

This study establishes permeabilized cells as a viable system in which to elucidate mechanisms for Insig-mediated, sterol-induced ubiquitination and subsequent dislocation of HMG CoA reductase from endoplasmic reticulum membranes into the cytosol for degradation by 26S proteasomes.
Abstract
The polytopic endoplasmic reticulum (ER)–localized enzyme 3-hydroxy-3-methylglutaryl CoA reductase catalyzes a rate-limiting step in the synthesis of cholesterol and nonsterol isoprenoids. Excess sterols cause the reductase to bind to ER membrane proteins called Insig-1 and Insig-2, which are carriers for the ubiquitin ligases gp78 and Trc8. The resulting gp78/Trc8-mediated ubiquitination of reductase marks it for recognition by VCP/p97, an ATPase that mediates subsequent dislocation of reductase from ER membranes into the cytosol for proteasomal degradation. Here we report that in vitro additions of the oxysterol 25-hydroxycholesterol (25-HC), exogenous cytosol, and ATP trigger dislocation of ubiquitinated and full-length forms of reductase from membranes of permeabilized cells. In addition, the sterol-regulated reaction requires the action of Insigs, is stimulated by reagents that replace 25-HC in accelerating reductase degradation in intact cells, and is augmented by the nonsterol isoprenoid geranylgeraniol. Finally, pharmacologic inhibition of deubiquitinating enzymes markedly enhances sterol-dependent ubiquitination of reductase in membranes of permeabilized cells, leading to enhanced dislocation of the enzyme. Considered together, these results establish permeabilized cells as a viable system in which to elucidate mechanisms for postubiquitination steps in sterol-accelerated degradation of reductase.
INTRODUCTION
The enzyme 3-hydroxy-3-methylglutaryl CoA (HMG CoA) reductase is a polytopic glycoprotein that is integrated into membranes of the endoplasmic reticulum (ER) through a hydrophobic N-terminal domain consisting of eight membrane-spanning helices separated by short hydrophilic loops (Roitelman et al., 1992). The membrane domain of reductase precedes a large hydrophilic C-terminal domain that projects into the cytosol and catalyzes reduction of HMG CoA to mevalonate (Liscum et al., 1985). This reaction has long been recognized as a rate-limiting step in the synthesis of cholesterol, as well as of essential nonsterol isoprenoids, such as heme A, ubiquinone, dolichol, and the farnesyl and geranylgeranyl groups that are found attached to many cellular proteins (Brown and Goldstein, 1980). Sterol-accelerated ubiquitination and subsequent ER-associated degradation (ERAD) of reductase from membranes constitutes one mechanism for feedback control of reductase, which ensures a constant supply of nonsterol isoprenoids while avoiding overproduction of cholesterol (Goldstein and Brown, 1990; Goldstein et al., 2006). The ERAD of reductase is initiated by the sterol-induced binding of its membrane domain to ER membrane proteins called Insig-1 and Insig-2 (Sever et al., 2003b; Goldstein et al., 2006). Insigs in turn associate with two membrane-bound ubiquitin ligases called gp78 and Trc8 that facilitate ubiquitination of reductase on two cytosolic lysine residues in the enzyme's membrane domain (Jo et al., 2011). This ubiquitination marks reductase for recognition by the ATPases associated with diverse cellular activities (AAA)–ATPase VCP/p97 and its cofactors, which mediate dislocation of reductase into the cytosol, where it is degraded by 26S proteasomes (Hartman et al., 2010). The reaction appears to be augmented by the 20-carbon nonsterol isoprenoid geranylgeraniol (GGOH) through a mechanism that remains to be fully elucidated (Sever et al., 2003a).
Cytosolic dislocation is a key step in the ERAD of substrates that are not only entirely soluble within the lumen of the ER, but also integrated into ER membrane through one or more transmembrane helices (Brodsky and Skach, 2011). It is widely accepted that after their selection, soluble ERAD substrates are transported across the ER membrane into the cytosol through a protein-conducting channel formed by Sec61, a component of the translocation channel that imports nascent polypeptides into the ER, the polytopic Derlin proteins, or the membrane domain of ubiquitin ligases that initiate substrate ubiquitination (Lilley and Ploegh, 2004; Ye et al., 2004; Meusser et al., 2005; Brodsky and Skach, 2011). The ER-to-cytosol dislocation of membrane-bound ERAD substrates, especially of those with multiple membrane-spanning segments like reductase, is less well understood. Complete resolution of mechanisms for cytosolic dislocation of polytopic proteins requires reconstitution of the reaction in a cell-free system that is amenable to biochemical manipulation.
In the present study, we examine the dislocation of reductase in a permeabilized cell system. Our results show that intact, full-length, as well as ubiquitinated, forms of reductase become dislocated from membranes of permeabilized cells through an Insig-mediated reaction requiring the in vitro addition of the oxysterol 25-hydroxycholesterol (25-HC) and rat liver cytosol. Moreover, the reaction is stimulated by compounds that mimic 25-HC in accelerating reductase degradation in intact cells and is augmented by the nonsterol isoprenoid geranylgeraniol. These results establish permeabilized cells as a viable system for the elucidation of mechanisms that mediate postubiquitination steps in reductase ERAD.
RESULTS
In the experiment shown in Figure 1A, SV-589 cells were transfected with duplexes of small interfering RNA (siRNA) targeting a control mRNA encoding vesicular stomatitis virus glycoprotein (VSV-G), which is not expressed in the cells, or the VCP/p97 mRNA. After transfection, the cells were depleted of sterols and subsequently incubated for 0.5 or 4 h with various combinations of 25-HC plus mevalonate (to provide nonsterol isoprenoids) and the proteasome inhibitor MG-132. The cells were then harvested for the preparation of detergent lysates that were immunoprecipitated with polyclonal anti-reductase antibodies. The resulting immunoprecipitates were subjected to SDS–PAGE and blotted with anti-ubiquitin (Figure 1A, panels 1 and 4) or anti-reductase (panels 2 and 5) monoclonal antibodies. Treatment of the cells with 25-HC plus mevalonate for 0.5 h caused a slight decrease in the amount of reductase (Figure 1A, panel 2, compare lanes 1 and 2), indicating accelerated degradation of the enzyme. Degradation of reductase was blocked by treatment of the cells with MG-132 (lanes 3 and 4) or RNA interference (RNAi)–mediated knockdown of VCP/p97 (panels 2 and 3, lanes 5 and 6). Stabilization of reductase in sterol-depleted VCP/p97 knockdown cells (panel 2, compare lanes 1 and 5) was likely due to slowed basal degradation of the enzyme. The sterol-accelerated degradation of reductase was more pronounced after 4 h of treatment (panel 5, compare lanes 7 and 8) and was blocked by either MG-132 (lanes 9 and 10) or VCP/p97 knockdown (panels 5 and 6, lanes 11 and 12). Of note, siRNAs targeting different regions of the VCP/p97 mRNA similarly blunted sterol-accelerated reductase degradation (Supplemental Figure S1A).
