

Abstract
Multisubunit DNA-dependent RNA polymerases synthesize RNA molecules thousands of nucleotides long. The reiterative reaction of nucleotide condensation occurs at rates of tens of nucleotides per second, invariably linked to the translocation of the enzyme along the DNA template, or threading of the DNA and the nascent RNA molecule through the enzyme. Reiteration of the nucleotide addition/translocation cycle without dissociation from the DNA and RNA requires both iso- and metamorphic conformational flexibility of a magnitude substantial enough to accommodate the requisite molecular motions. Here we review some of the more recently acquired insights into the structural flexibility and morphic fluctuations of RNA polymerases and their mechanistic implications.
Introduction
The central enzyme of gene expression, DNA-dependent RNA polymerase (RNAP), is one of the most processive of cellular enzymes. It catalyzes thousands of nucleotide condensation reactions while translocating along the DNA-template without dissociating from its RNA product. This reiterative mode of synthesis and translocation requires a complex mechano-chemical process of repetitively making and breaking a complicated network of contacts between the amino acid residues of the RNAP, the DNA/RNA scaffold and the NTP substrates. Many molecular details of RNAP action emerged from the biochemical and structural analyses that identified the individual essential elements of the transcriptional complex, such as the RNA-DNA hybrid and the catalytic center with its invariable and fixed triad of aspartic acid residues and mobile trigger loop (TL) and bridge helix (BH) domains (Fig.1). A number of recent reviews (including but not limited to[1-7])that synthesize the available data into mechanistic and structural models of transcription by multi-subunit RNAPs (including an animated nucleotide addition cycle[8]) have elucidated the details of this process well beyond the simple textbook depictions of a monotonous copying of genetic information, punctuated by initiation and termination. Here we offer our perspective on the current understanding of RNAP as a metamorphic protein complex with highly mobile elements and on the importance of structural information to the development of mechanistic models of transcriptional elongation.
Figure 1.

Mobile structural elements in the vicinity of the active site (T. thermophilus elongation complex, 205J): trigger loop (cartoon blue), bridge helix (cartoon green), catalytic triad (sticks purple), catalytic Mg++ ions (spheres, red and orange), substrate NTP (sticks, orange).
RNAP adds nucleotides to the 3′-end of the growing RNA and translocates reiteratively, in single nucleotide steps. However, reverse translocation (also known as backtracking) by a distance of one or more nucleotides disrupts the configuration of the catalytic center, leading to a temporary (spontaneously resolved) halt of the RNAP, called “pausing”, or to a transition into an irreversible arrested state[9, 10]. The latter can be restored to functionality by the endonucleolytic cleavage of the RNA or by “pushing” the backtracked complex from behind[11, 12]. Non-backtracked paused complexes are also described for bacterial RNAPs, where addition of the incoming NTP is hindered due to isomerization of the active site into an inactive conformation. The prominence and the precise mechanistic details of such pausing are a subject of some controversy and speculation[13, 14], but at least two types of such pauses have been characterized biochemically – RNA hairpin-dependent [15] and sequence-specific[16].
Endonucleolytic cleavage reactions occur in the same catalytic center of RNAP that catalyzes nucleotide addition and its reverse reaction, pyrophosphorolysis. An ensemble of 5 or more subunits (α2ββ’ω in case of bacterial RNAP) consisting of the approximately 3000 amino acids that constitute this enzyme carry out these catalytic activities and the coordinated translocation of RNAP along the DNA template. The enormous size of RNAP renders futile any ad initio predictions about, and severely limits the utility of popular homology-based methods to understand its structure. Extensive homology exists among RNAPs from bacteria, archaea, and eukaryotes, and between nuclear and chloroplast enzymes, with conserved regions scattered throughout the several subunits (reviewed in[17]), but the complex, and not completely understood constraints placed upon protein evolution by its function, structure and folding often restrict the conclusions of such analyses to an inferred “importance” of a given conserved region, limiting inferences concerning function or mechanism. It was not until 1999 that the spatial arrangement of the multitude of amino acids comprising RNAP was revealed for the first time as a 3.3 Å resolution structure of the Thermus aquaticus core enzyme[17], followed by structures of the eukaryotic (Saccharomyces cerevisiae[18]) and archaeal (Sulfolobus solfataricus[19]) RNAPs at similar resolutions.
