

DNA damage and DNA replication checkpoints regulate differently the G1-to-S phase transcriptional program, resulting in the repression or induction, respectively, of the same set of genes. When this signaling is disrupted, cells are unable to cope with DNA-damaging agents, leading to increased cell lethality.
Abstract
In fission yeast cells, Cds1 is the effector kinase of the DNA replication checkpoint. We previously showed that when the DNA replication checkpoint is activated, the repressor Yox1 is phosphorylated and inactivated by Cds1, resulting in activation of MluI-binding factor (MBF)–dependent transcription. This is essential to reinitiate DNA synthesis and for correct G1-to-S transition. Here we show that Cdc10, which is an essential part of the MBF core, is the target of the DNA damage checkpoint. When fission yeast cells are treated with DNA-damaging agents, Chk1 is activated and phosphorylates Cdc10 at its carboxy-terminal domain. This modification is responsible for the repression of MBF-dependent transcription through induced release of MBF from chromatin. This inactivation of MBF is important for survival of cells challenged with DNA-damaging agents. Thus Yox1 and Cdc10 couple normal cell cycle regulation in unperturbed conditions and the DNA replication and DNA damage checkpoints into a single transcriptional complex.
INTRODUCTION
Genomic integrity is constantly threatened by many processes that take place in any living cell. Processes like transcription and DNA replication and exposure to external or internal damaging agents represent for the cell increased risk of rearrangements in DNA or single-nucleotide substitutions, defects that are the hallmarks of cancer cells (Elledge, 1996; Ciccia and Elledge, 2010). To maintain genomic integrity, all eukaryotes have developed a highly conserved mechanism to detect, signal, and repair damage in DNA, known as the DNA damage response (Hartwell and Weinert, 1989; Elledge, 1996; Rhind and Russell, 1998). When DNA replication is challenged, cells activate a DNA replication checkpoint blocking cell cycle progression until they are able to overcome the replication defects (Murakami and Okayama, 1995; Boddy and Russell, 1999). Similarly, in response to damage to DNA, cell cycle must be arrested through the DNA damage checkpoint (Rhind and Russell, 2000). In the fission yeast Schizosaccharomyces pombe the effector kinases Cds1 and Chk1 are activated by the DNA replication and the DNA damage checkpoint, respectively. The main aim of these kinases is to block cell cycle progression before cells enter into mitosis by phosphorylating and inhibiting the phosphatase Cdc25, which fully prevents activation of Cdc2 (Walworth et al., 1993; Furnari et al., 1997).
In fission yeast, activation of Cds1 in response to a DNA replication defect also invokes a transcriptional response that ultimately increases the concentration of the deoxynucleotides required to complete DNA synthesis. This response is achieved by activating the transcription factor MluI-binding factor (MBF; Dutta et al., 2008), which in a normal—unperturbed—cell cycle is responsible for the transcription of a set of genes that are required for the S phase of the cell cycle (Lowndes et al., 1992). MBF, which is the functional homologue of mammalian RB/E2F, is a high–molecular weight complex whose core elements are the product of the Start gene cdc10 and Res1 and Res2, which form a heterodimeric DNA-binding domain (Simanis and Nurse, 1989; Lowndes et al., 1992; Tanaka et al., 1992; Miyamoto et al., 1994; Obara-Ishihara and Okayama, 1994; Ayte et al., 1995). Under replicative stress, the activation of MBF-dependent transcription is a consequence of phosphorylation of several components of the MBF complex, including Cdc10 (Dutta et al., 2008), the corepressor Nrm1 (de Bruin et al., 2008), the repressor Yox1 (Caetano et al., 2011; Gomez-Escoda et al., 2011; Purtill et al., 2011), and the coactivator Rep2 (Nakashima et al., 1995; Chu et al., 2007, 2009). Specifically, phosphorylation of Yox1 by Cds1 disrupts the binding of Yox1 to the MBF complex, activating MBF-dependent transcription. Mutants in which Yox1 cannot be phosphorylated lack the proper transcriptional response under replicative stress (Gomez-Escoda et al., 2011). In this work, we demonstrate that there is also a direct link between the DNA damage checkpoint and the MBF complex, which, contrary to what happens after the activation of the DNA replication checkpoint, is responsible for inactivating the transcription of S-phase genes. This is achieved by direct phosphorylation of Cdc10 at Ser-720 and Ser-732 by the effector kinase Chk1.
RESULTS
Cdc10 is targeted by the DNA damage checkpoint
While investigating the effect of the DNA replication checkpoint on the regulation of the transcription factor MBF, we noticed that when cells were treated with hydroxyurea (HU) on a Yox1 mutant background (Yox1.SATA) that cannot be phosphorylated by the DNA replication checkpoint effector kinase Cds1, MBF-dependent induction of transcription was abrogated (Figure 1A; Gomez-Escoda et al., 2011). Under these conditions, the core MBF element, Cdc10, was released from chromatin (Figure 1B), in parallel to the release of the repressor Yox1 (Figure 1C). We were able to observe this release independent of the presence of Cds1, since in cells lacking Cds1, Cdc10 was also released when they were treated with HU and to a similar extent as in the Yox1.SATA cells. Unexpectedly, this release of Cdc10 was abrogated in the absence of Chk1 (both Δchk1 and Δchk1Δcds1 strains) or when Chk1 could not be activated in cells that lack the sensor kinase (Δrad3 strain). Of interest, we were able to observe that Chk1 was phosphorylated (which is a hallmark of its activation) when either Yox1.SATA or Δcds1 cells were treated with HU, pointing to the fact that in these specific genetic backgrounds both the DNA replication and DNA damage checkpoints were activated by HU (Figure 1D).