FIGURE 1:
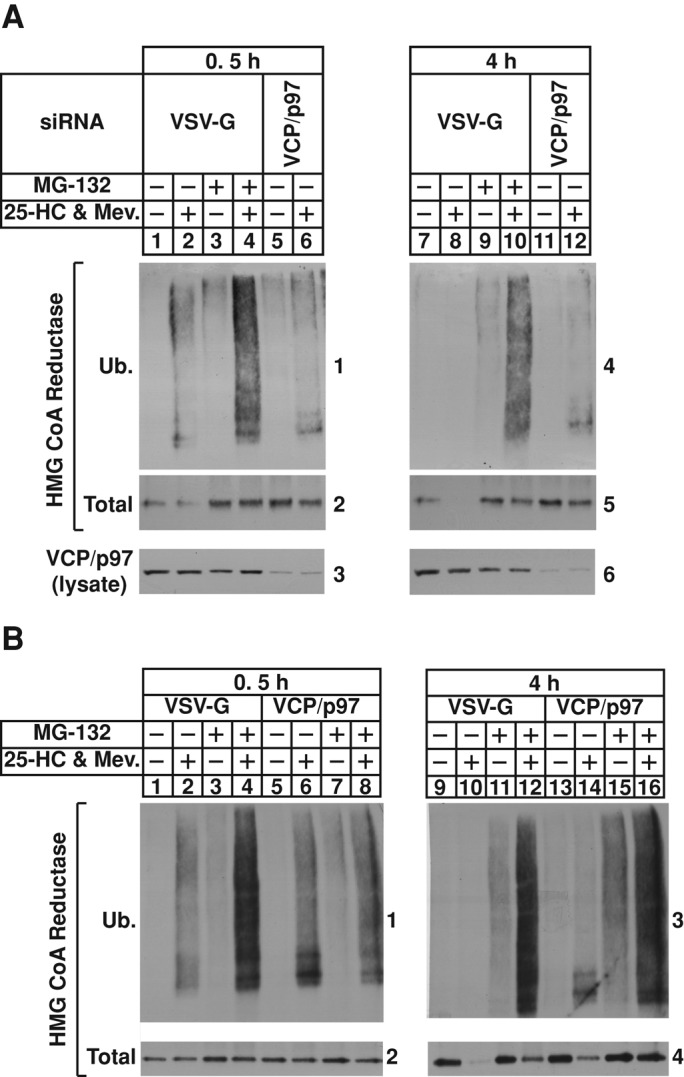
RNAi-mediated knockdown of VCP/p97 blocks sterol-accelerated degradation but not ubiquitination of HMG CoA reductase in SV-589 cells. SV-589 cells were set up on day 0 at 4 × 105 cells/100-mm dish in medium A supplemented with 10% FCS. On days 1 and 2, cells were transfected in medium A containing 10% FCS with siRNAs targeting the control mRNA, VSV-G, or the VCP/p97 mRNA as indicated and described in Materials and Methods. After the second transfection on day 2, the cells were depleted of sterols through incubation for 16 h at 37°C in medium A supplemented with 10% lipoprotein-deficient serum (LPDS), 10 μM sodium compactin, and 50 μM sodium mevalonate. The cells were subsequently treated with medium A containing 10% LPDS and 10 μM compactin in the absence or presence of 10 μM MG-132 as indicated for 0.5 h at 37°C. The cells were then treated for an additional 0.5 (A, lanes 1–6; B, lanes 1–8) or 4 h (A, lanes 7–12; B, lanes 9–16) in the absence or presence of 1 μg/ml 25-HC plus 10 mM mevalonate (Mev.). At the end of the incubations, the cells were harvested, lysed in detergent-containing buffer, and subjected to immunoprecipitation with polyclonal anti-reductase as described in Materials and Methods. Aliquots of immunoprecipitates and total lysates were subjected to SDS–PAGE, and immunoblot analysis was carried out with IgG-P4D1 (against ubiquitin), IgG-A9 (against reductase), or anti-VCP/p97 IgG. Numbers next to immunoblots are referred to as “panels” in the text.
After treatment for 0.5 h, 25-HC plus mevalonate caused reductase to become ubiquitinated, as indicated by the presence of high–molecular weight smears in the anti-ubiquitin immunoblots of the reductase immunoprecipitates (Figure 1A, panel 1, compare lanes 1 and 2). Treatment with MG-132 augmented sterol-induced ubiquitination of reductase (lane 4); the enzyme also became ubiquitinated in 25-HC plus mevalonate-treated VCP/p97-knockdown cells (lane 6). In the absence of MG-132, ubiquitinated forms of reductase were no longer detected in the anti-ubiquitin immunoblots of the reductase precipitates after 4 h of treatment with 25-HC plus mevalonate (Figure 1A, panel 4, lanes 7 and 8). This was likely due to accelerated degradation, as indicated by the presence of ubiquitinated reductase in sterol-treated cells that also received MG-132 (lanes 9 and 10) or subjected to VCP/p97 knockdown in the absence of MG-132 (lanes 11 and 12). Even though VCP/p97 knockdown stabilized reductase to a similar extent as that observed with MG-132 treatment, a similar buildup of ubiquitinated reductase was not observed. This may be due to proteasome-mediated degradation of ubiquitinated reductase owing to residual VCP/p97 in the knockdown cells. The repeat experiment of Figure 1B shows that after 4 h of treatment, ubiquitinated reductase accumulated in sterol-treated VCP/p97 knockdown cells but not in control-transfected cells (panel 3, compare lanes 10 and 14). However, treatment of the VCP/p97-knockdown cells with MG-132 enhanced the appearance of ubiquitinated reductase, indicating residual degradation (lane 16). Similar results were obtained in RNAi experiments conducted with Chinese hamster ovary cells (Supplemental Figure S2).
Sterol-induced dislocation of full-length reductase from ER membranes into the cytosol of intact cells has been reported (Leichner et al., 2009; Hartman et al., 2010). Regulated dislocation of reductase required its Insig-dependent ubiquitination, as well as the action of VCP/p97 (Hartman et al., 2010). The results of Figure 1 showing the accumulation of ubiquitinated reductase in sterol-treated VCP/p97-knockdown cells indicate that ubiquitinated forms of the enzyme are substrates for the ATPase. To examine this possibility, we began by initiating studies to reconstitute the reaction in a permeabilized cell system previously used to monitor sterol-induced ubiquitination of reductase (Song and DeBose-Boyd, 2004). SV-589 cells were depleted of sterols and subsequently harvested, washed, and permeabilized with buffer containing a low concentration of digitonin, an ATP-regenerating system, and protease inhibitors to prevent degradation of dislocated reductase. The samples were subjected to centrifugation to squeeze out the endogenous cytosol; pellets of permeabilized cells were resuspended in buffer containing the ATP-regenerating system and protease inhibitors, but no digitonin, and reisolated by centrifugation.
