

Abstract
A career in science is shaped by many factors, one of the most important being our tastes in research. These typically form early and are shaped by subsequent successes and failures. My tastes run to microscopes, chemistry, and spatial organization of cytoplasm. I will try to identify where they came from, how they shaped my career, and how they continue to evolve. My hope is to inspire young scientists to identify and celebrate their own unique tastes.
SCIENTIFIC TASTE AND THE ROLE OF EARLY INFLUENCES
What to work on? How to work on it? There are no right or wrong answers to these questions of taste, but our choices define our trajectories as scientists. Often, we make them early, without thinking, through the influence of opportunities and mentors who came our way as much by chance as choice. Only looking back do we realize their significance. My most important job in the past decade or two has been helping young scientists identify and develop their own tastes. Mine run to microscopes, chemistry, and spatial organization of cytoplasm. Where did they come from? In hindsight, I see the influence of an old brass microscope from the laboratory of my great-grandfather that I was given sometime around my seventh birthday, along with a Kipp's apparatus. The microscope introduced me to protozoa from the local pond, allowing me to see the insides of a living, moving cell. The Kipp's apparatus could produce spectacularly smelly or explosive gases, but was less used, because it required parental guidance. My love of chemistry, and its foundational role in my worldview, is better attributed to my high school chemistry teacher, Mr. Carleton. He provided my first glimpse of a rational underpinning for the natural world in the periodic table, and later explained how it arose from the Schrödinger equation. Neither of us understood the math, but what an inspiration to contemplate an abstract principle that explains reality! My interest in cytoplasm was kindled early, but it took the influence of my PhD mentor, Marc Kirschner, for me to develop a taste for interesting and tractable problems in how it is organized.
DISCOVERY OF DYNAMIC INSTABILITY
I was inspired to join Kirschner's lab at the University of California, San Francisco (UCSF), after hearing Kirschner give a series of lectures on space and time in biology. I felt then, and still do, that he aims at principles, although getting there entails a lot of wading through details. I chose to work on centrosomes, hoping they might be the brain of the cytoplasm, but our immediate goals were to purify them and figure out how they nucleate microtubules. This nucleation problem lies at the heart of cell organization and is still unsolved, although we probably do know the major protein players. I succeeded in purifying centrosomes, but the technologies then available were too insensitive to identify their components. Somewhat in desperation, I turned to study the assay I had been using, and ended up discovering dynamic instability, wherein individual microtubules exhibit large length fluctuations powered by GTP hydrolysis (Mitchison and Kirschner, 1984). This discovery defined my subsequent career and my taste in subsequent research. “You can always get a paper out of your assay” is something I tell students to this day. It came from analyzing individual microtubules, rather than average behavior, which was natural, given my taste for microscopy, but the key innovation was to freeze the tubulin in tiny aliquots. Every time I thawed one, it behaved the same way, so I could make measurements on multiple days and plot them on the same graph (Figure 1A). I think I learned that from Bruce Alberts. He favored 50% glycerol stocks at −20°C but drilled into me the idea that, if you are careful with your proteins, they will reward you with consistent and interesting behavior. Measuring individuals rather than averages turns out to be generally good taste. Systems biology, my current departmental affiliation, has prospered by quantifying the dynamics of individual cells, which is often more interesting than the population average.
FIGURE 1:
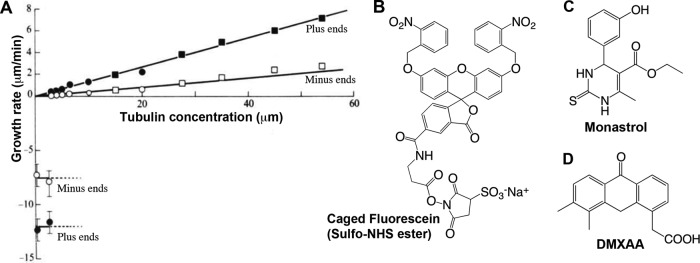
My taste in molecules. (A) The remarkable dynamics of tubulin. This graph shows microtubule growth rate as a function of soluble tubulin concentration. Microtubules shrink much faster than expected from extrapolating their growth rate to zero tubulin, because GTP hydrolysis destabilizes them. (Adapted from Mitchison and Kirschner, 1984.) (B) Caged fluorescein, used to measure microtubule sliding in mitotic spindles (Mitchison, 1989). The sulfo-NHS ester portion at the bottom is for labeling lysine residues. (C) Monastrol, the first small-molecule inhibitor of kinesin-5 (also known as Eg5, KSP, and Kif11; Mayer et al., 1999). (D) DMXAA, a drug that was effective for cancer treatment in mice but not humans. We and others recently found that it is a mouse STING agonist (Conlon et al., 2013; Kim et al., 2013). The normal function of STING is to activate an innate immune response to DNA or bacteria in the cytoplasm (reviewed in Paludan and Bowie, 2013).