FIGURE 1:
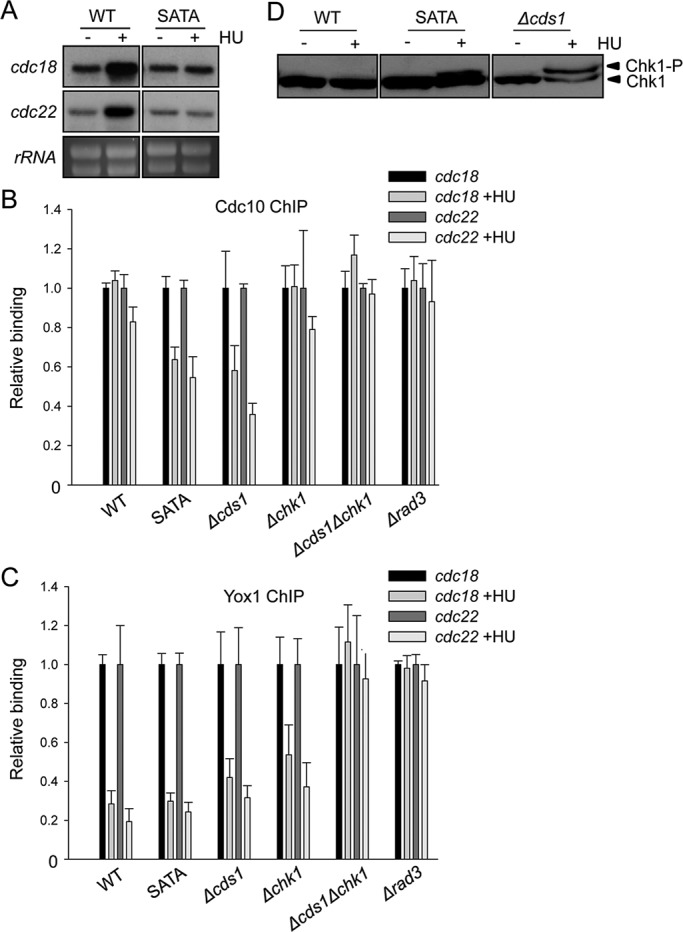
Cdc10 is targeted by the DNA damage response. (A) Total RNA was prepared from untreated (–) or HU-treated (+) cultures of wild-type (WT) and Yox1.SATA (SATA) cells and analyzed by hybridization to the probes indicated on the left. rRNA is shown as loading control. (B) Loading of Cdc10 on cdc22 and cdc18 promoters was measured by chromatin immunoprecipitation analysis of chromatin extracts isolated from untreated or HU-treated (10 mM HU, 4 h at 30°C) cultures of WT, SATA, ∆cds1, ∆chk1, ∆cds1∆chk1, or ∆rad3 cells. Endogenous Cdc10 is HA tagged, and the levels of binding are quantified on anti-HA immunoprecipitated DNA. (C) The same chromatin extracts analyzed for Yox1 binding with anti-Yox1 polyclonal antibodies. (D) Phosphorylation level of endogenous Chk1-HA in native extracts prepared from untreated (−) or HU-treated (+) cultures of WT, SATA, or ∆cds1 strains. Proteins were resolved in 8% SDS–PAGE and anti-HA Western blotted to detect Chk1.
It was previously shown that, besides Yox1, other components of the MBF complex (Nrm1 and Cdc10) could be phosphorylated by Cds1 when cells were under replicative stress (de Bruin et al., 2008; Dutta et al., 2008). However, our previous results (Figure 1) pointed to the possibility that some MBF components could also be targeted by Chk1 specifically when the DNA damage checkpoint was activated. To further investigate the signaling from this checkpoint to the MBF factor, we treated fission yeast cells with different DNA-damaging agents, such as MMS and γ-irradiation (Figure 2A). Indeed, both damaging agents were able to induce the release of Cdc10 from two of the better-characterized MBF-dependent promoters, cdc18 and cdc22. To determine whether Cdc10 was released alone or with other components of the MBF complex, we decided to test for coimmunoprecipitation between Cdc10 and Res2, which contains the DNA-binding activity of the MBF complex in its amino-terminal region (Miyamoto et al., 1994; Obara-Ishihara and Okayama, 1994). As shown in Figure 2B, the interaction between Cdc10 and Res2 was well preserved, if not improved, after treating fission yeast cells with MMS. In fact, both Res1 and Res2 are released from chromatin after treatment with MMS (Figure 2C), pointing to the possibility that the core elements of MBF (Res1, Res2, and Cdc10) are released as a complex from chromatin (and not as individual components) after the DNA damage checkpoint is induced.
FIGURE 2:
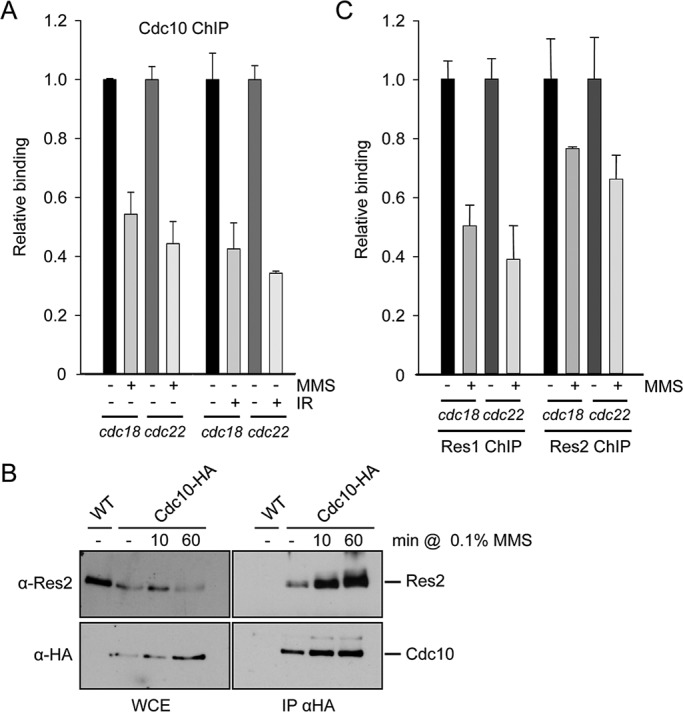
The DNA damage response releases intact MBF from its target promoters. (A) Loading of Cdc10 on cdc22 and cdc18 promoters was measured by chromatin immunoprecipitation (ChIP) analysis with anti-Cdc10 polyclonal antibodies of chromatin extracts isolated from untreated or MMS-treated (0.1% for 1 h at 30ºC) cultures of WT strain (left) or from irradiated cells (100 Gy; right). (B) The interaction between Cdc10 and Res2 is preserved in MMS-treated cells. Extracts (2.5 mg) from WT and Cdc10-HA strains (with or without MMS treatment for the time indicate on top) were immunoprecipitated with anti-HA antibody and analyzed for the presence of Res2 and Cdc10 with specific antibodies (monoclonal anti-Res2 and anti-HA, respectively). (C) Loading of Res1 (left) or Res2 (right) on cdc22 and cdc18 promoters was measured by ChIP analysis of chromatin extracts isolated from untreated or MMS-treated (0.1% for 1 h at 30ºC) cultures of a Res1-HA or Res2-HA strain, respectively.
