate 13-acetate (PMA) plus calcium ionophore ionomycin (Figure 3A and data not shown). Interestingly, when grown with concanavalin A (ConA), a stimulus acting downstream of receptor signaling, no significant difference was found between the proliferation rates of control and transgenic cells. Furthermore, no significant difference in viability between wild-type and transgenic cells was observed independently of the stimuli used (data not shown). When junD−/− T cells were analyzed using the same assays, a stronger proliferative response after induction with anti-CD3 and anti-CD28 or with PMA plus ionomycin was detected (Figure 3B). The proliferation of junD−/− T cells in response to ConA was again unaffected. These data indicate that JunD regulates the proliferation of T cells following specific mitogenic stimuli.
Figure 3.
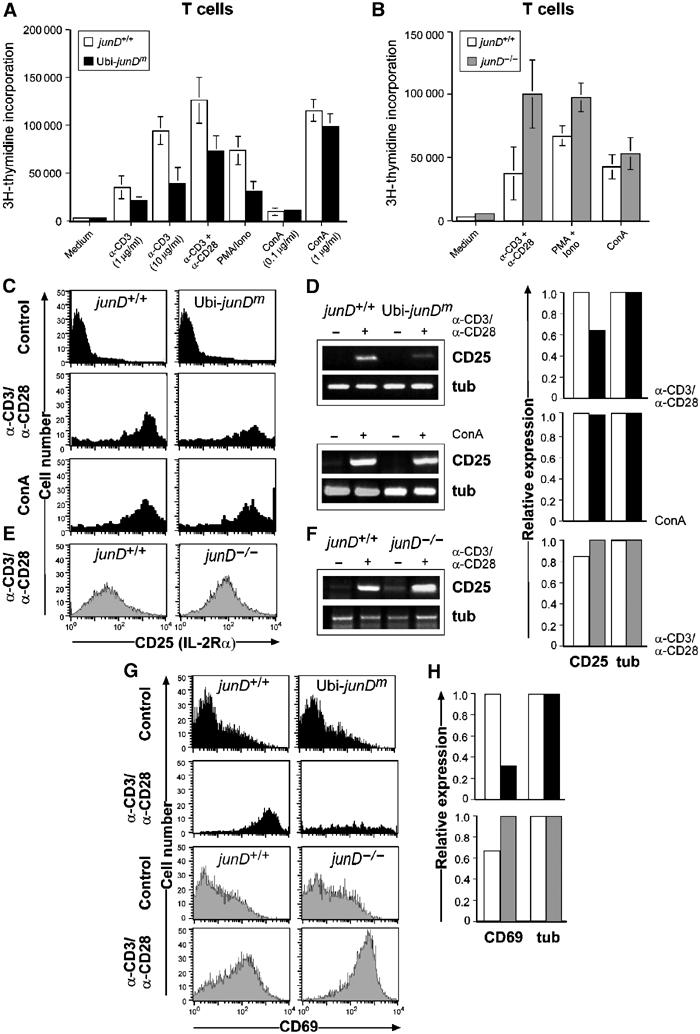
JunD and its role in T lymphocyte proliferation and activation. (A) Thy1.2+ junD+/+ (open bars) and Ubi-junDm (closed bars) T cells were stimulated with plate-bound anti-CD3 (1 or 10 μg/ml), anti-CD3 (1 μg/ml) with anti-CD28 (100 μg/ml), PMA (10 ng/ml) plus Ca2+ ionophore (100 ng/ml) (PMA+Iono) or with concanavalin A (0.1 or 1 μg/ml; ConA) for 48 h. (B) Proliferation of Thy1.2+ junD+/+ (open bars) and junD−/− (gray bars) T cells stimulated with plate-bound anti-CD3 (10 μg/ml) plus anti-CD28 (10 μg/ml), PMA (10 ng/ml) plus Ca2+ ionophore (100 ng/ml) (PMA+Iono) or concanavalin A (1 μg/ml; ConA) for 48 h. Results are mean±s.d. [3H]thymidine incorporation for quadruplet samples. (C) CD25 cell surface expression in junD+/+ and Ubi-junDm CD4+ T cells; untreated (Control); anti-CD3 (10 μg/ml) and anti-CD28 (10 μg/ml); and ConA (1 μg/ml) for 24 h. (D) CD25 RNA expression was determined by semiquantitative (left panel) and real-time (right panel) PCR: tub, tubulin (E, F) CD25 cell surface and RNA expression in junD+/+ and junD−/− CD4+ T cells activated and analyzed as described above. CD69 cell surface (G) and RNA expression (H) in junD+/+ and Ubi-junDm Thy1.2+ or in junD−/− CD4+ T cells (H) after 24 h stimulation with anti-CD3 and anti-CD28.
CD25 expression in CD4+ T cells is regulated by JunD
T cell receptor (TCR)-induced S-phase entry of resting T lymphocytes is promoted by transcriptional upregulation of the IL-2 gene, which has an AP-1 binding site and is an essential mediator of proliferation (Rooney et al, 1995b; Macian et al, 2000). Signaling through the IL-2Rα (CD25) plays a pivotal role in T cell proliferation. Interestingly, reduced expression of CD25 was detected in transgenic CD4+ T cells upon activation with anti-CD3 and anti-CD28 (Figure 3C) as measured by flow cytometric analysis (FACS). Real-time PCR further revealed that increased JunD expression resulted in decreased CD25, although CD25 expression was not changed when T cells were treated with ConA (Figures 3C and D). Consistently, increased expression levels of CD25 were detected in the junD−/− CD4+ T cell population analyzed either by FACS or real-time PCR (Figures 3E and F). In addition, expression of the cell surface CD69 receptor, another activation marker of T cell proliferation, was also found to be markedly decreased in activated transgenic T cells (Figures 3G and H), whereas expression was significantly increased in junD−/− T cells (Figures 3G and H). Thus, JunD is able to function as a negative regulator of TCR-induced lymphocyte proliferation.