In the experiment of Figure 2A, permeabilized SV-589 cells were subjected to in vitro treatments with FLAG-tagged ubiquitin in the absence or presence of the ATP-regenerating system, rat liver cytosol, and 25-HC. After incubation at 37°C, the cells were homogenized in the absence of detergents and subjected to 100,000 × g centrifugation. The pellet and supernatant fractions of this spin were mixed with detergent-containing buffer and immunoprecipitated with anti-reductase polyclonal antibodies. The precipitated material from both fractions was then analyzed by immunoblot with anti-FLAG (Figure 2A, panels 1 and 3) or anti-reductase (panels 2 and 4) monoclonal antibodies. Incubation of permeabilized cells with 25-HC led to the ubiquitination of reductase in the 100,000 × g pellet fraction when reactions were also supplemented with rat liver cytosol and the ATP-regenerating system (Figure 2A, panel 1, compare lanes 2, 4, and 6 with lane 8). The amount of total reductase immunoprecipitated from the pellet fractions remained constant throughout the assay (panel 2, lanes 1–8). Immunoblot analysis of reductase immunoprecipitates from the 100,000 × g supernatant fractions revealed that ubiquitinated, as well as intact, full-length forms of reductase became dislocated from membranes of permeabilized cells but only when reactions were supplemented with 25-HC, the ATP-regenerating system, and rat liver cytosol (Figure 2A, panels 3 and 4, lane 8). Figure 2B shows that the amount of ubiquitinated reductase in the pellet fraction of 25-HC–treated permeabilized cells rose with time, reaching a plateau after 30 min (panel 1, lanes 3–10). Dislocation of ubiquitinated and full-length reductase was observed after 30 min of incubation with 25-HC (Figure 2B, panels 3 and 4, lane 6), and this reached a maximum after 90 min (panels 3 and 4, lane 10). Finally, reductase ubiquitination and dislocation in permeabilized cells was proportional to the amount of rat liver cytosol in reactions (Figure 2C, panels 1–4, lanes 8 and 10) and 25-HC (Figure 2D, panels 1–4, lanes 2–6). Note that Insig-1 appears to become dislocated into the cytosol along with reductase (Leichner et al., 2009). We were unable to detect Insig-1 in the supernatant fractions of permeabilized cells due to insufficient sensitivity of anti–Insig-1 antibodies. However, experiments of Supplemental Figure S3 show that permeabilized SV-589 cells support dislocation of overexpressed Insig-1 in a manner dependent on ATP, rat liver cytosol, and time of incubation.
FIGURE 2:

Sterol-induced dislocation of HMG CoA reductase from membranes of permeabilized SV-589 cells. SV-589 cells were set up for experiments on day 0 at 2 × 105 cells/100-mm dish in medium A supplemented with 10% FCS. On day 4, cells were washed with PBS and depleted of sterols through incubation in medium A containing 10% LPDS, 10 μM compactin, and 50 μM mevalonate for 16 h at 37°C. The sterol-depleted cells were subsequently harvested into the medium, washed with PBS containing 0.9 mM CaCl2, and permeabilized with 0.025% digitonin as described in Materials and Methods. (A) Permeabilized cells were resuspended in permeabilization buffer containing protease inhibitors (10 μM MG-132, 5 μg/ml pepstatin, and 2 μg/ml aprotinin) and 0.1 mg/ml FLAG-ubiquitin in the absence or presence of 10 μg/ml 25-HC, the ATP-regenerating system, and 2 mg/ml rat liver cytosol as indicated. After 75 min at 37°C, the reactions were terminated; the samples were homogenized in the absence of detergents, and resulting lysates were subjected to centrifugation at 100,000 × g for 30 min at 4°C. The pellet and supernatant fractions of this spin were then immunoprecipitated with polyclonal anti-HMG CoA reductase IgG as described in Materials and Methods. Aliquots of the immunoprecipitates were subjected to SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with IgG-A9 (against reductase) or IgG-M2 (against FLAG-ubiquitin). (B–D) Permeabilized cells were resuspended in permeabilization buffer containing protease inhibitors, an ATP-regenerating system, and 0.1 mg/ml FLAG-ubiquitin. (B) Reactions received 2 mg/ml rat liver cytosol and were incubated in the absence or presence of 10 μg/ml 25-HC at 37°C. After the indicated period of time, reactions were terminated; the samples were lysed and separated into pellet and supernatant fractions by 100,000 × g centrifugation, followed by immunoprecipitation and immunoblot analysis as in A. (C) Rat liver cytosol was added to reactions at concentrations ranging from 0.1 to 3 mg/ml and incubated in the absence or presence of 10 μg/ml 25-HC as indicated. After incubation at 37°C for 75 min, samples were fractionated and subjected to immunoprecipitation and immunoblot as in A. (D) Reactions were incubated with 2 mg/ml rat liver cytosol and the indicated concentration of 25-HC. After incubation at 37°C for 75 min, samples were fractionated and subjected to immunoprecipitation and immunoblot as in A.
RNAi was next used to determine the Insig requirement for sterol-induced dislocation of reductase from membranes of permeabilized cells. Supplemental Figures S4 and S5 show that multiple siRNAs targeting different regions of the Insig-1 and Insig-2 mRNAs appropriately blunted reductase degradation in SV-589 cells. Sterol-depleted cells transfected with siRNA duplexes targeting the control mRNA green fluorescent protein (GFP) or the Insig-1 and Insig-2 mRNAs were permeabilized and subjected to the dislocation assay. The pellet and supernatant fractions of these reactions were then analyzed by immunoprecipitation and immunoblot. As expected, treatment of permeabilized cells with 25-HC caused reductase to become ubiquitinated in membranes (Figure 3, panel 1, lane 2); the sterol also triggered dislocation of ubiquitinated and full-length reductase into the supernatant (panels 3 and 4, lane 6). The RNAi-mediated knockdown of Insig-1 and Insig-2 (Figure 3, panel 5, lane 10) blunted both the 25-HC–stimulated ubiquitination of reductase in membranes (panel 1, lane 4) and dislocation of ubiquitinated and full-length forms of the enzyme into the supernatant (panels 3 and 4, lane 8). Considered together, results of Figure 2 and 3 demonstrate that permeabilized SV-589 cells support in vitro ubiquitination and dislocation of intact, full-length reductase stimulated by 25-HC through a reaction requiring the presence of Insigs, rat liver cytosol, and an exogenous energy source.