The effect of the DNA damage checkpoint on MBF is dose dependent
To further characterize the response to MMS, we treated cells with increasing concentrations of the drug (from 0.002 to 0.1%) for 60 min. At the lower doses, we could not observe any noticeable effect on Cdc10, since it remained bound to the canonical promoters that we tested. In fact, Cdc10 was not released from chromatin unless cells were treated with higher MMS concentrations (≥0.05%; Figure 3A). On the contrary, when we measured the effect of MMS on the repressor system (Nrm1 and Yox1), we clearly observed that both proteins were released from chromatin (and consequently from the MBF complex) already at the lower MMS concentrations (Figure 3A). This effect on Nrm1/Yox1 paralleled a noticeable induction of the transcription of the MBF genes at low MMS concentrations (Figure 3B). We detected further release of both Yox1 and Nrm1 when we treated cells with higher MMS concentrations, which paralleled the described release of Cdc10. This second wave of Nrm1 and Yox1 release at higher concentrations correlates with repression of MBF-dependent transcription at higher MMS doses (Figure 3B). To separate both events (Cdc10 release at higher concentrations and Nrm1/Yox1 release at lower concentrations) and determine whether the DNA damage checkpoint was indeed able to induce release of the MBF complex from chromatin (and its consequent down-regulation of the MBF-dependent transcription), we decided to repeat the MMS treatment in a strain lacking the repressor system (Δnrm1Δyox1 background strain). These cells, which have induced transcription of the MBF-dependent genes as their basal steady state, were exposed to increasing MMS concentration for 1 h. As shown in Figure 3C, clear repression of the two MBF-dependent genes (cdc18 and cdc22) was observed. To determine whether release of the MBF complex from chromatin was due to cell death, we measured the viability of the cells during the time of treatment. As shown in Figure 3D and Supplemental Figure S1, the MMS concentrations used (and even higher concentrations) barely affect cell viability during the time of treatment.
FIGURE 3:
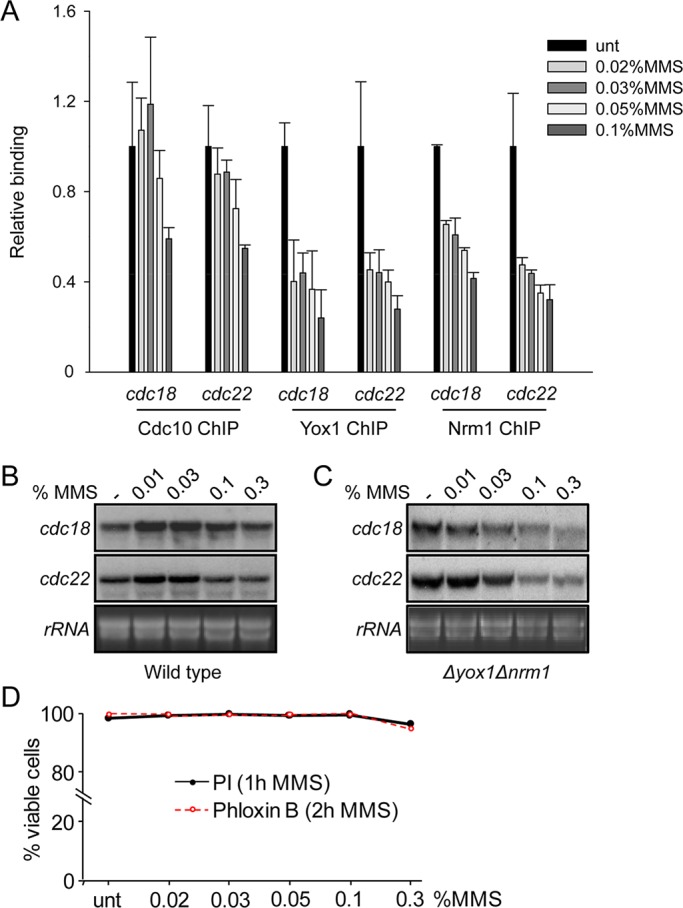
Chk1 effect on Cdc10 depends on MMS concentration. (A) Loading of Cdc10, Yox1, and Nrm1-HA on cdc22 and cdc18 promoters was measured by ChIP analysis of chromatin extracts isolated from untreated or MMS-treated cultures with the indicated concentrations (1 h at 30ºC). Cdc10 and Nrm1 are HA tagged, and the levels of binding are quantified on anti-HA immunoprecipitated DNA, whereas Yox1 is determined with anti-Yox1polyclonal antibodies. (B) Total RNA was prepared from untreated (–) or MMS-treated cultures of wild-type cells and analyzed by hybridization to the probes indicated on the left. rRNA is shown as loading control. (C) Total RNA was prepared from untreated or MMS-treated (increasing doses) cultures of Δyox1Δnrm1 cells and analyzed by hybridization with the probes indicated on the left. rRNA is shown as loading control. (D) Cell viability is unaffected at the used range of MMS concentrations. Viability test of wild-type cells treated with different concentrations of MMS, using propidium iodide or phloxine to measure viable cells, was performed by fluorescence-activated cell sorting analysis.