Molecular analysis of Ubi-junDm T cells
To examine the role of JunD in cytokine response, transgenic T cells were stimulated with anti-CD3 alone, or in combination with anti-CD28, and the production of cytokines was examined. Ubi-junDm Thy-1.2+ T cells secreted normal levels of IL-2 as well as IFN-γ, essential cytokines for Th1 lineage commitment (Figure 4A and data not shown). In contrast, transgenic Thy-1.2+ or CD4+ T cells produced significantly lower amounts of IL-4 and IL-10, two well-known Th2-specific cytokines (Figures 4B and C). To gain molecular insights into the transcriptional changes associated with altered JunD expression, splenic Thy-1.2+ T cells were analyzed by RT–PCR 24 h after stimulation with anti-CD3 and anti-CD28. While expression of IL-2 was comparable in transgenic and control T cells, expression of IL-4 and IL-10 was significantly reduced (Figures 4A–C). When Ubi-junDm CD4+ T cells were cultured with increasing amounts of recombinant IL-4, the proliferation defect of Ubi-junDm T cells was completely rescued in a dose-dependent manner (Figure 4D). Importantly, when cultured in the presence of neutralizing anti-IL-4 antibody, both wild-type and transgenic T cell populations exhibited the same proliferation as measured by thymidine incorporation (Figure 4D). This indicates that the reduced proliferation of transgenic T cells is most probably due to reduced IL-4 levels.
Figure 4.

Decreased IL-4 production in Ubi-junDm T cells. (A–C) Purified splenic T cells from junD+/+ and Ubi-junDm transgenic mice stimulated for 24 h with anti-CD3 (10 μg/ml) alone or with anti-CD28 (10 μg/ml). IL-2 (Thy1.2+, A), IL-4 (Thy1.2+, B) and IL-10 (CD4+, C) concentrations were determined in the supernatants by ELISA. Thy1.2+ T cells from junD+/+ and Ubi-junDm mice were stimulated for 24 h with anti-CD3 (10 μg/ml) and anti-CD28 (10 μg/ml). RNA expression of the indicated genes was analyzed by RT–PCR (lower panel): tub, tubulin (D) CD4+ junD+/+ (open bars) and Ubi-junDm (closed bars). T cells were stimulated with plate-bound anti-CD3 (1 μg/ml) alone, with increasing concentrations of recombinant murine IL-4 (1, 10, 100 U/ml) or with IL-4 (10 U/ml) in combination with anti-IL-4 (1 μg/ml) and cultured for 48 h. (E) Western blots from junD+/+ and Ubi-junDm CD4+ T cells stimulated for 48 h with ConA, anti-CD3/anti-CD28 or anti-CD3/anti-CD28/IL-4. Blots were probed with an anti-JunD antibody and reprobed with the actin antibody as loading control. Both wild-type JunD isoforms are recognized by the antibody (double arrow).
Further analysis on JunD expression during lymphocyte proliferation showed that endogenous and exogenous JunD levels remained at constant levels upon ConA and anti-CD3 and anti-CD28 stimulation (Figure 4E, upper and middle panel), whereas addition of IL-4 significantly induced exogenous and endogenous JunD levels (Figure 4E, lower panel).
Altered Th-cell differentiation and cytokine expression in JunD-modified T cells
Effector T cell populations from wild-type and JunD-modified CD4+ T cells were generated applying conditions that induce either a Th1 or Th2 phenotype. Upon restimulation, cytokine production was assayed by intracellular cytokine (ICC) staining and ELISA. Transgenic CD4+ T cells gave rise to significantly fewer IL-4-producing Th2 cells, as measured by ICC staining (Figure 5A). ELISA, semiquantitative and quantitative RT–PCR analyses further showed a reduction of IL-4 and IL-10 expression in Th2 cells from the Ubi-junDm CD4+ or Thy1.2+ T cell population (Figures 5A and B). When junD−/− CD4+ T cells were analyzed for differentiation of T cells into Th2 type, ICC analysis showed a marked increase in the number of IL-4-producing cells (Figure 5C). An increase in IL-4, and also IL-10 production, was further observed by ELISA analyses (Figure 5C). While expression levels of IL-4 were similar in junD−/− Th2 polarized CD4+ cells, a strong increase in IL-10 expression was found (Figure 5D). Induction of IL-10 mRNA expression under Th1-polarizing conditions was also seen, but to a slightly lesser extent (data not shown). The unaffected IL-4 mRNA expression, yet increased secretion might be explained by post-transcriptional regulation of IL-4 in junD−/− Th2 cells. Thus, under these conditions, JunD regulates Th2 cytokine production and Th cell differentiation.
Figure 5.