Atomic resolution structures of the core RNAPs from all three domains of life were soon complemented by X-ray structures of other functionally important states of these enzymes, such as the promoter-binding holoenzyme from Thermus thermophilus[20], reconstituted transcription elongation complexes[21-23], and RNAPs bound by small regulatory molecules, such as antibiotics [24-28]. The location of the catalytic center and the arrangement of the DNA template and RNA product, allowed the categorization of many of the amino acid substitutions that affect enzymatic activity into two classes, those positioned in proximity to the active site, where they could directly impact catalysis and translocation and those located further away, deemed to act allosterically. In retrospect, the notion of RNAP allostery could have focused studies of the mechanism and structure of RNAP on its requisite conformational mobility[29], but despite this and other evidence (such as reversible stalling of the elongation complex by the application of high pressure[30]) that suggested the importance of the structural flexibility and mobility of this enzyme, terms such as “allosteric effects”, “conformational change”, and “isomerization” remained largely convenient ad hoc explanations for otherwise unexplainable observations of mutational, kinetic and thermodynamic experiments[31].
“Bridge-helix”-based models of translocation
A comparative analysis of the bacterial and yeast RNAPs allowed Kornberg and co-workers to propose a model of the nucleotide addition-translocation cycle, centered around the structural oscillations of the BH[21]. The idea that the BH is structurally flexibility was based on observations of its two distinct conformations, the “bent” conformation, present the in T. aquaticus core structure[17], and the “straight” conformation observed in the yeast RNAP[18, 21]. The first BH-centric model proposed that “bending” of the BH induced translocation of RNAP along the nucleic acid scaffold by one nucleotide, whereas its subsequent relaxation would open the substrate-binding site for the incoming NTP[21]. The next significant attempt to consider the structural flexibility/mobility of RNAP in mechanistic studies of transcription was made by Goldfarb and colleagues. They proposed a so called “swing-gate” model of NTP entry into the RNAP active site in which the conformationally-flexible BH, a mostly α-helical region of the largest, β’ subunit located in the vicinity of the enzyme’s catalytic center, was assigned a central role [32]. Maps of RNA-RNAP chemical cross-links and structural studies appeared to indicate that oscillations between these two conformations correlate with lateral movements of the nascent RNA 3′ end during translocation, whereas structural modeling indicated that the “bent” conformation was incompatible with NTP binding in the active site[32]. These ideas were developed further as a “two-pawl ratchet” mechanism of transcript elongation in which RNAP oscillates between two functional states as a Brownian ratchet machine[33]. Presently, the idea of RNAP equilibrating between pre- and post-translocated states using only the energy of thermal (Brownian) molecular motion, which can be biased in favor of forward movement by the binding of the substrate NTP to the post-translocated state, is widely accepted, having received strong support from single-molecule analyses of transcription[34] and crystallographic studies[35, 36]. At the time of publication it was the first specific model of the mechano-chemical changes that accompany RNA synthesis that postulated alternate conformations of the coupled bridge helix and trigger loop domain as an explicit physical manifestation of a Brownian ratchet [33]. Flexibility of the BH was proposed to determine the rate and fidelity of NTP addition and the responsiveness of the elongation complex to pause and termination signals. The model was proposed to explain the effects of two dominant lethal amino acid substitutions in the adjacent TL that had drastically opposite effects on every elongation parameter indicated above. The amino acid substitutions also affected the contacts between the BH and 3′ terminus of RNA, consistent with their impact on the equilibrium between the “bent” and “straight” conformations of the BH [33].
“Trigger-loop”-based model of translocation
A number of “mutants” were created in E. coli RNAP in the BH and nearby TL, which appeared to be largely disordered or mobile in available RNAP structures, and their impact on enzyme activity was evaluated by in vitro transcriptional analyses[15, 23]. The data were interpreted as undermining the BH-centric models of transcription, and were used to question the reality of the instrumental “bent” conformation [15], resulting in the emergence of a new, alternate model centered on the function of the trigger loop. The TL was, for the first time, observed to adopt a new conformation, apparently dependent on the incoming NTP; in structures of both yeast and bacterial elongation complexes a part of it was observed to have transitioned from a disordered/mobile loop to a helical hairpin in close contact with the substrate NTP[23, 37]. The structural metamorphosis of the TL and its movement to and from the NTP insertion site also fit the Brownian ratchet formalism, albeit no connection of its transformations to the translocation process was evident in the structures of elongation complexes. The hypothetical conformation of the TL corresponding to the paused state[15] has not been observed in either backtracked[35] or non-backtracked paused complexes[38]). As the amount of available structural information and the scope of the “mutational” analyses continue to expand it seems prudent to remain cautions of the methodological disparities between the two types of data sets when attempting to integrate them into a single interpretive model.