Next we wanted to further characterize the signaling from the DNA damage checkpoint to the MBF complex. To confirm that release of Cdc10 was exclusively due to activation of the DNA damage checkpoint (and that the DNA replication checkpoint was not involved in this release), we analyzed the binding of Cdc10 and Yox1 to cdc18 and cdc22 promoters in cells with impaired signaling in each or both of these checkpoint-signaling pathways after treatment with MMS. As shown in Figure 4A, the release of Cdc10 in cells lacking Cds1 was similar to that in wild-type cells. However, in cells lacking either Chk1 or the upstream activating kinase, Rad3, Cdc10 was not released after treatment with MMS. Under these concentrations of MMS, Yox1 release from chromatin paralleled the release of Cdc10, pointing to the possibility that it was the MBF complex as a whole that was released from chromatin when only the DNA damage checkpoint was induced (Figure 4B).
FIGURE 4:
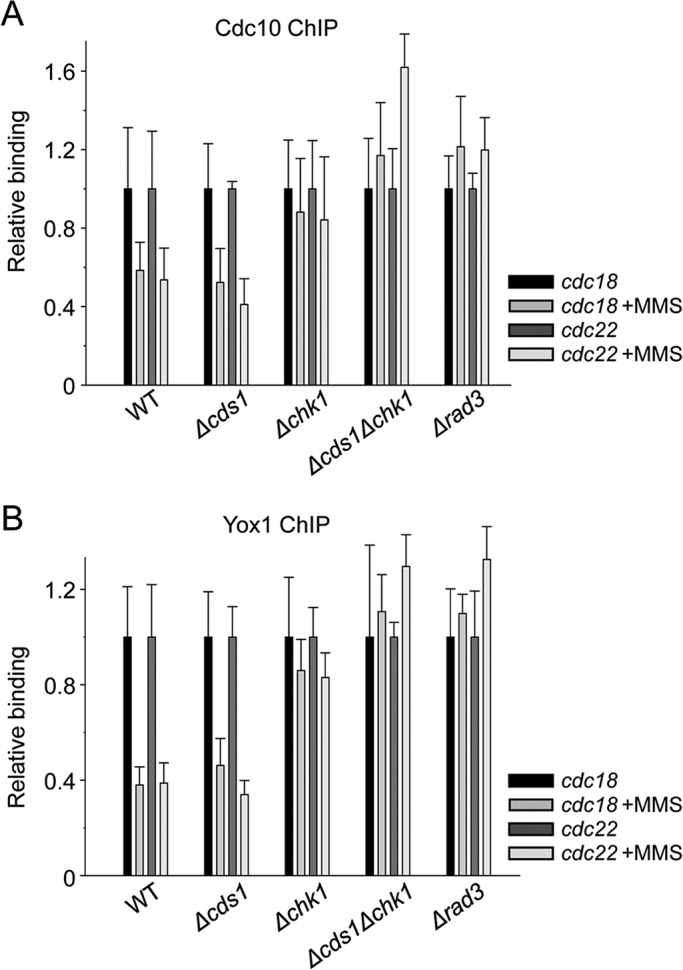
Cdc10 is released from chromatin upon DNA damage. (A) Loading of Cdc10 on cdc22 and cdc18 promoters was measured by ChIP analysis of chromatin extracts isolated from untreated or treated (0.1% MMS, 1 h at 30ºC) cultures of WT, ∆cds1, ∆chk1, ∆cds1∆chk1, or ∆rad3 cells. Endogenous Cdc10 is HA tagged, and the levels of binding are quantified on anti-HA immunoprecipitated DNA. (B) The same chromatin extracts were analyzed for Yox1 binding with anti-Yox1 polyclonal antibodies.
Chk1 phosphorylates Cdc10 and inactivates MBF-dependent transcription
We then decided to focus on the possibility that Cdc10 itself could be a direct target of Chk1. In fact, Cdc10 was already described as a target for Cds1, although no clear phenotype was associated with Cdc10 mutants in the residues that are phosphorylated in vitro by Cds1 (Dutta et al., 2008), that is, Ser-720 and Thr-723. Because Cds1 and Chk1 can phosphorylate similar target sequences (O'Neill et al., 2002; Seo et al., 2003; Xu and Kelly, 2009), we set out to determine whether Cdc10 was in vitro a bona fide target for Chk1 phosphorylation.
Cdc10 has four putative sites that can be phosphorylated by Chk1 (Ser-563, Thr-603, Ser-720, and Ser-732). Whereas the first two are in close proximity to the ankyrin domain, which mediates protein–protein interactions, the last two residues are in the C-terminal region of Cdc10, which is essential for loading the Yox1/Nrm1 repressor system onto chromatin (Supplemental Figure S2). In fact, and as a first approach, we noticed that Cdc10 release after MMS treatment was not observed in a strain that lacks the last 61 amino acids of Cdc10, cdc10-C4 (Figure 5A). Thus we decided to focus on this carboxy-terminal domain of Cdc10 as a potential substrate of Chk1 phosphorylation. In fact, in our in vitro Chk1 kinase assays, a Cdc10 construct lacking the last 61 amino acids was not phosphorylated. Conversely, a construct containing only the carboxy-terminal 61 amino acids (and thus containing the last two putative phosphorylation sites) was consistently phosphorylated (Figure 5, B and C). When Ser-720 or Ser-732 was mutated to alanine, the extent of phosphorylation was diminished. Furthermore, in the double mutant, Cdc10 phosphorylation by Chk1 was completely abolished (Figure 5C). It is worth noting that these phosphorylation sites are partially different from the described as Cds1-phosphorylation sites in Cdc10, where the authors noticed that only when both Ser-720 and Thr-723 were mutated to glutamic acid was MBF-dependent transcription induced (Dutta et al., 2008). In fact, in this mutant background, the Nrm1/Yox1 repressor system is unable to bind the MBF complex (unpublished data). Next, to determine whether in vivo Chk1 was able to phosphorylate Cdc10 on Ser-720 and Ser-732, we used an anti-phosphoserine antibody. As shown in Figure 5D, Cdc10 is phosphorylated when cells are treated with MMS. However, when Ser-720 and Ser-732 were replaced by alanines, we were unable to detect this phosphorylation. In fact, and confirming the notion that the DNA damage checkpoint could be regulating the MBF complex, we were able to detect direct interaction between Chk1 and Cdc10 by coimmunoprecipitation (Figure 5E)
FIGURE 5:

Cdc10 Ser-720 and Ser-732 are phosphorylated by Chk1, inactivating MBF-dependent transcription. (A) Chk1 signals MBF through the C-terminal region of Cdc10. Loading of Cdc10 on cdc22 and cdc18 promoters was measured in untreated or MMS-treated (0.1% MMS, 1 h at 25ºC) cultures of WT and cdc10-C4 strain by ChIP. Average of three individual experiments (±SD). (B) Amino acid sequence of the Cdc10 region phosphorylated by Chk1. The phosphorylation consensus is indicated at the bottom. (C) Chk1 in vitro kinase activity (in arbitrary units) was assayed using GST, WT, Cdc10, or the Cdc10 mutants indicated on top as substrates. Coomassie staining of the gel is shown at the bottom. (D) Cdc10 phosphorylation was determined on extracts prepared from untreated (unt.) or MMS-treated (0.1%) cells from WT (Cdc10) or Cdc10.2A strains. Immunoprecipitates were analyzed by Western blot with anti-phosphoserine (α-P-Ser) or anti-Cdc10 antibodies (α-Cdc10). (E) Extracts from the tagged strains indicated on top, untreated (unt) or treated with 0.1% MMS for 60 min, were immunoprecipitated with anti-Myc antibody and analyzed for the presence of Chk1 and Cdc10 with specific antibodies (HA and Myc, respectively). Left, Western blot of the whole-cell extracts used in the immunoprecipitations.