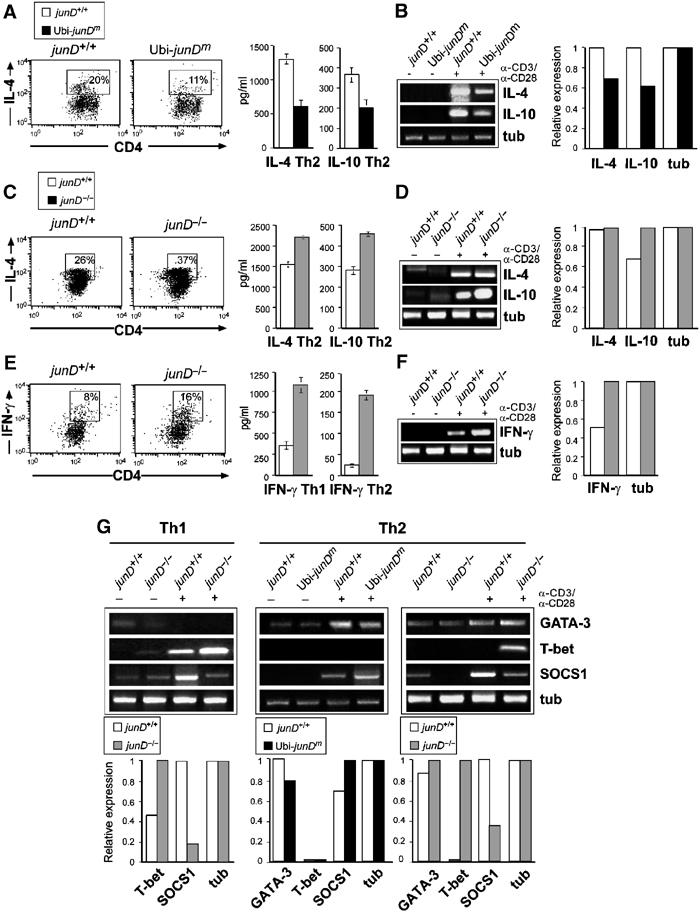
JunD and regulation of Th differentiation. (A) CD4+ T cells from junD+/+ and Ubi-junDm mice were cultured under Th2 conditions for 5 days. Intracellular staining for IL-4 was performed and the cells were analyzed by FACS (left panel). Cytokine levels were determined by ELISA (Thy1.2+ for IL-4; right panel). (B) RNA expression levels were analyzed by semiquantitative (left panel) and real-time (right panel) PCR. (C, D) CD4+ junD+/+ or junD−/− T cells were induced to differentiate to the Th2 subsets for 5 days. Intracellular staining for IL-4, IL-4 and IL-10 cytokine levels measured by ELISA (Thy1.2+ T cells for IL-4) and semiquantitative and quantitative RT–PCR expression analysis are shown. (E, F) CD4+ junD+/+ or junD−/− T cells were cultured under Th1 conditions and after differentiation, the intracellular IFN-γ concentration was measured. IFN-γ concentration in the supernatants of either Th1 or Th2 cells was determined by ELISA and RT–PCR. (G) CD4+ junD+/+, Ubi-junDm or junD−/− CD4+ T cells were induced to differentiate either to the Th1 or the Th2 subsets for 5 days. RNA expression of the indicated genes was analyzed by semiquantitative and quantitative RT–PCR.
To examine whether JunD is also involved in Th1 lineage progression, wild-type and junD−/− CD4+ T cells were cultured under Th1-polarizing conditions. ICC analysis revealed a significant increase in Th1 IFN-γ-producing cells generated from junD−/− CD4+ T cell population (Figure 5E). Moreover, JunD-deficient cells cultured under both Th-polarizing conditions secreted increased levels of IFN-γ (Figure 5F). The expression of IFN-γ mRNA was also found to be increased in both junD−/− Th1 and Th2 cells (Figure 5F and data not shown). Thus, JunD not only controls the expression of Th2-specific cytokines but also affects Th1 helper cell differentiation.
To further study the possible mechanism of how JunD regulates Th differentiation, the expression of additional regulators was analyzed. The expression of GATA-3, a potent transcription factor promoting Th cells into Th2 lineage differentiation (Zhang et al, 1997, 1998; Zheng and Flavell, 1997), was slightly affected in Ubi-junDm or junD−/− Th2 cells (Figure 5G). However, the expression of T-bet, an important regulator of the Th1 lineage controlling IFN-γ production (Szabo et al, 2000, 2002), was significantly increased in junD−/− CD4+ T cells, and a specific induction of T-bet expression was also seen in the junD−/− Th2 population (Figure 5G). Taken together, JunD appears to be involved in regulating the expression of key molecules implicated in the control of Th cell differentiation.
SOCS1 expression is modulated by JunD in vitro and in vivo
It was reported that JunD is a potent protector against numerous apoptotic insults, for example, TNFα-mediated apoptosis in LPS-induced hepatitis (Weitzman et al, 2000). LPS rapidly induces SOCS1 expression, which in an autoregulatory manner negatively regulates LPS signaling (Krebs and Hilton, 2001; Kinjyo et al, 2002; Nakagawa et al, 2002) as well as cytokine signaling, including IL-4 and IFN-γ (Alexander, 2002; Fujimoto et al, 2002). As elevated levels of IFN-γ and IL-4 were observed in junD−/− CD4+ polarized T cells, the expression level of SOCS1 during Th differentiation was analyzed next. When compared to wild-type Th cells, SOCS1 expression was drastically reduced in the junD−/−Th1 and Th2 cell populations (Figure 5G). Conversely, SOCS1 expression was found to be increased in Ubi-junDm transgenic CD4+ polarized Th2 cells (Figure 5G).
To investigate whether JunD can regulate SOCS1 expression in vivo, Ubi-junDm transgenic and junD−/− mice were challenged intraperitoneally with LPS and after 6 h SOCS1 expression was examined. RNA analyses revealed a slightly increased SOCS1 expression in Ubi-junDm splenocytes, whereas induction of SOCS1 expression was abolished in cells from junD−/− mice (Figure 6A). The toxic effect of LPS is believed to be mediated in part by TNF-α expression (Beutler et al, 1985; Pfeffer et al, 1993); therefore expression of TNF-α was measured by RNA analyses of total spleen and in serum after LPS administration. Whereas serum TNF-α levels were found to be decreased in Ubi-junDm transgenic mice, the production of TNF-α was comparable in wild-type and junD−/− mice after LPS challenge (Figure 6B), although a slight increase in TNF-α mRNA expression was found in the junD−/− splenocytes (Figure 6A). To test whether Ubi-junDm transgenic mice are more resistant to LPS treatment than controls, mice were treated with LPS at a dose that killed control mice after 48 h. Ubi-junDm transgenic mice were found to be more resistant to LPS treatment than controls (Figure 6C), as wild-type animals died approximately 40 h earlier than the first Ubi-junDm transgenic mice. These data demonstrate that JunD does affect SOCS1/TNF-α expression in vivo, underscoring the importance of JunD in modulating LPS-induced endotoxicity.