FIGURE 3:
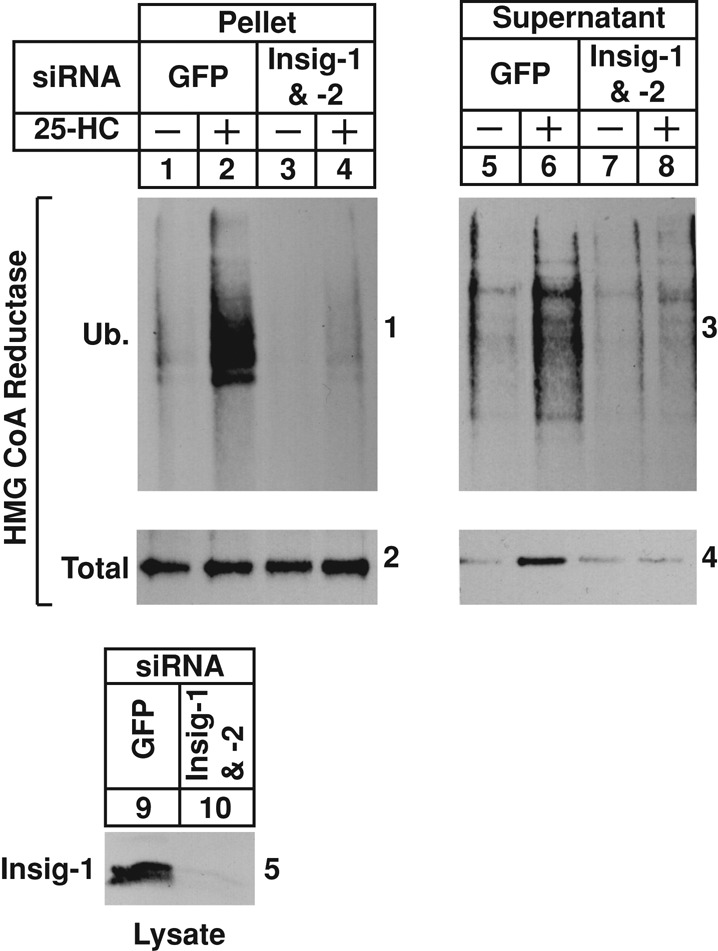
RNAi-mediated knockdown of Insigs blunts sterol-induced dislocation of HMG CoA reductase from membranes of permeabilized SV-589 cells. SV-589 cells were set up on day 0 at 1 × 105 cells/100-mm dish in medium A supplemented with 10% FCS. On day 3, the cells were transfected with siRNAs targeting the control mRNA, GFP, or mRNAs encoding Insig-1 and Insig-2 as indicated and depleted of sterols as described in the legend to Figure 1. After sterol depletion, the cells were harvested and permeabilized as described in the legend to Figure 2. Pellets of permeabilized cells were resuspended in permeabilization buffer containing protease inhibitors, the ATP regenerating system, 0.1 mg/ml FLAG-ubiquitin, and 2 mg/ml rat liver cytosol in the absence or presence of 10 μg/ml 25-HC. After incubation for 90 min at 37°C, reactions were terminated, and samples were subjected sequentially to fractionation and anti-reductase immunoprecipitation; the resulting pellet and supernatant fractions, along with aliquots of the cell lysates, were analyzed by immunoblot with IgG-A9 (against reductase), IgG-M2 (against FLAG-ubiquitin), and IgG-17H1 (against Insig-1).
The recombinant catalytic domain of ubiquitin specific protease 2 (USP2-cd), which efficiently removes ubiquitin from polyubiquitin chains attached to substrates (Ryu et al., 2006), was next used to demonstrate that sterols trigger dislocation of ubiquitinated reductase from membranes of permeabilized cells. In the experiment of Figure 4A, permeabilized cells were incubated with rat liver cytosol and ATP in the absence or presence of 25-HC to allow for the ubiquitination and subsequent dislocation of reductase. After 75 min, reactions were supplemented with various amounts of USP2-cd for an additional 15 min. The reactions were then terminated, and pellet and supernatant fractions of the permeabilized cells were analyzed sequentially by immunoprecipitation and immunoblot. The results show that, as expected, 25-HC stimulated ubiquitination of reductase in the pellet fraction of control reactions (Figure 4A, panel 1, compare lanes 1 and 2); this ubiquitination was reduced by USP2-cd in a concentration-dependent manner (lanes 4, 6, 8, and 10). Ubiquitinated forms of reductase appeared in the supernatant of 25-HC–treated permeabilized cells (panel 3, lane 12); this appearance was reduced by USP2-cd treatment (lanes 14, 16, 18, and 20). The amount of full-length reductase in the pellet fraction of permeabilized cells remained constant throughout the assay (Figure 4A, panel 2, lanes 1–10); however, treatment with USP2-cd before immunoprecipitation and immunoblot analysis markedly enhanced the amount of full-length reductase that was detected in the supernatant (panel 4, compare lane 12 with lanes 14, 16, 18, and 20).
FIGURE 4:

Sterols trigger the dislocation of ubiquitinated forms of HMG CoA reductase from membranes of permeabilized SV-589 cells. (A–C) SV-589 cells were set up on day 0, depleted of sterols on day 4, and permeabilized with 0.025% digitonin as described in the legend to Figure 2. Pellets of permeabilized cells were then resuspended in permeabilization buffer containing protease inhibitors, the ATP-regenerating system, 0.1 mg/ml FLAG-ubiquitin, and 2 mg/ml rat liver cytosol in the absence or presence of 10 μg/ml 25-HC as indicated. Some of the reactions in B and C also received the indicated amount of ubiquitin aldehyde. After incubation for 75 min at 37°C, the reactions were supplemented with either increasing amounts of recombinant USP2-cd (A) or a constant amount of the enzyme (1.7 μg; B and C) and incubated for an additional 15 min at 37°C. Reactions were then terminated, and the samples were fractionated and subjected to anti-reductase immunoprecipitation, followed by immunoblot analysis with IgG-A9 (against reductase) or IgG-M2 (against FLAG-ubiquitin).