To test whether Cdc10 phosphorylation by Chk1 is essential for in vivo regulation of Cdc10/MBF binding to its target promoters upon activation of the DNA damage checkpoint, we introduced serine-to-alanine mutations in fission yeast, replacing the endogenous copy of cdc10. When treated with MMS, the strains that carry single mutations (including those next to the ankyrin domain) responded in a similar manner to a wild-type strain, that is, Cdc10 was released from its target promoters (Figure 6A and Supplemental Figure S3). However, in a strain that carries the double mutation S720AS732A (here Cdc10.2A) and cannot be phosphorylated in vitro and in vivo by Chk1, the release of Cdc10 was impaired from cdc18 promoter after treatment with MMS (Figure 6A). Of interest, we could observe only a small effect on the regulation of its binding activity to cdc22 promoter, pointing to the fact that Chk1 might differentially regulate the binding of Cdc10 to only a subset of MBF-dependent genes. A similar effect was observed when cells were irradiated (Figure 6B).
FIGURE 6:

Cdc10 phosphorylation after DNA damage is essential for viability. (A) Loading of Cdc10 on cdc22 and cdc18 promoters was measured by ChIP analysis of chromatin extracts isolated from untreated or treated (0.1% MMS, 1 h at 30ºC) cultures of WT Cdc10 or the mutants indicated at the bottom. (B) Phosphorylation of S720 and S732 after ionizing radiation (IR) induces the release of Cdc10 from chromatin. Loading of Cdc10 on cdc22 and cdc18 promoters was measured by ChIP analysis of chromatin extracts isolated from untreated or IR (100 Gy) cultures of WT and Cdc10.2A cells. Average of three individual experiments (±SD). (C) Total RNA was prepared from untreated or MMS-treated (increasing doses) cultures of a cdc10.2AΔyox1Δnrm1 strain and analyzed by hybridization with the probes indicated on the left. rRNA is shown as loading control. (D) RNA was prepared from wild-type (Cdc10) or Cdc10.2A cells exponentially growing or treated with 0.1% MMS for 1 h. cdc18, cdt2, cdc22, and mik1 were quantitated by RT-qPCR. Results are shown as fold induction over untreated wild-type cells as the average of three individual experiments (±SD). (E) Survival was performed by spotting 10–105 cells of the indicated strains (in a Δyox1Δnrm1 background) onto YE5S plates in the absence or presence of MMS or HU. Plates were incubated at 30°C for 3–4 d.
To test the consequences of the Chk1-mediated regulation of Cdc10 binding to chromatin, we measured the effect on transcription. As expected, when a strain in which the two Chk1 phosphorylation sites were mutated to alanine (Cdc10.2A) was treated with increasing doses of MMS, cdc18 transcription was steadily maintained, whereas cdc22 decreased to a similar extent as in the wild-type strain counterpart (Figure 6C; compare with Figure 3C). This different response between two MBF-dependent genes led us to expand our set of analyzed genes and in a more quantitative manner by reverse transcriptase-quantitative PCR (RT-qPCR). As shown in Figure 6D, whereas phosphorylation of Cdc10 by Chk1 is responsible for the down-regulation of cdc18 and cdt2, regulation of cdc22 and mik1 might be mediated by some other, overlapping mechanisms. Finally, we hypothesized that a strain in which the transcriptional response of the DNA damage checkpoint was abolished should have survival problems when confronted with a damaging agent, like MMS. As shown in Figure 6E, a strain carrying the double mutation (Cdc10.2A) was sensitive to MMS. Of interest, this strain has a wild-type level of survival when confronted with drugs that block DNA replication (HU), indicating that these two residues are not regulated by the DNA replication checkpoint.