Figure 6.
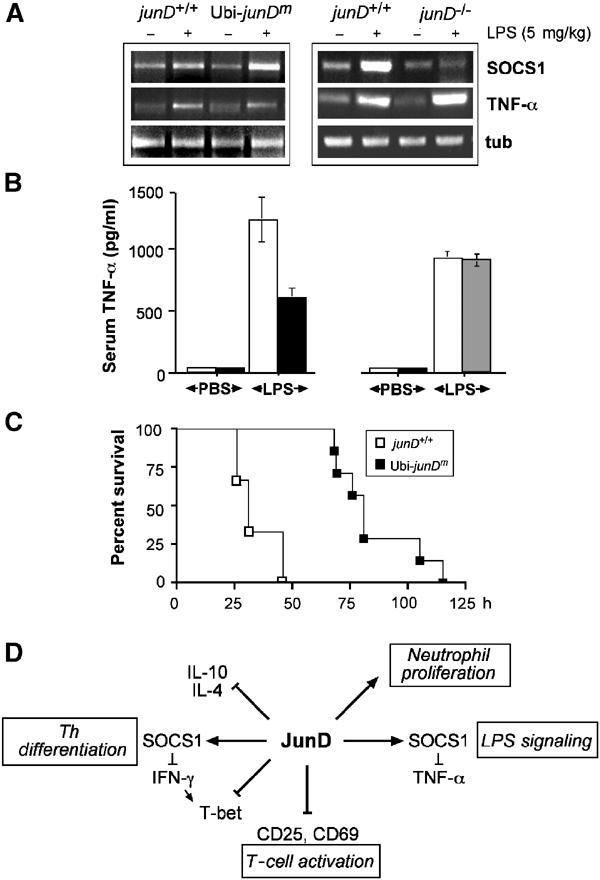
LPS-induced SOCS1 expression in Ubi-junDm and junD−/− mice. (A) Wild-type, Ubi-junDm (left panel) or junD−/− mice (right panel) were injected with 5 mg per kg body weight of E.coli LPS and total spleen was isolated 6 h later and analyzed for SOCS1 and TNF-α expression by RT–PCR. (B) Serum TNF-α levels from wild-type (n=5), Ubi-junDm (n=5) and junD−/− mice (n=5) were measured at 3 h after intraperitoneal injection of PBS or LPS (2.5 mg/kg). (C) Survival curve for wild-type (open squares) and Ubi-junDm mice (closed squares) following intraperitoneal injection of LPS (25 mg/kg). (D) Proposed molecular functions of JunD in T cell activation, Th differentiation, neutrophil proliferation and LPS signaling. We propose that JunD is involved in T cell activation through repression of CD25 and CD69. We also provide evidence that JunD negatively affects Th differentiation by controlling SOCS1, IFN-γ and T-bet expression and by preventing IL-4 and IL-10 expression. Finally, JunD appears to modulate the sensitivity to TNF-α via SOCS1.
Discussion
JunD is the most abundantly expressed Jun family member, which was proposed to act as a ‘protector' for cells from stress-induced apoptosis and negative regulator of cell proliferation. Here we show that JunD modified mice, either transgenic or knock-out, exhibit profound defects in the lymphoid system (Figure 6D). In transgenic mice, lymphocyte numbers are diminished at the expense of increased neutrophils and the animals suffer from spontaneous infections. Whereas Ubi-junDm transgenic B and T lymphoid cells respond normally to death-inducing signals, these cells exhibit a severe proliferation defect. Conversely, lymphocyte proliferation was enhanced in the absence of JunD.
As cells and mice lacking JunD are sensitive to p53-dependent stress and TNF-α-induced apoptosis, JunD has been postulated to function as an antiapoptotic gene (Weitzman et al, 2000). Increased antiapoptotic activity has also been linked to a collaboration of the JNK/JunD pathway with NF-κB (Lamb et al, 2003). Moreover, a protective role of JunD was also shown to involve paracrine factors using a chronic kidney disease mouse model (Pillebout et al, 2003). As treatment of thymocytes, B or T lymphocytes with death-promoting agents as well as activation-induced cell death revealed no differences in apoptosis between Ubi-junDm mice and control littermates, both in vitro and in vivo, JunD does not appear to be a ‘general' protector in all cell types. In addition, junD−/− thymocytes were not sensitive to anti-CD3-induced cell death in vivo, suggesting that JunD cannot protect lymphoid cells from apoptotic responses. Further support comes from experiments using retinal photoreceptors lacking JunD, which are also not sensitive to apoptosis (Hafezi et al, 1999).
A function of JunD as a negative regulator of T cell activation was revealed, as overexpression of JunD reduced proliferation, whereas the loss of JunD caused an acceleration of lymphoid proliferation. A similar inhibitory effect of JunD on proliferation was also observed in transgenic B lymphocytes (data not shown). JunD seems to regulate key molecules involved in T cell signaling and cell proliferation, as both IL-2Rα (CD25) and CD69 are less efficiently upregulated in transgenic T lymphocytes upon TCR-mediated stimulation. Conversely, increased IL-2Rα and CD69 expression was found in the absence of JunD, suggesting that both surface markers are regulated by JunD in T lymphocytes. Given normal CD25 expression and proliferation upon ConA stimulation in transgenic T cells, JunD seems to be critically involved in CD25-dependent T lymphocyte activation. Tyrosine phosphorylation and ERK activation were unchanged in Ubi-junDm T cells, indicating that ectopic JunD is apparently not affecting these signaling events (data not shown). However, the inhibitory effect of JunD on CD3/CD28-induced proliferation could be rescued by high doses of IL-4. This demonstrates that JunD regulates IL-4 expression in T cells directly or indirectly. JunD protein might bind directly to the IL-4 promoter and downregulate its activity, or alternatively, might control the expression of other transcriptional regulators of the IL-4 gene in T cells, such as GATA-3 or JunB. These observations support a role for JunD as a regulator of T cell activation rather than being a modulator of apoptosis for TCR-mediated signaling.