Evaluation of the translocation models
Presently, the available high-resolution structures of the multi-subunit RNAPs have all been obtained by means of X-ray crystallography, which limits their use in the interpretation of biochemical data, especially in the interpretation of data obtained with “mutants”, i.e. amino acid substitutions within the enzyme, and their impact on its function. In case of eukaryotes, the available structures are those of the same enzyme that was used in most of the relevant biochemical studies involving “mutants”, i.e. the yeast S. cerevisiae RNAP. The sources of the molecules used to obtain the major structures of the bacterial enzyme are thermophiles of the genus Thermus[17, 20], which are only distantly related to the mesophilic enterobacteriumE. coli, which was the source of the enzyme used in the majority of the biochemical investigations (including single molecule studies[39], fast kinetic studies[40], and surface plasmon resonance studies[41]). This disparity is routinely disregarded during analyses, merely for the purpose of convenience, without any demonstration that such unqualified projection of the structural data onto the biochemical data and vice versa is justified.
X-ray crystallography is a powerful tool for obtaining information regarding the spatial arrangement of amino acids, metals, and substrates in a protein. With high enough resolution it also allows the inference of electrostatic, van der Waals, and hydrophobic interactions, and H-bonding. However, other important factors that affect the rigorous interpretation of the structural data, such as entropy, solvation, and conformational strain, are not directly reported by the crystallographic analysis, requiring their explicit determination by means of molecular dynamics or molecular mechanics [42]. Failure to consider the enzyme’s conformational mobility can introduce errors as large as 20 kcal/mol [42], which is often substantially larger than the predicted effects of amino acid substitutions. In their analysis of the functions of the BH and TL, Landick and colleagues estimated the effect of an Ala to Gly substitution in the “straight” BH to be 2 kcal/mol (based on the guest-host behavior of Ala-based isolated helices), and speculated that it would facilitate the adoption of the “bent” (sometimes referred to as “flipped-out”) conformation[15]. We used a molecular mechanics analysis [43] to predict the free energy changes associated with this substitution in the “straight” and “bent” conformations of the bridge helix of T. thermophilus RNAP and found that such a substitution would destabilize both conformations by 0.9 and 0.4 kcal/mol, respectively, and also cause the flipped-out region of the “bent” helix to contract by one residue in the most stable conformation (V.S. and E.N., unpublished observations). Both the free energy change and the structure resulting from this amino acid substitution differ from those inferred in the original analysis[15].
The mechanistic dominance of the TL refolding was illustrated by the severe defects exhibited by a double L930P,T931P mutant that was assumed to affect only the trigger loop refolding, but not its contacts [15]. However, our molecular mechanics analysis of the TL-BH group predicts that the effect of this double mutant would not be limited only to the folding efficiency; in addition to a free energy differential of ca.3 kcal/mol, these amino acid substitutions lead to a steric clash of the Arg1239 with the position of the incoming NTP, a 3.2 Å displacement of the His1242 residue implicated in the catalysis of NTP addition[37, 44], and a distortion of the bridge helix conformation relative to the “wild type” molecule (Fig. 2). Furthermore, a rigorous mechanistic interpretation of every given amino acid substitution requires not only an explicit treatment of the physical factors that affect protein structure and/or mobility in one of the functional states (such as RNAP holoenzyme or the post-translocated elongation complex, as above), but also a survey of all the distinct transcriptional states and the transitions between them. The most complete model of the nucleotide addition cycle proposed by Cramer and colleagues, postulates at least three different conformational states of the trigger loop, disordered (post-translocated), refolded into a trigger helical hairpin (NTP insertion), and “wedged” (during translocation), as well as a transient “flipped-out” conformation of the bridge helix during translocation [8]. Concerted action of the TL and BH is expected according to the assumption that the “wedging” conformation of the former promotes the “flipping-out” of the latter [8, 36], whereas similar, but not identical, conformations were reported recently by Kornberg and co-workers for a backtracked yeast RNAP elongation complex with a 12 nucleotide long RNA[35]. Backtracking over a longer distance appears to disfavor BH-TL interactions, when interactions of the BH with other elements of the elongation complex, such as RNA and a patch of amino acids from the second largest subunit, begin to dominate[35] (Fig. 3). An additional layer of complexity is added by the implications of the BH together with the new conformation of the fork loop 2 in resulting from the fraying of the 3′ end of the RNA in non-backtracked, non-translocated elongation complexes[38] and by the movement of the fork loop 1 in translocation[45]. Any given amino acid substitution may affect the structure, stability, mobility, and interaction network formed by the structural element to which it belongs.