DISCUSSION
The MBF complex is an essential transcription factor that fission yeast cells need for the normal and controlled expression of the S-phase transcription program. When DNA replication is challenged (e.g., after treatment of cells with HU), fission yeast cells activate their effector kinase (Cds1) and, among many other effects, are able to maintain a high level of MBF-dependent transcription (Gomez-Escoda et al., 2011). Because ribonucleotide reductase (Cdc22) is the target of HU and its expression is directly regulated by MBF (Lowndes et al., 1992), hyperactivation of the complex might help to overcome the block to DNA replication inflicted by the drug. Similar processes have been described in the distantly related budding yeast (de Bruin et al., 2008; Bastos de Oliveira et al., 2012; Travesa et al., 2012) and might be conserved to some extent in higher eukaryotes. Yox1, the repressor of the MBF complex, is the main MBF target of fission yeast Cds1 (Aligianni et al., 2009; Gomez-Escoda et al., 2011). Yox1 phosphorylation by Cds1 results in its inactivation (Caetano et al., 2011; Gomez-Escoda et al., 2011; Ivanova et al., 2011; Purtill et al., 2011). We now report here that the DNA damage checkpoint exerts a new layer of control on the MBF complex. However, instead of exerting a positive effect on MBF, Chk1, the effector kinase of the DNA damage checkpoint, is responsible for inactivating MBF-dependent transcription (Figure 7). This is achieved by direct phosphorylation of one of the core components of the MBF complex, Cdc10, at two different sites on its carboxy-terminal domain. This phosphorylation induces the exit of Cdc10 from the chromatin and thus the repression of the transcription of the MBF-dependent genes. Of interest, low doses of MMS are able to induce MBF-dependent transcription (probably through Cds1-dependent phosphorylation of Yox1), whereas high doses repress the same set of genes by directly phosphorylating Cdc10. In fact, under such severe damage there is no active MBF complex associated with the corresponding promoters, since Res1 and Res2 are also released from chromatin (Figure 2, B and C). Our hypothesis is that cells that have to cope with severe DNA damage must stop any attempt to initiate DNA synthesis, which will worsen its situation; this is achieved by switching off the S-phase transcriptional program. However, fission yeast cells sense discrete or minor DNA damage (low MMS concentration, HU) at least partly as a block to DNA synthesis, activating the DNA replication checkpoint. Consequently, these cells need to maintain activated the transcriptional S-phase program until they manage to fully complete the duplication of its genome. In conclusion, MBF would be doubly targeted by the DNA replication and the DNA damage checkpoints with outcomes that go in opposite directions: whereas the DNA damage checkpoint targets Cdc10 and causes repression, the DNA replication checkpoint phosphorylates Yox1 and induces activation of transcription. Of interest, whereas all of the MBF-dependent genes are induced upon a challenge to DNA replication (Dutta et al., 2008; Gomez-Escoda et al., 2011), only a subset seems to be under the control of the DNA damage checkpoint (Figure 6). We do not know how this is achieved, but it has long been known that not all MBF-dependent genes are regulated in the same manner; for example, in synchronized cultures, transcription of cdc18 is induced in anaphase, whereas induction of cig2 takes place later during the G1-to-S transition (Baum et al., 1997). Thus the differential regulation of the MBF-dependent genes by the DNA damage checkpoint may be due to intrinsically differences in the chromatin structure of the two groups of MBF dependent genes; alternatively, we have not excluded that other components or regulators of the MBF complex can be overlapping targets for Chk1 and play a role in only a subset of MBF-dependent genes. Further work is required to characterize this differential regulation.
FIGURE 7:
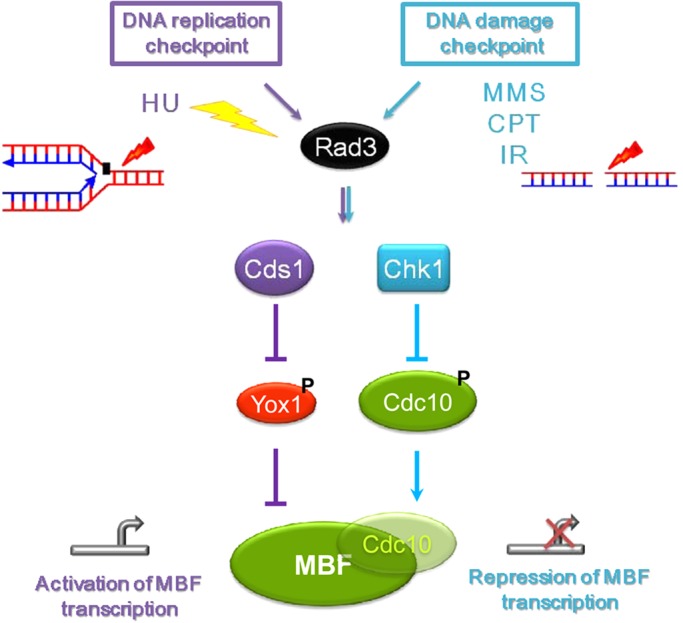
A model for the integration of the DNA damage and the DNA replication checkpoints on the MBF complex. On replicative stress, fission yeast cells activate the effector kinase Cds1. Among its targets, the repressor Yox1 is phosphorylated and no longer can bind the MBF complex, alleviating the transcriptional repression of genes required for DNA synthesis. On DNA damage, the effector kinase Chk1 phosphorylates Cdc10, which is a core component of the MBF complex. The outcome of this phosphorylation is, contrary to what happens under replicative stress, release of Cdc10 from its target promoters and repression of MBF-dependent transcription.
Although a clear link had not been demonstrated in higher eukaryotes between the DNA replication checkpoint and the regulation of the expression of S-phase genes, previous reports implied a connection between the DNA damage checkpoint and E2F, which, to some extent, is the functional homologue of fission yeast MBF and budding yeast MBF/SBF (Stevens et al., 2003; Inoue et al., 2007; Zalmas et al., 2008). Initially it was reported that E2F-1 was phosphorylated and activated in response to DNA damage, resulting in cells being directed to apoptosis (Stevens et al., 2003). However, another report demonstrated that irradiation might also cause phosphorylation of Rb (by Chk1/2) on a site that is also phosphorylated by CDK/cyclins in unperturbed cell cycle. Intriguingly, phosphorylation of Rb on this site induces repression of E2F-dependent transcription (Inoue et al., 2007). We propose that the checkpoint regulation of transcription through Cdc10 might be distantly conserved across eukaryotes, with the same final outcome (repression of transcription after DNA damage), but using highly divergent mechanisms: whereas in higher eukaryotes phosphorylation tethers the repressor (Rb) to the transcription factor (E2F-1), in fission yeast it decreases the binding of the transcription factor to its cognate promoters. It will be interesting to know whether similar mechanisms are conserved in the distantly related budding yeast.