Dynamic instability occurs when two chemicals, tubulin and GTP, self-organize—a complex process that is lifelike in the sense that it generates form and motion. I owe a huge debt to Marc Kirschner for pointing me at this problem and for his obsessive work on treadmilling theory with Terrell Hill, which set up a large applecart for me to overturn. I acknowledge the pioneering work of Marie-France Carlier on the biochemistry of GTP hydrolysis by tubulin, and her prior idea of a kinetic lag between polymerization and hydrolysis. The American Society for Cell Biology also plays an important role. I will never forget preparing for my first big talk at the 1983 meeting in San Antonio. Marc and I were still debating what to call it the night before. “Dynamic Instability” was his idea, and he prevailed, thank goodness! My idea was “Microtubule Jerking”—think on- and off-rate. Only later, when I became friends with Ted Salmon, did I realize to what an extent both dynamic instability and treadmilling were presaged by the work of Shinya Inoue and his students on mitotic spindle dynamics in living cells. I joined the Inoue School when I started working summers with Ted at the Marine Biological Laboratory at Woods Hole, next door to Inoue and his big polarization microscope. MBL has been an inspiring place to pursue cytoskeleton dynamics and an important relief from faculty tedium at Harvard Medical School (HMS).
CHEMISTRY FOR RESEARCH AND THERAPEUTICS
My taste for organic chemistry was subdued by boring university lectures but reawakened when I moved to the Medical Research Council at Mill Hill in London after my PhD. There, I tried to synthesize a photoactivated fluorescent probe for spindle dynamics, encouraged by David Trentham. I finished caged fluorescein (Figure 1B) after moving back to UCSF, and used it to prove that spindle microtubules slide poleward, with important implications for chromosome segregation mechanism (Mitchison, 1989). Synthesizing fluorescent probes is great fun, but chemistry applied to perturb biology is more important, especially since the introduction of green fluorescent protein. I became interested in drug development during a sabbatical visit to Stuart Schreiber's lab at Harvard in 1996, and moved to HMS to start the Institute of Chemistry and Cell Biology with him and Rebecca Ward in 1997. Back then, we had few tools for perturbing dynamical processes in human cells. Stuart and I imagined that cell-permeable small-molecule “tool compounds” could fill this void. We set out to develop lots of them, using combinatorial chemistry and high-throughput screening, imagining one for every protein. In retrospect, that was ludicrously overambitious, but we did generate some useful tools, notably the kinesin-5 inhibitor monastrol (Figure 1C) and the myosin II inhibitor blebbistatin. In the early 2000s, RNA interference (RNAi) emerged as a general tool for knocking out protein function. It does not replace what you can do with a good drug, but it weakened the rationale for large-scale tool compound development for basic research. Post-RNAi, and post my realization of how much effort is needed, I now feel that tool compound development is most justified when it will test a therapeutic hypothesis.
What is a therapeutic hypothesis, and how might you test one? These are application-oriented questions that I have come to find fascinating, although, like many of my contemporaries, I worry that the drive to “translate” basic discoveries into disease treatments is overwhelming the more important long-term mission of advancing fundamental knowledge. My emerging tastes are strongly influenced by a failed experiment. After monastrol, several companies made potent and specific kinesin-5 inhibitors in the hope of broad-spectrum anticancer drugs that would kill cancer cells by blocking mitosis but that would lack the neurotoxicity caused by microtubule-targeting drugs like Taxol. In hindsight, this was an invalid therapeutic hypothesis. As I discussed in a 2012 Molecular Biology of the Cell essay (Mitchison, 2012), mitosis-specific drugs have strong anti-proliferative action in the human body. They cause neutropenia and lack strong anticancer activity at the exposure limit, presumably because preneutrophils in the bone marrow divide much more frequently than cancer cells. Taxol was developed on the basis of an older and simpler therapeutic hypothesis: drugs that efficiently kill cancer cells in culture may work in man. Many important therapeutics were developed this way, but the hypothesis is far from universally valid—most poisons lack selectivity for killing cancer versus noncancer cells. We are now looking for mechanisms by which Taxol kills cancer cells in interphase that are either absent in standard tissue culture or masked by the high proliferation rate. We are also investigating mechanisms for converting tumor-resident leukocytes from pro- to anti-tumor phenotypes, following up on target ID of the failed cancer drug DMXAA (Figure 1D). Time will tell whether these choices of direction in the therapeutic arena are good taste.
FIND YOUR OWN TASTE!
A life in science is a long and interesting path, and our trajectories are determined as much by our tastes as by skill and luck. For those of you near the start of this path, I don't have advice for what kinds of questions you should ask or what techniques you should learn to answer them. I rather suggest you identify your own tastes, celebrate their uniqueness, and trust them to guide you.

T. J. Mitchison
Abbreviations used:
- HMS
Harvard Medical School
- MBL
Marine Biological Laboratory
- RNAi
RNA interference
- UCSF
University of California, San Francisco
Footnotes
Timothy J. Mitchison is the 2013 Porter Lecturer for the American Society for Cell Biology.
REFERENCES
- Conlon J, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Li L, Maliga Z, Yin Q, Wu H, Mitchison TJ. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem Biol. 2013;8:1396–1401. doi: 10.1021/cb400264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ. Polewards microtubule flux in the mitotic spindle: evidence from photoactivation of fluorescence. J Cell Biol. 1989;109:637–652. doi: 10.1083/jcb.109.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]