Figure 2.

Molecular mechanics simulation of the L930P, T931P substitutions in the T. thermophilus elongation complex (205J): “wild type” (WT) TL (cartoon,teal), “PP” TL (cartoon, red), WT BH (cartoon, green), “PP” BH (cartoon, yellow), catalytic Mg++ ions (spheres, red and orange), substrate NTP (sticks, orange), Pro930&931(sticks, black), WT Arg1239 (sticks, teal), “PP” Arg1239 (sticks, red), WT His1242 (sticks, blue), “PP” His1242 (sticks, dark red).
Figure 3.

Interactions of the BH (cartoon, teal) in the backtracked yeast elongation complex (3GTG) with TL (cartoon, purple), RNA (cartoon, red), DNA (cartoon, green) and Rpb2 fragments (cartoon, sand).
Conformational plasticity of the active center
The conformational plasticity of the BH, TL and nearby structural elements of the RNAP, evident from crystallographic studies, is in good agreement with the anisotropic network model analyses of yeast and bacterial RNAPs [46]. According to these findings the locally flexible residues in both the yeast and bacterial enzymes cluster in the mobile clamp region and in the BH, with some high-scoring outliers in the trigger loop and nearby “switch” regions (Fig.4). Remarkably, the maximum flexibility in the BH of the yeast RNAP was mapped to the same residue, Val829, that was observed five years later to be the focal point of the BH “bend” in the backtracked elongation complex[35, 46] and the residue “wedged” by the TL in the α-amanitin inhibited RNAP[36](Fig. 5). The same study also suggested a mechanism for the allosteric effects of the σ subunit binding to the core enzyme that resulted in restricted mobility/deformability of the residues in and around the BH [46]. It is possible that other allosteric RNAP-binding transcriptional factors, such as the virulence regulator RfaH[47], can act in a similar fashion, by modulating the mobility of the flexible regions involved in catalysis and translocation (RfaH, in particular, even binds to a region of RNAP overlapping σ-binding site[48]). An allosteric pathway that implicates RNAP mobile structural elements was also proposed for the mechanism of factor-independent, intrinsic transcription termination[49].
Figure 4.

Location of the flexible residues within the T. thermophilus elongation complex (2O5J): RNAP (cartoon, grey), RNA (cartoon, chartreuse), DNA (cartoon, limegreen), BH (cartoon, teal), TL (cartoon, yellow), NTP (sticks, blue), catalytic Mg++ (spheres, green and blue), flexible residues (spheres, orange, except: within BH – red, Leu1086 (Val829 in yeast) – black, within TL – yellow).
Figure 5.
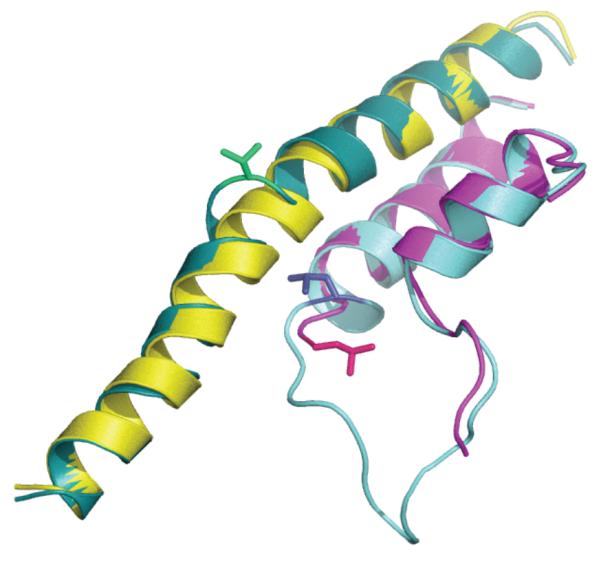
“Bent” (“looped-out”) conformation of the BH (cartoon, teal) in yeast backtracked (3GTG) vs. “straight” (cartoon, yellow) in the yeast pre-translocated (1Y77) elongation complexes. Also shown Val829 (sticks, green), ordered TL (cartoon, light blue) and L1081 (stick, dark blue) forming “wedge” to BH, disordered TL (cartoon, magenta) and L1081 (stick, hotpink) flipped away from the BH.