MATERIALS AND METHODS
Strains and media
All S. pombe strains are isogenic to wild-type 972h-. The strains used in this work are listed in Supplemental Table S1. Media were prepared as previously described (Moreno et al., 1991). HU (10 mM), MMS, and γ-irradiation treatment were carried out on mid-log-grown cultures ([3–4] × 106 cells/ml) in YE5S media. Liquid cultures were treated with HU for 4 h and to MMS for 60 min, unless otherwise indicated. To analyze sensitivity to HU and MMS on plates, S. pombe strains were grown in liquid YE5S media to an OD600 of 0.5. Cells were then diluted in YE5S and spotted onto YE5S media agar plates. Plates were incubated at 30°C for 3–4 d.
Viability assays
For viability tests, cells were grown in liquid YE5S media to an OD600 of ∼0.3, and the cultures were treated with MMS. For propidium iodine staining, cells were centrifuged, washed twice with phosphate-buffered saline (PBS), and incubated with 3 μg/ml dye for 40 min on ice in darkness. For phloxine B staining, cells were incubated with 5 μg/ml dye for 2 h with shaking at 30°C in darkness, centrifuged, and washed twice with PBS. Ten thousand cells from each sample were scanned using channel FL3 for propidium iodide and channel FL2 for phloxine B with FACSCalibur (BD Biosciences, San Jose, CA).
Protein extraction
Extracts were prepared in NET-N buffer (20 mM Tris HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP40, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, protease inhibitor cocktail [Sigma-Aldrich, St. Louis, MO], 2 mM NaF, 0.2 mM Na3VO4, 2 mM β-glycerophosphate). Cells were broken with glass beads in a Minibeadbeater (BioSpec, Bartlesville, OK). Immunoprecipitations (1–3 mg of whole-cell lysate) were performed with 10 μl of protein G–Sepharose previously cross-linked with anti-hemagglutinin (HA) monoclonal antibody. Immunoprecipitates were washed after 1 h of incubation three times with NET-N buffer and resolved in SDS–PAGE, transferred to nitrocellulose membranes, and blotted with the indicated antibody. In Figure 5D, the detection of phosphorylated Cdc10 was performed using purified mouse anti-phosphoserine/threonine monoclonal antibody (612548; BD Biosciences).
In vitro Chk1 kinase assay
Substrates were prepared as glutathione S-transferase fusion proteins in Escherichia coli as described (Dutta et al., 2008). Protein extracts (300 μg) from MMS-treated cultures of a strain with HA-tagged Chk1 were immunoprecipitated as described (Ayte et al., 2001), followed by three washes with NET-N buffer and one wash with kinase buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 20 mM MgCl2, 4 mM EGTA, 2 mM DTT). Immunoprecipitates were incubated in kinase buffer containing 2 μg of substrate and 10 μCi of [γ-32P]ATP for 30 min at 30°C. Labeled proteins were resolved in 12% SDS–PAGE and detected by autoradiography.
Gene expression analysis
RNA extraction was performed as described (Moldon et al., 2008), and 10 μg of extracted RNA was loaded on agarose gels and analyzed by Northern blot. cdc18, cdc22, and tfb2 probes contained the complete open reading frames of the genes. For the RT-qPCR, RNA was digested with DNase I for 30 min at 37°C, phenol extracted, and precipitated. Eight micrograms of total RNA was denatured at 65°C for 10 min and then chilled on ice. Reverse transcriptase reactions were carried out (60 min at 42°C, 30 min at 52°C, and 3 min at 94°C) following the manufacturer's guidelines (Promega, Madison, WI) in the presence or absence of the enzyme. One microliter of the cDNA was used in the RT-qPCR with specific oligonucleotides.
Chromatin immunoprecipitation
Chromatin immunoprecipitation experiments were performed as described (Moldon et al., 2008). All experiments are plotted as the average of at least three different biological replicates ± SD and represented as relative binding with respect to untreated wild-type cells to facilitate comparison between different strains.
Supplementary Material
Acknowledgments
We thank Paul Nurse and Paul Russell for strains, Gabriel Gil for reagents and critical reading of the manuscript, and members of the Oxidative Stress and Cell Cycle group for helpful discussions. We acknowledge the technical support of Mercè Carmona. This work was supported by grants from the Spanish Ministry of Science and Innovation (BFU2009–07453 and BFU2012-31939), PLAN E and FEDER, Consolider-Ingenio 2007–0020, and SGR2009-195 from the Generalitat de Catalunya. J.A. and E.H. are recipients of ICREA Academia Awards (Generalitat de Catalunya).
Abbreviations used:
- MBF
MluI-binding factor
- MMS
methyl methanesulfonate
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-05-0257) on September 4, 2013.
*These authors contributed equally to this work.