The results of Figure 4A are consistent with a scenario in which removal of polyubiquitin chains from dislocated reductase enhanced detection of nonubiquitinated, full-length reductase in the supernatant of permeabilized cells. However, an alternative scenario in which removal of polyubiquitin from membrane-associated reductase stimulates its dislocation cannot be ruled out. To differentiate between these possibilities, we used ubiquitin aldehyde, a cell-impermeable reagent that inhibits deubiquitinating enzymes (Hershko and Rose, 1987). Inclusion of ubiquitin aldehyde in reactions of permeabilized cells augmented both sterol-induced ubiquitination of reductase in the pellet fraction (Figure 4B, panel 1, compare lane 2 with lanes 4 and 6) and sterol-induced dislocation of the ubiquitinated enzyme into the supernatant (panel 3, compare lane 2 with lanes 4 and 6). Full-length reductase appeared in the supernatant of 25-HC–treated, permeabilized cells (panel 4, lane 2), and this appearance was noticeably increased by treatment with USP2-cd after the ubiquitination/dislocation reaction (lane 8). In the presence of ubiquitin-aldehyde, high–molecular weight forms of reductase were observed in the anti-reductase immunoblot of supernatants from permeabilized cells subjected to treatment with 25-HC (panel 4, lanes 4 and 6). Figure 4C shows an experiment in which permeabilized cells were first treated with increasing amounts of ubiquitin aldehyde to enhance sterol-mediated ubiquitination of reductase. The reactions were then subjected to treatment with USP2-cd to collapse ubiquitinated forms of reductase that became dislocated into the supernatant during the initial treatment, thereby maximizing detection of full-length reductase in subsequent immunoblots. The results show that 25-HC–induced ubiquitination of reductase in the pellets of permeabilized cells was enhanced by incubation with ubiquitin aldehyde as expected (Figure 4C, panel 1, compare lane 2 with lanes 4, 6, and 8). The degree to which ubiquitin aldehyde enhanced ubiquitination of reductase was noticeably reduced compared with that observed in Figure 4B, which likely results from USP2-cd mediated removal of ubiquitin molecules that became attached to reductase. Ubiquitin aldehyde also enhanced the amount of ubiquitinated reductase that became dislocated into the supernatant of permeabilized cells (panel 3, compare lane 2 with lanes 4, 6, and 8). In reactions supplemented with USP2-cd but not ubiquitin aldehyde, full-length reductase appeared in the supernatant of 25-HC–treated permeabilized cells (Figure 4C, panel 4, lane 2); this appearance was augmented by 0.5 μg of ubiquitin aldehyde (lane 4). Supplementation of reactions with higher amounts (1 and 3 μg) of ubiquitin aldehyde in the presence of 25-HC resulted in the appearance of high–molecular weight forms of reductase (lanes 6 and 8), indicating that the deubiquitination inhibitor partially blocked USP2-cd activity under these conditions. Taken together, the results of Figure 4 argue that 25-HC triggers dislocation of polyubiquitinated forms of intact, full-length reductase in the permeabilized cell system.
In cultured cells, the 1,1-bisphosphonate esters Apomine and SR-12813 mimic 25-HC in stimulating Insig-mediated ubiquitination and subsequent degradation of reductase in intact cells (Roitelman et al., 2004; Sever et al., 2004). Having optimized the assay for sterol-induced dislocation of reductase in permeabilized cells using either ubiquitin aldehyde or USP2-cd (Figure 4), we next designed experiments to determine whether SR-12813 and Apomine trigger the reaction. The results show that, in a dose-dependent manner, both Apomine and SR-12813 stimulated ubiquitination of reductase in the pellet fraction of permeabilized cells (Figure 5, A and B, panel 1, compare lane a with lanes b–g). The bisphosphonate esters also triggered the dislocation of ubiquitinated and full-length forms of the enzyme into the supernatant fraction (Figure 5, A and B, panels 3 and 4, lanes b–g).
FIGURE 5:
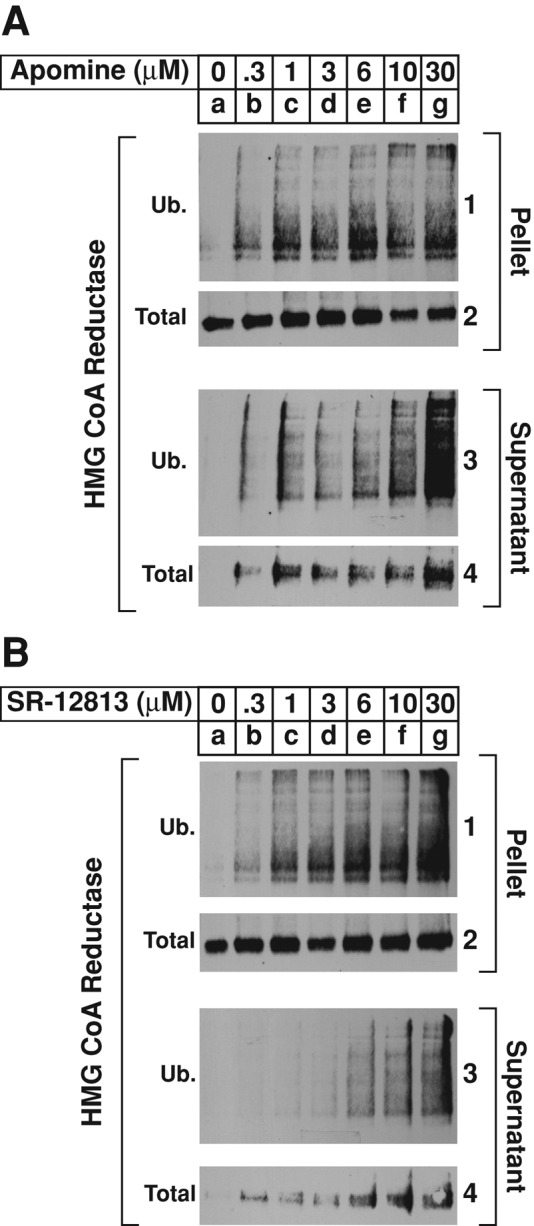
The 1,1-bisphosphonate esters Apomine and SR-12813 stimulate ubiquitination and dislocation of HMG CoA reductase from membranes of permeabilized SV-589 cells. SV-589 cells were set up on day 0, depleted of sterols on day 4, and permeabilized with 0.025% digitonin as described in the legend to Figure 2. Pellets of permeabilized cells were resuspended in permeabilization buffer containing protease inhibitors, the ATP-regenerating system, 0.1 mg/ml FLAG-ubiquitin, and 2 mg/ml rat liver cytosol. (A, B) Reactions were supplemented with the indicated concentration of Apomine (A) or SR-12813 (B). After incubation for 75 min at 37°C, 1.7 μg of USP2-cd was added, and reactions were incubated for an additional 15 min at 37°C. Reactions were then stopped, and samples were subjected sequentially to fractionation, immunoprecipitation, and immunoblot analysis with IgG-A9 (against reductase) or IgG-M2 (against FLAG-ubiquitin).
In intact cells, GGOH augments sterol-accelerated degradation and cytosolic dislocation of reductase (Sever et al., 2003a). The experiment shown in Figure 6A was designed to determine whether GGOH also enhanced 25-HC–induced dislocation of reductase in permeabilized cells. In the absence of GGOH, 25-HC stimulated the ubiquitination of reductase in the pellet fraction (Figure 6A, panel 1, lane b); this was marginally enhanced by the addition of increasing concentrations of GGOH (lanes d, f, h, and j). Treatment of the permeabilized cells with 25-HC also stimulated dislocation of ubiquitinated, as well as intact, full-length reductase into the supernatant (panels 3 and 4, lane b), and the reaction was augmented by GGOH in a dose-dependent manner (panels 3 and 4, lanes d, f, h, and j).