The flexibility (low deformation energies) of many structural elements within RNAP is necessary for reiterative NTP addition and translocation, and is dependent on low energy barriers between distinct conformations that can be interconverted with energy input largely from thermal molecular motions according to the Brownian ratchet model[8, 33, 34, 36]. Flexibility is also the basis of action of many transcription inhibitors, including antibiotics, which interfere with domain movements or force flexible/deformable structural elements to adopt non-productive conformations. The fungal toxin α-amanitin was proposed to inhibit enzymatic activity of its target, RNAP II, by interfering with the action of the TL during NTP addition/catalysis [50] and/or translocation[36]. The inhibitory effect of the antibiotic streptolydigin on transcription is also mediated through the TL, whose displaced and distorted conformation appears to be stabilized by binding to the drug[23]. Another inhibitor of bacterial RNAP, myxopyronin, interferes with DNA template melting by inducing refolding of another flexible element, the so called switch-2 segment, into a non-productive conformation[51]. An allosteric component was also proposed in the mechanism of inhibition caused by the rifamycin class of antibiotics , whose binding may lead to the loss of the catalytic magnesium ion from the active site[25]. A similar loss/displacement of this essential catalytic element[44, 52] was recently observed in the inhibited for NTP addition paused state of the yeast RNAP elongation complex[38].
Consideration of the conformational mobility/flexibility of transcriptional complexes became essential to understand the mechanistic aspects of transcription and to bridge the gap between the structural, biochemical and biophysical data. Further progress in the exploration of the catalytic and regulatory mechanisms of RNAP action depends critically on further structural studies of the many RNAP functional states and of different RNAPs (such as E. coli), as well as on further biochemical studies of RNAP for which extensive structural data are currently available, such as RNAP from T. thermophilus. Also of critical importance the development of simulations of the molecular mechanics/molecular dynamics of the transcription process, which these authors believe is the single, most substantial impediment to a rigorous interpretation of the vast amount of biochemical knowledge and its relation to the growing structural dataset.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kornberg RD. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A. 2007;104:12955–12961. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brueckner F, Armache KJ, Cheung A, Damsma GE, Kettenberger H, Lehmann E, Sydow J, Cramer P. Structure-function studies of the RNA polymerase II elongation complex. Acta Crystallogr D Biol Crystallogr. 2009;65:112–120. doi: 10.1107/S0907444908039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nudler E. RNA polymerase active center: the molecular engine of transcription. Annu Rev Biochem. 2009;78:335–361. doi: 10.1146/annurev.biochem.76.052705.164655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borukhov S, Nudler E. RNA polymerase: the vehicle of transcription. Trends Microbiol. 2008;16:126–134. doi: 10.1016/j.tim.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Landick R. The regulatory roles and mechanism of transcriptional pausing. Biochem Soc Trans. 2006;34:1062–1066. doi: 10.1042/BST0341062. [DOI] [PubMed] [Google Scholar]
- 6.Roberts JW, Shankar S, Filter JJ. RNA polymerase elongation factors. Annu Rev Microbiol. 2008;62:211–233. doi: 10.1146/annurev.micro.61.080706.093422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Werner F. Structural evolution of multisubunit RNA polymerases. Trends Microbiol. 2008;16:247–250. doi: 10.1016/j.tim.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Brueckner F, Ortiz J, Cramer P. A movie of the RNA polymerase nucleotide addition cycle. Curr Opin Struct Biol. 2009;19:294–299. doi: 10.1016/j.sbi.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- 10.Komissarova N, Kashlev M. Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3′ end of the RNA intact and extruded. Proc Natl Acad Sci U S A. 1997;94:1755–1760. doi: 10.1073/pnas.94.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park J, Marr MT, Roberts JW. E. coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 12.Epshtein V, Nudler E. Cooperation between RNA polymerase molecules in transcription elongation. Science. 2003;300:801–805. doi: 10.1126/science.1083219. [DOI] [PubMed] [Google Scholar]