REFERENCES
- Aligianni S, Lackner DH, Klier S, Rustici G, Wilhelm BT, Marguerat S, Codlin S, Brazma A, de Bruin RA, Bahler J. The fission yeast homeodomain protein Yox1p binds to MBF and confines MBF-dependent cell-cycle transcription to G1-S via negative feedback. PLoS Genet. 2009;5:e1000626. doi: 10.1371/journal.pgen.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayte J, Leis JF, Herrera A, Tang E, Yang H, De Caprio JA. The Schizosaccharomyces pombe MBF complex requires heterodimerization for entry into S phase. Mol Cell Biol. 1995;15:2589–2599. doi: 10.1128/mcb.15.5.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayte J, Schweitzer C, Zarzov P, Nurse P, DeCaprio JA. Feedback regulation of the MBF transcription factor by cyclin Cig2. Nat Cell Biol. 2001;3:1043–1050. doi: 10.1038/ncb1201-1043. [DOI] [PubMed] [Google Scholar]
- Baum B, Wuarin J, Nurse P. Control of S-phase periodic transcription in the fission yeast mitotic cycle. EMBO J. 1997;16:4676–4688. doi: 10.1093/emboj/16.15.4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Russell P. DNA replication checkpoint control. Front Biosci. 1999;4:D841–D848. doi: 10.2741/boddy. [DOI] [PubMed] [Google Scholar]
- Caetano C, Klier S, de Bruin RA. Phosphorylation of the MBF repressor Yox1p by the DNA replication checkpoint keeps the G1/S cell-cycle transcriptional program active. PLoS One. 2011;6:e17211. doi: 10.1371/journal.pone.0017211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Eshaghi M, Poon SY, Liu J. A Cds1-mediated checkpoint protects the MBF activator Rep2 from ubiquitination by anaphase-promoting complex/cyclosome-Ste9 at S-phase arrest in fission yeast. Mol Cell Biol. 2009;29:4959–4970. doi: 10.1128/MCB.00562-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Li J, Eshaghi M, Peng X, Karuturi RK, Liu J. Modulation of cell cycle-specific gene expressions at the onset of S phase arrest contributes to the robust DNA replication checkpoint response in fission yeast. Mol Biol Cell. 2007;18:1756–1767. doi: 10.1091/mbc.E06-10-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Harris MR, Brazauskas P, de Bruin RA, Smolka MB. Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 2012;31:1798–1810. doi: 10.1038/emboj.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RA, Kalashnikova TI, Aslanian A, Wohlschlegel J, Chahwan C, Yates JR, Russell P, 3rd, Wittenberg C. DNA replication checkpoint promotes G1-S transcription by inactivating the MBF repressor Nrm1. Proc Natl Acad Sci USA. 2008;105:11230–11235. doi: 10.1073/pnas.0801106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta C, Patel PK, Rosebrock A, Oliva A, Leatherwood J, Rhind N. The DNA replication checkpoint directly regulates MBF-dependent G1/S transcription. Mol Cell Biol. 2008;28:5977–5985. doi: 10.1128/MCB.00596-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- Gomez-Escoda B, Ivanova T, Calvo IA, Alves-Rodrigues I, Hidalgo E, Ayte J. Yox1 links MBF-dependent transcription to completion of DNA synthesis. EMBO Rep. 2011;12:84–89. doi: 10.1038/embor.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Kitagawa M, Taya Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007;26:2083–2093. doi: 10.1038/sj.emboj.7601652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova T, Gomez-Escoda B, Hidalgo E, Ayte J. G1/S transcription and the DNA synthesis checkpoint: common regulatory mechanisms. Cell Cycle. 2011;10:912–915. doi: 10.4161/cc.10.6.14963. [DOI] [PubMed] [Google Scholar]
- Lowndes NF, McInerny CJ, Johnson AL, Fantes PA, Johnston LH. Control of DNA synthesis genes in fission yeast by the cell-cycle gene cdc10+ Nature. 1992;355:449–453. doi: 10.1038/355449a0. [DOI] [PubMed] [Google Scholar]
- Miyamoto M, Tanaka K, Okayama H. res2+, a new member of the cdc10+/SWI4 family, controls the ‘start’ of mitotic and meiotic cycles in fission yeast. EMBO J. 1994;13:1873–1880. doi: 10.1002/j.1460-2075.1994.tb06456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldon A, Malapeira J, Gabrielli N, Gogol M, Gomez-Escoda B, Ivanova T, Seidel C, Ayte J. Promoter-driven splicing regulation in fission yeast. Nature. 2008;455:997–1000. doi: 10.1038/nature07325. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of the fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374:817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- Nakashima N, Tanaka K, Sturm S, Okayama H. Fission yeast Rep2 is a putative transcriptional activator subunit for the cell cycle ‘start’ function of Res2-Cdc10. EMBO J. 1995;14:4794–4802. doi: 10.1002/j.1460-2075.1995.tb00161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara-Ishihara T, Okayama H. A B-type cyclin negatively regulates conjugation via interacting with cell cycle “start” genes in fission yeast. EMBO J. 1994;13:1863–1872. doi: 10.1002/j.1460-2075.1994.tb06455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill T, Giarratani L, Chen P, Iyer L, Lee CH, Bobiak M, Kanai F, Zhou BB, Chung JH, Rathbun GA. Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J Biol Chem. 2002;277:16102–16115. doi: 10.1074/jbc.M111705200. [DOI] [PubMed] [Google Scholar]
- Purtill FS, Whitehall SK, Williams ES, McInerny CJ, Sharrocks AD, Morgan BA. A homeodomain transcription factor regulates the DNA replication checkpoint in yeast. Cell Cycle. 2011;10:664–670. doi: 10.4161/cc.10.4.14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Russell P. Mitotic DNA damage and replication checkpoints in yeast. Curr Opin Cell Biol. 1998;10:749–758. doi: 10.1016/s0955-0674(98)80118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Russell P. Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J Cell Sci. 2000;113:3889–3896. doi: 10.1242/jcs.113.22.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Kim SE, Lee YM, Lee JW, Lee JR, Hahn MJ, Kim ST. Determination of substrate specificity and putative substrates of Chk2 kinase. Biochem Biophys Res Commun. 2003;304:339–343. doi: 10.1016/s0006-291x(03)00589-8. [DOI] [PubMed] [Google Scholar]
- Simanis V, Nurse P. Characterization of the fission yeast cdc10+ protein that is required for commitment to the cell cycle. J Cell Sci. 1989;92(Pt 1):51–56. doi: 10.1242/jcs.92.1.51. [DOI] [PubMed] [Google Scholar]
- Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol. 2003;5:401–409. doi: 10.1038/ncb974. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Okazaki K, Okazaki N, Ueda T, Sugiyama A, Nojima H, Okayama H. A new cdc gene required for S phase entry of Schizosaccharomyces pombe encodes a protein similar to the cdc 10+ and SWI4 gene products. EMBO J. 1992;11:4923–4932. doi: 10.1002/j.1460-2075.1992.tb05599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travesa A, et al. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012;31:1811–1822. doi: 10.1038/emboj.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- Xu YJ, Kelly TJ. Autoinhibition and autoactivation of the DNA replication checkpoint kinase Cds1. J Biol Chem. 2009;284:16016–16027. doi: 10.1074/jbc.M900785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalmas LP, Zhao X, Graham AL, Fisher R, Reilly C, Coutts AS, La Thangue NB. DNA-damage response control of E2F7 and E2F8. EMBO Rep. 2008;9:252–259. doi: 10.1038/sj.embor.7401158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.