T helper cell differentiation into Th1 and Th2 subsets is an important process that determines the immune response against intra- or extracellular pathogens via cytokine secretion (Mosmann and Coffman, 1989; Paul and Seder, 1994). JunD promotes proliferation and differentiation of Th2 cells, as transgenic T cells showed a strong reduction in secretion of the Th2-cytokines IL-4 and IL-10. Conversely, in junD−/− T cells IL-4 and IL-10 secretion was elevated and therefore T cells are biased toward Th2 differentiation. The involvement of JunD in Th2 differentiation was further supported by the observation that Ubi-junDm transgenic mice displayed IgG1 subclass deficiency and that these mice were susceptible to infections. The second Ig subtype associated with the Th2 pathway, IgE, was not found to be changed in JunD transgenic mice.
The transcription factors c-maf and GATA-3 are critical for the commitment of Th2 lineage (Ho et al, 1996; Zheng and Flavell, 1997), whereas Th1 cell differentiation is dependent on T-bet expression (Szabo et al, 2000). Among the Jun-family members, JunB has been implicated in Th2 but not in Th1 differentiation (Rincon et al, 1997; Li et al, 1999; Hartenstein et al, 2002). JunB is selectively expressed by Th2 cells; it can activate IL-4 promoter activity, and JunB transgenic mice have increased IL-4 levels in Th1 cells (Li et al, 1999). c-Jun is also involved in IL-4 promoter activity, whereas even high amounts of JunD did not activate IL-4 promoter activity (Li et al, 1999); however, this does not exclude the possibility that JunD may repress IL-4 transcription in vivo. Itch, an E3 ubiquitin ligase, was found to ubiquitinate JunB and to a lesser extent c-Jun, but not JunD (Fang et al, 2002). As a consequence, Th2 cells of itch–/– mice exhibited enhanced levels of JunB, and IL-4 and IL-5 production was enhanced (Fang et al, 2002).
Recently, JunB was identified as a regulator of myeloid and B cell proliferation using Ubi-junB transgenic mice (Passegue et al, 2001; Szremska et al, 2003) as well as of bone cell differentiation (Kenner et al, 2004).
We propose that a major function of JunD together with JunB is to regulate IL-4 expression in Th2 differentiation. JunD negatively affects Th2 differentiation and proliferation by preventing IL-4 expression. In contrast, in the absence of JunD or when JunB is overexpressed, JunB induces IL-4 transcription and biases Th cells toward the Th2 lineage. Thus, the Th differentiation model provides an excellent example for the antagonistic effects of JunD and JunB.
In the absence of JunD, Th2 cells also produced high levels of the Th1-specific cytokine IFN-γ, which was also increased in polarized Th1 cells. Unchanged IFN-γ levels were observed in Thy1.2+ transgenic splenic T cells, which could be caused by the presence of both CD4+ and CD8+ cells. The enhanced levels of IFN-γ in the Th1 cells may explain the aberrant expression of T-bet in junD−/− Th1 and Th2 cells, as it has recently been shown that IFN-γ is a potent inducer of T-bet expression (Lighvani et al, 2001). The loss of a JunD-mediated T-bet repression could further explain the altered cytokine profile observed in junD−/− cells. As T-bet has been reported to induce IFN-γ expression and to repress IL-4 in fully differentiated Th2 cells, JunD might also serve as a regulator of T-bet gene expression. Defective cytokine expression of junD−/− mice might also be a consequence of insufficient SOCS1 expression. Increased IFN-γ expression accompanied by increased T-bet expression was also observed in JunB-deficient Th2 cells, but not in Th1 cells (Hartenstein et al, 2002), further supporting the antagonistic effects of JunD and JunB during Th differentiation.
The ability of JunD to control SOCS1 expression, a negative regulator of IFN-γ and TNF-α signaling, may also explain why junD−/− mice are sensitive to LPS-induced hepatitis (Weitzman et al, 2000). SOCS1−/− mice display increased sensitivity to IFN-γ and contain elevated serum levels of this cytokine (Alexander, 2002). As a result, SOCS1−/− mice are sensitive to LPS-induced endotoxin shock (Nakagawa et al, 2002). Proinflammatory cytokines, in particular, TNF-α secreted from macrophages and IFN-γ produced from T cells, play key roles in the pathogenesis of septic shock (Hack et al, 1997). Serum levels of TNF-α were reduced to half in LPS treated transgenic JunD mice, whereas comparable amounts were detected in junD−/− mice. Thus, the sensitivity to LPS-treatment in the absence of JunD (Weitzman et al, 2000) might not be due to increased TNF-α production, but is probably a result of defective IFN-γ and IL-4 signaling in T cells, which express receptors for both cytokines. It is further possible that different T cell, populations, such as natural killer T cells are critically involved, as these cells might receive simultaneous signals from both IL-4 and IFN-γ and are responsible for the sensitivity to LPS-induced endotoxin shock. As Ubi-junDm mice are also more resistant to LPS treatment probably due to less TNF-α production modulation of JunD levels might be a potential strategy for the treatment of Th-associated diseases including septic shock.