FIGURE 6:

The nonsterol isoprenoid geranylgeraniol augments sterol-induced dislocation of HMG CoA reductase from membranes of permeabilized SV-589 cells. (A–C) SV-589 cells were set up on day 0 and depleted of sterols on day 4 as described in the legend to Figure 2. (A) Sterol-depleted cells were harvested and permeabilized with 0.025% digitonin as described in the legend to Figure 2. The permeabilized cells were resuspended in permeabilization buffer containing protease inhibitors, the ATP regeneration system, 0.1 mg/ml FLAG-ubiquitin, and 2 mg/ml rat liver cytosol in the absence or presence of 10 μg/ml 25-HC and the indicated concentration of GGOH. After incubation for 75 min at 37°C, 1.7 μg of USP2-cd was added and reactions were incubated for an additional 15 min at 37°C. Reactions were then stopped, and samples were subjected sequentially to fractionation, immunoprecipitation, and immunoblot analysis with IgG-A9 (against reductase) or IgG-M2 (against FLAG-ubiquitin). (B, C) Sterol-depleted cells were pretreated for 1 h at 37°C in medium A containing 10% LPDS, 10 μM compactin, 50 μM mevalonate, and 10 μM MG-132; the cells were subsequently switched to identical medium in the absence or presence of 1 μg/ml 25-HC. After 2 h at 37°C, the cells were harvested and permeabilized with 0.025% digitonin as described in the legend to Figure 2. The permeabilized cells were resuspended in permeabilization buffer containing protease inhibitors, the ATP regeneration system, 2 mg/ml rat liver cytosol, and GGOH or FOH as indicated. The amount of GGOH used in C was 15 μM. After incubation for 75 min at 37°C, 1.7 μg of USP2-cd was added, and reactions were incubated for an additional 15 min at 37°C. Reactions were then stopped, and samples were subjected sequentially to fractionation, immunoprecipitation, and immunoblot analysis with IgG-A9 (against reductase) and IgG-P4D1 (against ubiquitin).
We previously postulated that in intact cells, GGOH augments a postubiquitination step in sterol-accelerated degradation of reductase (Sever et al., 2003a). This notion is further supported by results of the experiment shown in Supplemental Figure S1B, which shows that GGOH does not augment sterol-induced ubiquitination of reductase in control or VCP/p97-knockdown cells. To further explore GGOH-mediated regulation of reductase, we subjected intact, sterol-depleted cells to treatment with MG-132 in the absence or presence of 25-HC. The cells were then harvested, permeabilized, and incubated with ATP and rat liver cytosol in the absence or presence of GGOH. The results show that whereas in vitro treatment with GGOH had little, if any, effect on the 25-HC–mediated ubiquitination of reductase that occurred in intact cells (Figure 6B, panel 1, compare lane b with lanes d and f), the treatment significantly enhanced sterol-mediated dislocation of ubiquitinated, full-length reductase from membranes of the permeabilized cells (panels 3 and 4, compare lane b with lanes d and f). As a control, we treated permeabilized cells with the 15-carbon isoprenoid farnesol (FOH), which does not combine with sterols, to stimulate reductase degradation (Sever et al., 2003a). The results show that in vitro treatment of permeabilized cells with FOH neither enhanced 25-HC–mediated ubiquitination of reductase in membranes (Figure 6B, panel 1, compare lane b with lanes h and j) nor augmented its 25-HC–induced dislocation into supernatant of permeabilized cells (panels 3 and 4, compare lane b with lanes h and j). The experiment of Figure 6C shows that in the absence of rat liver cytosol, GGOH enhanced sterol-induced dislocation of reductase from membranes of permeabilized cells (panel 4, compare lanes 10 and 12). Dislocation of reductase was further enhanced by rat liver cytosol when added to reactions together with GGOH (panel 4, lane 16). Thus both membrane-associated and soluble factors contribute to this sterol-regulated reaction.
DISCUSSION
We previously established a permeabilized cell system to examine the sterol-induced ubiquitination of HMG CoA reductase (Song and DeBose-Boyd, 2004). These studies revealed that ubiquitination of reductase in permeabilized cells exhibited a strict dependence on the action of Insigs, and the reaction was stimulated by in vitro addition of sterols and rat liver cytosol, which provided a source of activated ubiquitin. The establishment of a cell-free system that precisely reconstitutes sterol-induced ubiquitination of reductase offers the opportunity to dissect subsequent steps in the enzyme's degradation. In the present report, we describe the analysis of sterol-induced membrane extraction and dislocation of reductase from membranes of permeabilized cells. Our initial characterization revealed that ubiquitinated, as well as intact, full-length forms of reductase became dislocated into the supernatant of permeabilized cells through a reaction that was stimulated by the oxysterol 25-HC, rat liver cytosol, and an energy source (Figure 2A). This dislocation occurred in a time-dependent manner (Figure 2B); the amount of dislocated reductase was proportional to the amount of rat liver cytosol (Figure 2C) or 25-HC (Figure 2D) used in the assay. Finally, sterol-induced dislocation of ubiquitinated and full-length reductase in permeabilized cells required the action of Insigs (Figure 3) and was enhanced by the 20-carbon nonsterol isoprenoid geranylgeraniol (Figure 6), which also enhances sterol-accelerated reductase dislocation and degradation in intact cells (Sever et al., 2003a; Hartman et al., 2010). Together these results indicate that sterol-induced dislocation of reductase in the permeabilized cell system is reflective of a physiologically relevant event.
The 1,1-bisphosphonate esters Apomine and SR-12813 mimic 25-HC in stimulating ubiquitination and subsequent degradation of reductase in intact cells (Roitelman et al., 2004; Sever et al., 2004). Both compounds activate farnesoid and pregnane X receptors (FXR and PXR, respectively; Jones et al., 2000; Niesor et al., 2001). Thus the possibility that the nuclear receptors modulate transcription of gene(s) required for reductase ubiquitination and degradation cannot be excluded. The experiments of Figure 5, A and B, show that supplementing reactions of permeabilized cells with rat liver cytosol and either Apomine or SR-12813 led to ubiquitination of reductase in the membrane pellet of permeabilized cells. In addition, the 1,1-bisphosphonate esters triggered dislocation of ubiquitinated and full-length forms of reductase into the supernatant. These findings argue that Apomine and SR-12813 stimulate reductase ubiquitination through direct interactions with the ER membrane rather than by modulating gene expression. An important goal for future studies will be to determine whether these interactions involve direct binding of Apomine and SR-12813 to reductase or an unknown factor that triggers association of reductase with Insigs, resulting in ubiquitination, cytosolic dislocation, and subsequent degradation of the enzyme.