- 13.Landick R. Transcriptional pausing without backtracking. Proc Natl Acad Sci U S A. 2009;106:8797–8798. doi: 10.1073/pnas.0904373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Depken M, Galburt EA, Grill SW. The origin of short transcriptional pauses. Biophys J. 2009;96:2189–2193. doi: 10.1016/j.bpj.2008.12.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toulokhonov I, Zhang J, Palangat M, Landick R. A central role of the RNA polymerase trigger loop in active-site rearrangement during transcriptional pausing. Mol Cell. 2007;27:406–419. doi: 10.1016/j.molcel.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Kireeva ML, Kashlev M. Mechanism of sequence-specific pausing of bacterial RNA polymerase. Proc Natl Acad Sci U S A. 2009;106:8900–8905. doi: 10.1073/pnas.0900407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- 18.Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 19.Hirata A, Klein BJ, Murakami KS. The X-ray crystal structure of RNA polymerase from Archaea. Nature. 2008;451:851–854. doi: 10.1038/nature06530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- 21.Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 22.Kettenberger H, Armache K, Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell. 2004;16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 23.Vassylyev DG, Vassylyeva MN, Zhang J, Palangat M, Artsimovitch I, Landick R. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007;448:163–168. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- 24.Mariani R, Maffioli SI. Bacterial RNA polymerase inhibitors: an organized overview of their structure, derivatives, biological activity and current clinical development status. Curr Med Chem. 2009;16:430–454. doi: 10.2174/092986709787315559. [DOI] [PubMed] [Google Scholar]
- 25.Vassylyev DG, Svetlov V, Vassylyeva MN, Perederina A, Igarashi N, Matsugaki N, Wakatsuki S, Artsimovitch I. Structural basis for transcription inhibition by tagetitoxin. Nat Struct Mol Biol. 2005;12:1086–1093. doi: 10.1038/nsmb1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuske S, Sarafianos SG, Wang X, Hudson B, Sineva E, Mukhopadhyay J, Birktoft JJ, Leroy O, Ismail S, Clark ADJ, et al. Inhibition of bacterial RNA polymerase by streptolydigin: stabilization of a straight-bridge-helix active-center conformation. Cell. 2005;122:541–552. doi: 10.1016/j.cell.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Temiakov D, Zenkin N, Vassylyeva MN, Perederina A, Tahirov TH, Kashkina E, Savkina M, Zorov S, Nikiforov V, Igarashi N, et al. Structural basis of transcription inhibition by antibiotic streptolydigin. Mol Cell. 2005;19:655–666. doi: 10.1016/j.molcel.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 28.Artsimovitch I, Vassylyeva MN, Svetlov D, Svetlov V, Perederina A, Igarashi N, Matsugaki N, Wakatsuki S, Tahirov TH, Vassylyev DG. Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell. 2005;122:351–363. doi: 10.1016/j.cell.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 30.Erijman L, Clegg RM. Reversible stalling of transcription elongation complexes by high pressure. Biophys J. 1998;75:453–462. doi: 10.1016/S0006-3495(98)77533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erie DA. The many conformational states of RNA polymerase elongation complexes and their roles in the regulation of transcription. Biochim Biophys Acta. 2002;1577:224–239. doi: 10.1016/s0167-4781(02)00454-2. [DOI] [PubMed] [Google Scholar]
- 32.Epshtein V, Mustaev A, Markovtsov V, Bereshchenko O, Nikiforov V, Goldfarb A. Swing-gate model of nucleotide entry into the RNA polymerase active center. Mol Cell. 2002;10:623–634. doi: 10.1016/s1097-2765(02)00640-8. [DOI] [PubMed] [Google Scholar]