Materials and methods
Generation of Ubi-junDm transgenic mice
A 1.0-kb cDNA fragment containing the coding region of mouse junD was amplified by PCR using oligos with a 5′-SpeI and 3′-HindIII overhang and fused into a vector containing the human ubiquitin C promoter and an in-frame COOH-terminal myc-tag, amino acid sequence, EQKLISEEDLN, containing a variable linker of additional three amino acids (Schorpp et al, 1996; Togel et al, 1998). The transgene was released by digestion with NotI and SalI and microinjected into the pronuclei of one-cell embryos (C57BL/6 × CBA F1) using standard techniques. Transgenic progeny were identified by Southern blotting. Founder males were mated to C57BL/6 females to generate lines that were maintained as heterozygotes. Transgenic offspring were identified by PCR analysis of tail DNA using the following primers: JunDA, CTCAAGGACGAGCCGC; AM9, CCTGCAGGAATTCGATAAGCTTAAGTCA; and RTmyc, GGAAACACACACTCAACACGCAACCAAC. Amplified fragments of 617 bp (transgenic) and about 900 bp (wild type) were obtained. JunD−/− mice used in this study have been previously described (Thepot et al, 2000). All mice were housed under pathogen-free conditions and were handled according to the Austrian laws governing the use of animals for research.
Serum Ig measurements
The concentrations of the different immunoglobulin subclasses in mouse sera were measured from at least 8-week-old mice by ELISA according to the manufacturer's instructions (BD PharMingen). ELISA for IgE was performed using rat anti-mouse IgE (clone 23G3; Southern Biotechnology Associates) and biotinylated-anti-mouse Ig, followed by horseradish peroxidase-conjugated streptavidin (BD PharMingen).
Flow cytometry
Single-cell suspensions of thymi and spleens were prepared and 1 × 106 cells were stained in PBS/1% FCS with the respective antibodies for 30 min at 4°C. Monoclonal antibodies were FITC-conjugated (CD8, IgM, IgD, CD11b, Mac1), PE-conjugated (CD4, Thy-1.2, CD3, Gr-1, CD19) or APC-conjugated (TCR-β, CD45R (B220) CD44) from BD PharMingen. Expression of CD25 (IL-2Rα) and CD69 was determined by cell surface staining using PE-conjugated anti-CD25 or anti-CD69 antibody. Analyses were performed on a FACScalibur flow cytometer (Becton Dickinson) using CELL Quest™ software.
In vitro and in vivo apoptosis assays
Single-cell suspension of thymocytes and splenocytes were maintained at 37°C in an atmosphere at 5% CO2. A total of 2 × 106 cells/ml were treated with various apoptotic stimuli. Apoptosis was induced by the addition of various concentrations of anti-Fas antibody (Jo-2 clone; BD PharMingen), anti-CD3 antibody (clone 145-2C11; BD PharMingen), 10 ng/ml TNF-α (R&D Systems) or by UVC irradiation (40 J/m2) using a Stratagene crosslinker. Cells were harvested 24 h after treatment, and cell viability was determined by staining with PE-conjugated AnnexinV (BD PharMingen) and propidium iodide and analyzed with a FACScan™ cytometer as described above. For in vivo experiments, 6–12-week-old mice were injected i.p. with PBS or anti-CD3 (BD PharMingen) antibody (20 or 40 μg). After 48 h, thymi were removed and the viability of thymocytes was assessed as described before.
T lymphocyte proliferation
Spleens were removed from 6–8-week-old mice and single-cell suspensions were prepared. T cell isolation was performed by incubating splenocytes with anti-mouse Thy-1.2 or anti-CD4 microbeads (Miltenyi Biotec) and T cells were separated by using an autoMACS magnetic cell sorter (Miltenyi Biotec). Purified T cells were cultured at 0.5 × 105 cells per well in 100 μl medium, pre-coated with anti-CD3 antibody (clone 145-2C11; BD PharMingen), anti-CD28 (clone 37.51; BD PharMingen), concanavalin A (Sigma) or PMA (Sigma) plus Ca2+ ionophore A23617 (Sigma) as indicated in the figures. Cells were stimulated in quadruplets for 48 h and proliferation was assessed by the [3H]thymidine incorporation added (1 μCi/well) during the last 10 h of culture. Cells were collected using a cell harvester and [3H]thymidine incorporation was quantified by scintillation counting.
RT–PCR and Western analyses
Purified splenic T cells were incubated with or without the indicated mitogenic stimuli and cultured for the time points indicated. Total RNA was isolated using Trizol reagent (GIBCO-BRL); cDNA synthesis was performed as recommended by the Ready-To-Go™-You-Prime-First-Strand Beads Kit from Amersham Biosciences. PCR amplifications were performed under standard conditions. Primer sequences are available upon request. Expression of gene markers and controls were analyzed by real-time PCR using DNA Engine Opticon 2 Lightcycler™. Protein extracts and Western blot analyses were performed according to standard procedures using 20 μg of whole-cell extracts. The following antibodies were used in this study: anti-myc (a kind gift from F Propst, University of Vienna, Austria), anti-JunD antibody (a kind gift from D Lallemand) and anti-actin (A2066; Sigma). Proteins were visualized by ECL (Amersham Biosciences).
Cytokine measurements
Thy1.2+ T cells were cultivated for 24 h and culture supernatants were harvested to measure IL-2, IL-4, IL-10, or IFN-γ levels by ELISA (R&D Systems). For Th differentiation cytokine measurement, CD4+ T cells were cultivated under Th1- or Th2-differentiating conditions. After 4 days medium was changed. Cells were collected after 5 days, washed and 5 × 105 cells were restimulated with plate-bound anti-CD3/anti-CD28 in the absence of any additional cytokines. Supernatants were collected 24 h later and subjected to ELISA. For intracellular staining, CD4+ T cells were cultivated 5 days under Th1- or Th2-differentiating conditions, restimulated with anti-CD3/anti-CD28 and 0.01% Brefeldin A for 6 h. Cells were then further processed according to the manufacturer's protocol (BD PharMingen) and stained with PE-coupled anti-IL4 or APC-coupled anti-IFN-γ for 30 min. After washing, cells were analyzed by FACScalibur flow cytometer (Becton Dickinson) using CELL Quest™ software.