In intact cells, the sterol-induced cytosolic dislocation of reductase requires ubiquitination of the protein on lysine residues in the membrane domain (Sever et al., 2003a). The reaction also requires the action of VCP/p97, a member of the hexameric AAA family of proteins that uses energy from ATP hydrolysis to drive membrane extraction and cytosolic dislocation of ubiquitinated substrates. This is consistent with the observation in Figure 1 that knockdown of VCP/p97 blocked sterol-accelerated degradation of reductase and caused ubiquitinated forms of the protein to accumulate. However, it is becoming appreciated that VCP/p97 also mediates reactions that modulate the ubiquitination status of ERAD substrates. For example, VCP/p97 associates with an E4 polyubiquitin chain elongation factor called UBE4B that extends polyubiquitin chains (Meyer et al., 2012). Conversely, the ATPase binds to several deubiquitinating enzymes, including VCIP135, Otu1, Yod1, and ataxin-3; the significance of these interactions is indicated by recent studies that suggest deubiquitination plays a key role in VCP/p97-mediated cytosolic dislocation of some ERAD substrates (Ernst et al., 2011; Claessen et al., 2012). The establishment of an in vitro assay for reductase dislocation offers the opportunity to use reagents that are not permeable to intact cells to determine whether ubiquitinated forms of the enzyme become extracted from ER membranes or whether the protein is deubiquitinated before dislocation.
Two lines of evidence suggest that ubiquitinated forms of reductase are substrates for VCP/p97-mediated dislocation. First, treatment of reactions with the catalytic domain of the ubiquitin-specific protease-2 (USP2-cd) enhanced the amount of intact, full-length reductase detected in the supernatant fraction of permeabilized cells (Figure 4A). Similar results have been obtained in studies reconstituting the cytosolic dislocation of the yeast reductase homologue Hmg2p (Garza et al., 2009), but the effects of sterols and other end products of mevalonate metabolism on the in vitro reaction were not assessed. Experiments using ubiquitin aldehyde, a cell-impermeable inhibitor of deubiquitinating enzymes (Hershko and Rose, 1987), provide the second line of evidence that ubiquitinated reductase becomes dislocated from membranes. Inclusion of ubiquitin aldehyde in reactions of permeabilized cells led to a dramatic increase in the amount of reductase that became ubiquitinated in the pellet fraction (Figure 4B). The treatment also led to the appearance of high–molecular weight forms of reductase in the supernatant fraction of permeabilized cells incubated in the presence of 25-HC. These high–molecular weight species are likely ubiquitinated forms of reductase, as indicated by their collapse upon treatment with USP2-cd (Figure 4C).
The successful development of a cell-free system that faithfully reconstitutes sterol-induced dislocation of reductase represents a significant technical advance in the elucidation of mechanisms for the enzyme's degradation. With this system in place, we are now poised to biochemically dissect postubiquitination steps of the pathway. For example, studies will be undertaken to determine how the nonsterol isoprenoid GGOH modulates reductase dislocation, using procedures and reagents that cannot be used in intact cells. The results of Figure 6C indicate that proteins present in rat liver cytosol combine with membrane-associated proteins to maximally stimulate dislocation of ubiquitinated reductase. Thus we are poised in future studies to use the permeabilized cell system to identify these cytosolic factors and determine the minimal requirements for reductase dislocation.
MATERIALS AND METHODS
Materials
We obtained MG-132 from BostonBiochem (Cambridge, MA) and Peptides International (Osaka, Japan); digitonin from Calbiochem (San Diego, CA); horseradish peroxidase–conjugated donkey anti-mouse (affinity-purified) from Jackson ImmunoResearch Laboratories (West Grove, PA); FLAG-ubiquitin, ubiquitin aldehyde, and recombinant human ubiquitin-specific protease catalytic domain (USP2-cd) from BostonBiochem (Cambridge, MA); and 25-hydroxycholesterol (25-HC) from Steraloids (Wilton, NH). SR-12813, FOH, and GGOH were purchased from Sigma-Aldrich (St. Louis, MO). Apomine was synthesized by the Core Medicinal Chemistry Laboratory in the Department of Biochemistry at University of Texas Southwestern Medical Center. Stock solutions of digitonin were prepared by dissolving 1 g of solid in 10 ml of boiling H2O. After cooling to room temperature, the solutions were filtered, aliquoted, and stored at −20°C until use. Lipoprotein-deficient serum (d > 1.215 g/ml) was prepared from newborn calf serum by ultracentrifugation as described previously (Goldstein et al., 1983). Rat liver cytosol was obtained from adult male Sprague–Dawley rats as previously described (Song and DeBose-Boyd, 2004). Other reagents were obtained from sources described previously (Sever et al., 2003b).
Cell culture
Stock cultures of SV-589 cells, an immortalized line of human fibroblasts expressing the SV40 large T antigen (Yamamoto et al., 1984), were maintained in medium A (DMEM containing 1000 mg glucose/l, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate) supplemented with 10% fetal calf serum (FCS) at 37°C in 5% CO2.
Dislocation of HMG CoA reductase in permeabilized SV-589 cells
The protocol used to analyze dislocation of reductase in permeabilized cells was adapted from procedures used to monitor sterol-induced ubiquitination of reductase in permeabilized cells (Song and DeBose-Boyd, 2004). The conditions of incubations before harvesting of cells are described in the figure legends. SV-589 cells were harvested into the medium by scraping and collected by centrifugation, after which pooled cell pellets from triplicate dishes were washed with phosphate-buffered saline (PBS) containing 0.9 mM CaCl2. Cells were then resuspended in 0.5 ml of prechilled permeabilization buffer (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]-KOH at pH 7.3, 115 mM potassium acetate, 5 mM sodium acetate, 2.5 mM MgCl2, and 0.5 mM sodium ethylene glycol tetraacetic acid [EGTA]) containing 0.025% (wt/vol) digitonin, an ATP-regenerating system (2 mM HEPES-KOH at pH 7.3, 1 mM magnesium acetate, 1 mM ATP, 30 mM creatine phosphate, and 0.05 mg/ml creatine kinase), and protease inhibitors (10 μM MG-132, 5 μg/ml pepstatin A, and 2 μg/ml aprotinin). After rotation for 10 min at 4°C, the cells were collected by centrifugation for 10 min at 4000 rpm at 4°C, resuspended in 0.5 ml of permeabilization buffer containing protease inhibitors, the ATP-regenerating system, but no digitonin, and subjected to a second round of centrifugation for 10 min at 4000 rpm at 4°C. The resulting pellets of permeabilized cells were then subjected to dislocation assays in a final volume of 0.3 ml of permeabilization buffer containing the ATP-regenerating system, 0.1 mg/ml FLAG-ubiquitin, and 0.1–3 mg/ml rat liver cytosol. 25-HC was added to reactions in a final concentration of 1% (vol/vol) ethanol. In typical experiments, reactions were carried out at 37°C for 60–90 min unless otherwise stated in the figure legends. Reactions were placed on ice and terminated by passing the suspension of permeabilized cells through a 22.5-gauge needle 20 times. The resulting lysates were diluted with 1 volume of permeabilization buffer and subjected to centrifugation at 100,000 × g for 30 min at 4°C. The supernatants of this spin were transferred to fresh tubes, and the volume was adjusted to 1 ml with immunoprecipitation buffer (PBS containing 1% NP-40, 1% sodium deoxycholate, 5 mM EDTA, 5 mM EGTA, 0.1 mM leupeptin, the protease inhibitor cocktail, and 10 mM N-ethylmaleimide). The 100,000 × g pellets were resuspended in immunoprecipitation buffer and passed through a 22.5-gauge needle 15 times, followed by rotation for 30 min at 4°C. The samples were then clarified by centrifugation at 20,000 × g for 10 min, and detergent-solubilized material was subjected to immunoprecipitation as described later.