- 33.Bar-Nahum G, Epshtein V, Ruckenstein AE, Rafikov R, Mustaev A, Nudler E. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 34.Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438:460–465. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang D, Bushnell DA, Huang X, Westover KD, Levitt M, Kornberg RD. Structural basis of transcription: backtracked RNA polymerase II at 3.4 angstrom resolution. Science. 2009;324:1203–1206. doi: 10.1126/science.1168729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brueckner F, Cramer P. Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation. Nat Struct Mol Biol. 2008;15:811–818. doi: 10.1038/nsmb.1458. [DOI] [PubMed] [Google Scholar]
- 37.Wang D, Bushnell DA, Westover KD, Kaplan CD, Kornberg RD. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006;127:941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sydow JF, Brueckner F, Cheung ACM, Damsma GE, Dengl S, Lehmann E, Vassylyev D, Cramer P. Structural basis of transcription: mismatch-specific fidelity mechanisms and paused RNA polymerase II with frayed RNA. Mol Cell. 2009;34:710–721. doi: 10.1016/j.molcel.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Herbert KM, Greenleaf WJ, Block SM. Single-molecule studies of RNA polymerase: motoring along. Annu Rev Biochem. 2008;77:149–176. doi: 10.1146/annurev.biochem.77.073106.100741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kireeva M, Nedialkov YA, Gong XQ, Zhang C, Xiong Y, Moon W, Burton ZF, Kashlev M. Millisecond phase kinetic analysis of elongation catalyzed by human, yeast, and Escherichia coli RNA polymerase. Methods. 2009 doi: 10.1016/j.ymeth.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greive SJ, Weitzel SE, Goodarzi JP, Main LJ, Pasman Z, von Hippel PH. Monitoring RNA transcription in real time by using surface plasmon resonance. Proc Natl Acad Sci U S A. 2008;105:3315–3320. doi: 10.1073/pnas.0712074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mobley DL, Dill KA. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benedix A, Becker CM, de Groot BL, Caflisch A, Böckmann RA. Predicting free energy changes using structural ensembles. Nat Methods. 2009;6:3–4. doi: 10.1038/nmeth0109-3. [DOI] [PubMed] [Google Scholar]
- 44.Castro C, Smidansky ED, Arnold JJ, Maksimchuk KR, Moustafa I, Uchida A, Götte M, Konigsberg W, Cameron CE. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nat Struct Mol Biol. 2009;16:212–218. doi: 10.1038/nsmb.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suenaga A, Okimoto N, Futatsugi N, Hirano Y, Narumi T, Ohno Y, Yanai R, Hirokawa T, Ebisuzaki T, Konagaya A, et al. Structure and dynamics of RNA polymerase II elongation complex. Biochem Biophys Res Commun. 2006;343:90–98. doi: 10.1016/j.bbrc.2006.02.124. [DOI] [PubMed] [Google Scholar]
- 46.Yildirim Y, Doruker P. Collective motions of RNA polymerases. Analysis of core enzyme, elongation complex and holoenzyme. J Biomol Struct Dyn. 2004;22:267–280. doi: 10.1080/07391102.2004.10507000. [DOI] [PubMed] [Google Scholar]
- 47.Svetlov V, Belogurov GA, Shabrova E, Vassylyev DG, Artsimovitch I. Allosteric control of the RNA polymerase by the elongation factor RfaH. Nucleic Acids Res. 2007;35:5694–5705. doi: 10.1093/nar/gkm600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sevostyanova A, Svetlov V, Vassylyev DG, Artsimovitch I. The elongation factor RfaH and the initiation factor sigma bind to the same site on the transcription elongation complex. Proc Natl Acad Sci U S A. 2008;105:865–870. doi: 10.1073/pnas.0708432105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Epshtein V, Cardinale CJ, Ruckenstein AE, Borukhov S, Nudler E. An allosteric path to transcription termination. Mol Cell. 2007;28:991–1001. doi: 10.1016/j.molcel.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 50.Kaplan CD, Larsson K, Kornberg RD. The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin. Mol Cell. 2008;30:547–556. doi: 10.1016/j.molcel.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belogurov GA, Vassylyeva MN, Sevostyanova A, Appleman JR, Xiang AX, Lira R, Webber SE, Klyuyev S, Nudler E, Artsimovitch I, et al. Transcription inactivation through local refolding of the RNA polymerase structure. Nature. 2009;457:332–335. doi: 10.1038/nature07510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci U S A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]