In vitro Th differentiation
For in vitro T cell differentiation assays, CD4+ T cells (1 × 106/ml) were cultured in Iscoves's modified Dulbecco's medium and stimulated with each 10 μg/ml plate-bound anti-CD3 and anti-CD28 in the presence of IL-12 (4 ng/ml; R&D Systems) for Th1 differentiation or 1000 U/ml IL-4 and anti-IL12 (5 ng/ml; R&D Systems) for Th2 differentiation.
LPS induction in vivo
Matched mice (8–12 weeks old) of each genotype were injected i.p. with the indicated amounts of LPS (Escherichia coli serotype 0111:B4, Sigma), and blood or spleens were collected at the indicated time. Animals were monitored for lethality every 12 h.
Acknowledgments
We thank C Theussl for DNA injections into fertilized eggs; S Jungwirth for maintaining the mouse colony; H Tkadletz for help with preparing the illustrations; and G Schaffner for DNA sequencing. We thank D Lallemand for providing us with anti-JunD antibody and J Weitzman for providing the junD−/− mice. We also thank L Bakiri, B Hartenstein, J Hess, L Klein, D Maurer, J Penninger, K Sabapathy, J Smolen and J Weitzman for critical reading of the manuscript and helpful discussions. The IMP is funded by Boehringer Ingelheim and this work was supported by the Austrian Industrial Research Promotion Fund.
References
- Agarwal S, Guru S, Heppner C, Erdos M, Collins R, Park S, Saggar S, Chandrasekharappa S, Collins F, Spiegel A, Marx S, Burns A (1999) Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 96: 143–152 [DOI] [PubMed] [Google Scholar]
- Alexander WS (2002) Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol 2: 410–416 [DOI] [PubMed] [Google Scholar]
- Beutler B, Milsark IW, Cerami AC (1985) Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229: 869–871 [DOI] [PubMed] [Google Scholar]
- Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A (2001) A critical role of NF-κB in GATA3 expression and TH2 differentiation. Nat Immunol 2: 45–50 [DOI] [PubMed] [Google Scholar]
- Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath I, Guertl B, Zenz R, Wagner EF, Zatloukal K (1999) Functions of c-Jun in liver and heart development. J Cell Biol 145: 1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R, Wagner EF (2003) AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer 3: 859–868 [DOI] [PubMed] [Google Scholar]
- Fang D, Elly C, Gao B, Fang N, Altman Y, Joaerizo C, Hunter T, Copeland N, Jenkins N, Liu YC (2002) Dysregulation of T lymphocyte function in itchy mice: a role for Itch in Th2 differentiation. Nat Immunol 3: 281–287 [DOI] [PubMed] [Google Scholar]
- Ferreira V, Sidenius N, Tarantino N, Hubert P, Chatenoud L, Blasi F, Korner M (1999) In vivo inhibition of NF-kappa B in T-lineage cells leads to a dramatic decrease in cell proliferation and cytokine production and to increased cell apoptosis in response to mitogenic stimuli, but not to abnormal thymopoiesis. J Immunol 162: 6442–6450 [PubMed] [Google Scholar]
- Fujimoto M, Tsutsui H, Yumikura-Futatsugi S, Ueda H, Xingshou O, Abe T, Kawase I, Nakanishi K, Kishimoto T, Naka T (2002) A regulatory role for suppressor of cytokine signaling-1 in T(h) polarization in vivo. Int Immunol 14: 1343–1350 [DOI] [PubMed] [Google Scholar]
- Glimcher LH, Murphy KM (2000) Lineage commitment in the immune system: the T helper lymphocytes grows up. Genes Dev 14: 1693–1711 [PubMed] [Google Scholar]
- Hack CE, Aarden LA, Thijs LG (1997) Role of cytokines in sepsis. Adv Immunol 66: 101–195 [DOI] [PubMed] [Google Scholar]
- Hafezi F, Grimm C, Wenzel A, Weitzman J, Abegg M, Yaniv M, Remé CR (1999) Retinal photoreceptors are apoptosis-competent in the absence of JunD/AP-1. Cell Death Differ 6: 934–936 [DOI] [PubMed] [Google Scholar]
- Hartenstein B, Teurich S, Hess J, Schenkel J, Schorpp-Kistner M, Angel P (2002) Th2 cell specific cytokine expression and allergen-induced airway inflammation depend on JunB. EMBO J 21: 6321–6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilberg F, Aguzzi A, Howells N, Wagner EF (1993) c-jun is essential for normal mouse development and hepatogenesis. Nature 365: 179–181 [DOI] [PubMed] [Google Scholar]
- Ho IC, Glimcher LH (2002) Transcription: tantalizing times for T cells. Cell 109: S109–S120 [DOI] [PubMed] [Google Scholar]
- Ho IC, Hodge MR, Rooney JW, Glimcher LH (1996) The protooncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell 85: 973–983 [DOI] [PubMed] [Google Scholar]
- Jochum W, Passegué E, Wagner EF (2001) AP-1 in mouse development and tumorigenesis. Oncogene 20: 2401–2412 [DOI] [PubMed] [Google Scholar]
- Johnson RS, van Lingen B, Papaioannou VE, Spiegelman BM (1993) A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev 7: 1309–1317 [DOI] [PubMed] [Google Scholar]
- Kenner L, Hoebertz A, Beil T, Keon N, Karreth F, Eferl R, Scheuch H, Szremska A, Amling M, Schorpp-Kistner M, Angel P, Wagnez EF (2004) Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J cell Biol 164: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Ho IC, Grusby MJ, Glimcher LH (1999) The transcription factor c-Maf controls the production of interleukin-4 but not other Th2 cytokines. Immunity 10: 745–751 [DOI] [PubMed] [Google Scholar]
- Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A (2002) SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity 17: 583–591 [DOI] [PubMed] [Google Scholar]
- Krebs DL, Hilton DJ (2001) SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19: 378–387 [DOI] [PubMed] [Google Scholar]
- Lamb JA, Ventura JJ, Hess P, Flavell RA, Davis RJ (2003) JunD mediates survival signaling by the JNK signal transduction pathway. Mol Cell 11: 1479–1489 [DOI] [PubMed] [Google Scholar]