RNA interference
Duplexes of siRNAs targeting human VCP/p97 (AAUAGAGUUGUUCGGAAUUU), Insig-1 (CCCACAAAUUUAAGAGAGAUU), Insig-2 (CUAAAGUGGAUUUCGAUAAUU and UGGCAAUGUACGAAUGUAAUU), and the irrelevant control genes, VSV-G (GGCUAUUCAAGCAGACGGUUU) and GFP (CAGCCACAACGUCUAUAUCUU), were synthesized by Dharmacon/Thermo Fisher Scientific (Lafayette, CO). RNAi experiments were carried out as described previously (Sever et al., 2003a; Hartman et al., 2010).
HMG CoA reductase immunoprecipitation and immunoblot analysis
Immunoprecipitation of HMG CoA reductase from detergent lysates of intact cells or supernatant and pellet fractions of permeabilized cells was carried out with polyclonal antibodies directed against the C-terminal domain of human reductase as previously described (Sever et al., 2003a). Aliquots of the immunoprecipitates were subjected to SDS–PAGE on 8 or 10% gels calibrated with prestained molecular mass markers (Bio-Rad, Hercules, CA), after which proteins were transferred to Hybond C-Extra filters (Amersham Biosciences, Piscataway, NJ) and subjected to immunoblot analysis.
Primary antibodies used for immunoblotting were as follows: immunoglobulin G (IgG)-A9 (IgG1), a mouse monoclonal antibody against the catalytic domain of hamster reductase (amino acids 450–887; Liscum et al., 1983); IgG-P4D1, a mouse monoclonal antibody against bovine ubiquitin (Santa Cruz Biotechnology, Dallas, TX); IgG-M2, a mouse monoclonal against the FLAG epitope (Sigma-Aldrich); monoclonal anti-VCP/p97 IgG (BD Transduction Laboratories, San Diego, CA); and IgG-17H1, a mouse monoclonal antibody against Insig-1 (Jo et al., 2011). Bound antibodies were visualized with peroxidase-conjugated, affinity-purified donkey anti-mouse IgG using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL) according to manufacturer's instructions. Filters were exposed to film at room temperature.
Supplementary Material
Acknowledgments
We thank Michael S. Brown and Joseph L. Goldstein for their continued encouragement and insightful advice. We also thank Ijeoma Onwuneme and Nimisha Jacob for help with tissue culture. This work was supported in whole or in part by National Institutes of Health Grants HL20948 and GM090216. R.A.D.-B. is an Early Career Scientist of the Howard Hughes Medical Institute.
Abbreviations used:
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- FCS
fetal calf serum
- FOH
farnesol
- GGOH
geranylgeraniol
- 25-HC
25-hydroxycholesterol
- HMG CoA
3-hydroxy-3-methylglutaryl CoA
- LPDS
lipoprotein-deficient serum
- RNAi
RNA interference
- siRNA
small interfering RNA
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-03-0157) on September 11, 2013.
REFERENCES
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Claessen JH, Mueller B, Sanyal S, Spooner E, van der Veen AG, Kirak O, Schlieker CD, Weihofen WA, Ploegh HL. Enzymatic blockade of the ubiquitin-proteasome pathway. PLoS Biol. 2011;8:e1000605. doi: 10.1371/journal.pbio.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Sato BK, Hampton RY. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Hartman IZ, Liu P, Zehmer JK, Luby-Phelps K, Jo Y, Anderson RG, DeBose-Boyd RA. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J Biol Chem. 2010;285:19288–19298. doi: 10.1074/jbc.M110.134213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Rose IA. Ubiquitin-aldehyde: a general inhibitor of ubiquitin-recycling processes. Proc Natl Acad Sci USA. 1987;84:1829–1833. doi: 10.1073/pnas.84.7.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y, Lee PC, Sguigna PV, DeBose-Boyd RA. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc Natl Acad Sci USA. 2011;108:20503–20508. doi: 10.1073/pnas.1112831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, et al. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- Leichner GS, Avner R, Harats D, Roitelman J. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell. 2009;20:3330–3341. doi: 10.1091/mbc.E08-09-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- Liscum L, Finer-Moore J, Stroud RM, Luskey KL, Brown MS, Goldstein JL. Domain structure of 3-hydroxy-3-methylglutaryl coenzyme A reductase, a glycoprotein of the endoplasmic reticulum. J Biol Chem. 1985;260:522–530. [PubMed] [Google Scholar]
- Liscum L, Luskey KL, Chin DJ, Ho YK, Goldstein JL, Brown MS. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and its mRNA in rat liver as studied with a monoclonal antibody and a cDNA probe. J Biol Chem. 1983;258:8450–8455. [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- Niesor EJ, Flach J, Lopes-Antoni I, Perez A, Bentzen CL. The nuclear receptors FXR and LXRalpha: potential targets for the development of drugs affecting lipid metabolism and neoplastic diseases. Curr Pharm Des. 2001;7:231–259. doi: 10.2174/1381612013398185. [DOI] [PubMed] [Google Scholar]
- Roitelman J, Masson D, Avner R, Ammon-Zufferey C, Perez A, Guyon-Gellin Y, Bentzen CL, Niesor EJ. Apomine, a novel hypocholesterolemic agent, accelerates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase and stimulates low density lipoprotein receptor activity. J Biol Chem. 2004;279:6465–6473. doi: 10.1074/jbc.M308094200. [DOI] [PubMed] [Google Scholar]
- Roitelman J, Olender EH, Bar-Nun S, Dunn WA, Jr, Simoni RD. Immunological evidence for eight spans in the membrane domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for enzyme degradation in the endoplasmic reticulum. J Cell Biol. 1992;117:959–973. doi: 10.1083/jcb.117.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KY, Baker RT, Kopito RR. Ubiquitin-specific protease 2 as a tool for quantification of total ubiquitin levels in biological specimens. Anal Biochem. 2006;353:153–155. doi: 10.1016/j.ab.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Sever N, Lee PC, Song BL, Rawson RB, Debose-Boyd RA. Isolation of mutant cells lacking Insig-1 through selection with SR-12813, an agent that stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2004;279:43136–43147. doi: 10.1074/jbc.M406406200. [DOI] [PubMed] [Google Scholar]
- Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003a;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell. 2003b;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- Song BL, DeBose-Boyd RA. Ubiquitination of 3-hydroxy-3-methylglutaryl-CoA reductase in permeabilized cells mediated by cytosolic E1 and a putative membrane-bound ubiquitin ligase. J Biol Chem. 2004;279:28798–28806. doi: 10.1074/jbc.M402442200. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russell DW. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.