- Li B, Tournier C, Davis RJ, Flavell RA (1999) Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J 18: 420–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O'Shea JJ (2001) T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci USA 98: 15137–15142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F, Garcia-Rodriguez C, Rao A (2000) Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J 19: 4783–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F, Lopez-Rodriguez C, Rao A (2001) Partners in transcription: NFAT and AP-1. Oncogene 20: 2476–2489 [DOI] [PubMed] [Google Scholar]
- Mechta-Grigoriou F, Gerald D, Yaniv M (2001) The mammalian Jun proteins: redundancy and specificity. Oncogene 20: 2378–2389 [DOI] [PubMed] [Google Scholar]
- Mosmann T, Coffman R (1989) TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7: 145–173 [DOI] [PubMed] [Google Scholar]
- Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, Kishimoto T (2002) SOCS-1 participates in negative regulation of LPS responses. Immunity 17: 677–687 [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, Gadina M, Schreiber RD (2002) Cytokine signaling in 2002: new suprises in the Jak/Stat pathway. Cell 109: S121–S131 [DOI] [PubMed] [Google Scholar]
- Passegue E, Jochum W, Schorpp-Kistner M, Mohle-Steinlein U, Wagner EF (2001) Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell 104: 21–32 [DOI] [PubMed] [Google Scholar]
- Paul W, Seder RA (1994) Lymphocyte responses and cytokines. Cell 76: 241–251 [DOI] [PubMed] [Google Scholar]
- Pfarr C, Mechta F, Spyrou G, Lallemand D, Carillo S, Yaniv M (1994) Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell 76: 747–760 [DOI] [PubMed] [Google Scholar]
- Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW (1993) Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 7: 457–467 [DOI] [PubMed] [Google Scholar]
- Pillebout E, Weitzman JB, Burtin M, Martino C, Federici P, Yaniv M, Friedlander G, Terzi F (2003) JunD protects against chronic kidney disease by regulating paracrine mitogens. J Clin Invest 112: 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon M, Derijard B, Chow CW, Davis RJ, Flavell RA (1997) Reprogramming the signalling requirement for AP-1 (activator protein-1) activation during differentiation of precursor CD4+ T-cells into effector Th1 and Th2 cells. Genes Funct 1: 51–68 [DOI] [PubMed] [Google Scholar]
- Rooney JW, Hoey T, Glimcher LH (1995a) Coordinate and cooperative roles for NF-AT and AP-1 in the regulation of the murine IL-4 gene. Immunity 2: 473–483 [DOI] [PubMed] [Google Scholar]
- Rooney JW, Sun YL, Glimcher LH, Hoey T (1995b) Novel NFAT sites that mediate activation of the interleukin-2 promoter in response to T-cell receptor stimulation. Mol Cell Biol 15: 6299–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp M, Jager R, Schellander K, Schenkel J, Wagner E, Weiher H, Angel P (1996) The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res 24: 1787–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp-Kistner M, Wang ZQ, Angel P, Wagner EF (1999) JunB is essential for mammalian placentation. EMBO J 18: 934–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4: E131–E136 [DOI] [PubMed] [Google Scholar]
- Shi Y, Bissonette RP, Parfrey N, Szalay M, Kubo RT, Green DR (1991) In vivo administration of mononuclear antibodies to the CD3T cell receptor complex induces cell death (apoptosis) in immature thymocytes. J Immunol 146: 3340–3346 [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100: 655–669 [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH (2002) Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8T cells. Science 295: 338–342 [DOI] [PubMed] [Google Scholar]
- Szremska AP, Kenner L, Weisz E, Ott RG, Passegue E, Artwohl M, Freissmuth M, Stoxreiter R, Theussl HC, Parzer SB, Moriggl R, Wagner EF, Sexl V (2003) JunB inhibits proliferation and transformation in B-lymphoid cells. Blood 102: 4159–4165 [DOI] [PubMed] [Google Scholar]
- Thepot D, Weitzman JB, Barra J, Segretain D, Sinnakre M, Babinet C, Yaniv M (2000) Targeted disruption of the murine junD gene results in multiple defects in male reproductive system. Development 127: 143–153 [DOI] [PubMed] [Google Scholar]
- Togel M, Wiche G, Propst F (1998) Novel features of the light chain of microtubule-associated protein MAP1B: microtubule stabilization, self interaction, actin filament binding, and regulation by the heavy chain. J Cell Biol 143: 695–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman JB, Fiette L, Matsuo K, Yaniv M (2000) JunD protects cells from p53-dependent senescence and apoptosis. Mol Cell 6: 1109–1119 [DOI] [PubMed] [Google Scholar]
- Wurster AL, Tanaka T, Grusby MJ (2000) The biology of Stat4 and Stat6. Oncogene 19: 2577–2584 [DOI] [PubMed] [Google Scholar]
- Zhang DH, Cohn L, Ray P, Bottomly K, Ray A (1997) Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th-2 specific expression of the interleukin-5 gene. J Biol Chem 272: 21597–21603 [DOI] [PubMed] [Google Scholar]
- Zhang DH, Yang L, Ray A (1998) Differential responsiveness of the IL-5 and IL-4 genes to transcription factor GATA-3. J Immunol 161: 3817–3821 [PubMed] [Google Scholar]
- Zhang DH, Yang L, Cohn L, Parkyn L, Homer R, Ray P, Ray A (1999) Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity 11: 473–482 [DOI] [PubMed] [Google Scholar]
- Zheng W, Flavell RA (1997) The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4T cells. Cell 89: 587–596 [DOI] [PubMed] [Google Scholar]