

Abstract
In the first part of a two-phase pursuit of highly oxidized members of the ent-kaurane and beyerane diterpenoid families, steviol was identified as the ideal cyclase phase terminus. Accordingly, a synthesis of steviol has been developed. This synthesis features a polyene cyclization precursor designed to directly yield oxidation on the axial C19 methyl group. Construction of the necessary [3.2.1]bicyclic system found in the ent-kaurane skeleton was realized with two overbred intermediates. The resulting [3.2.1]bicyclic system undergoes Wagner–Meerwein rearrangement to yield the beyerane skeleton of isosteviol.
Keywords: total synthesis, diterpenes, ent-kaurane, beyerane, overbred intermediates
The ent-kauranes present highly varied oxidation patterns (3, 4, Figure 1A), undergo intriguing skeletal rearrangements, and possess antibacterial, antitumor, and anti-malarial activity.1 These attributes make ent-kauranes interesting candidates for two-phase terpene total synthesis.2 Steviol (1) was chosen as the lowest oxidized logical target due to the known conversion of such structures to beyeranes3 (isosteviol, 2) and the useful functionality present for an “oxidase phase.” The first total synthesis reported by Mori et al. provides steviol (1) in 35 steps and 0.013% overall yield.4a,b,c A 19-step synthesis of steviol methyl ester was reported subsequently by Ziegler et al., but it relied on a key step that gave only a 3% yield of the [3.2.1]bicyclic system and 0.015% overall yield.4d An elegant approach to isosteviol (2) was reported by Snider et al. in 13–18 steps, 0.37–1.2% overall yield.5 Conversion of isosteviol (2) to steviol (1), however, is unknown. In this communication, an efficient synthesis of (±)-steviol (1) is presented.
Figure 1.

A. Truncated oxidation pyramid for ent-kauranes and beyeranes. B. Cyclase phase retrosynthetic strategy. C. Polycyclization methods for installation of C18 or C19 methyl group oxidation.
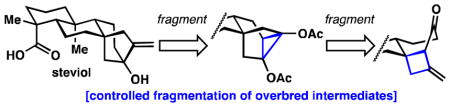
In 2009, R. W. Hoffmann formalized the concept of “overbred intermediates” in synthesis design as intermediates having one or more excess C–C bonds that must be subsequently cleaved.6 The route to the [3.2.1]bicyclic system of steviol (1) relies on the controlled fragmentation of two overbred intermediates. Cyclopropane 5 (Figure 1B) would require preferential cleavage of the C12–C16 bond over the C12–C13 bond. Such a fragmentation would be ambitious because these two bonds appear to be nearly indistinguishable.7 Indeed, a similar system fragmented with only modest diastereoselectivity (2:1).4c Cyclobutane 6, would then arise from a [2+2] photocycloaddition with allene. This strategy should install a very hindered quaternary center with high diastereo-selectivity.8 It is known that strained cyclobutanones in similar systems will open upon nucleophilic attack to break the analogous C13–C14 bond.9
Tricyclc system 7 presents a challenge when the issues of stereo- and regioselectivity are considered. In particular, the required axial C19 methyl oxidation and para-regioselectivity are not adequately addressed by known approaches (Figure 1C). Radical-mediated methods give the undesired ortho-methoxy product.10 C–H activation reactions directed from C3 preferentially oxidize the C18 methyl group.11,12 Cyclizations initiated from a terminal epoxide (i.e. 10, Scheme 1) provide oxidation on the equatorial C18 methyl group.13 Consequentially, a unique cyclization precursor (8) was designed with the following considerations: (i) the polycyclization should be Lewis acid-initiated (rather than radically initiated) to give the correct para-regioselectivity; (ii) the epoxide should be internal (rather than terminal) to give the required C19 oxidation;13 and (iii) the Z-stereochemistry of this internal epoxide is imperative to give C19 oxidation.14
Scheme 1. Total synthesis of (±)-steviol (1).

Reagents and Conditions: (a) LiNEt2 (3 equiv), THF, 60 °C, 2 h (92%); (b) VO(acac)2 (0.25 equiv), t-BuOOH (5 M in decane) (1.6 equiv), benzene, 6 °C, 2 h (74% or 83% BRSM); (c) CBr4 (3 equiv), PPh3 (2.9 equiv), i-Pr2NEt (3.3 equiv), CH2Cl2, −10 °C, 12 h (81%); (d) NaOt-Bu (2.4 equiv) benzyl alcohol (solvent), 100 °C, 3 h (92%); (e) FeCl3 (2 equiv), CH2Cl2, rt, 3 h (52%); (f) Li0 (50 equiv), NH3, THF; t-BuOH, −78 °C to −45 °C, 2 h; 4M HCl in dioxane, rt, 30 min (79%); (g) DEAD (5 equiv), PPh3 (5 equiv), THF, 70 °C, 5 h (91%); (h) H2, Pd/C (10 wt%)(10 mol%), EtOAc, rt, 7 h (93%); (i) allene, CH2Cl2, rt, 450 W Hg-lamp, pyrex, 12 h (82%); (j) O3, MeOH, −78 °C, 5 min; Me2S, rt, 30 min; AcOH: PPA (9:1), 110 °C, 12 h (62%); (k) HCl(g), Ac2O (solvent), act. Zn0 (60 equiv), 0 °C, 45 min; (l) AcCl (3 M in MeOH), 0–6 °C, 12 h (79% 18 and 11% 17); (m) PPh3 (6.6 equiv), RhCl(PPh3)3 (5 mol%), THF, i-PrOH; TMSCHN2 (20 equiv), 48 h (63%); (n) PDC (5 equiv), DMF, rt, 18 h, (92%); (o) NaClO2 (6 equiv), NaH2PO4(10 equiv), 2-methyl-2-butene (10 equiv), THF/t-BuOH, 0 °C to rt, 16 h (85%). (DEAD = diethyl azodicarboxylate, PPA = polyphosphoric acid, PDC = pyridinium dichromate, DMF = N,N-dimethylformamide).
The pursuit of cyclization precursor 8 began from epoxide 9 (see Scheme 1). Elimination to open epoxide 9 followed by vanadium-directed epoxidation gave the erythro-product 10 in 68% overall yield (5.3 gram scale). The secondary alcohol in 10 was inverted to give the threo-bromide 11 in 81% yield (7.2 gram scale). Nucleophilic addition of benzyloxide to open the epoxide followed by closure of the bromohydrin provided the cyclization precursor 8 (7.1 gram scale). The polycyclization was most efficiently effected by iron trichloride to give tricyclic system 12 (1.1 gram scale). Compound 12 was converted to crystalline enone 13 by Birch reduction-deprotection and isomerization. X-ray analysis confirmed the para-regiochemistry, the crucial C19 axial methyl group oxidation and the correct stereochemistry at the C9 methine group.
Next, the neopentyl alcohol was eliminated followed by hydrogenation to furnish compound 7 (2.1 gram scale). Birch reduction and isomerization proceeded to give enone 14 (1.3 gram scale). Allene [2+2] photocycloaddition with 14 formed the hindered C8 quaternary center in overbred cyclobutane 6 (1.1 gram scale).8 This overbred intermediate was enabling because all other attempts to form this quaternary center failed including: copper, indium, and tin mediated 1,4-additions, Sakurai and Keck allylations, as well as intramolecular bond formations via sigmatropic rearrangements. Cyclobutane 6 was transformed to 15 in a one-pot sequence (1.0 gram scale): ozonolysis, selective fragmentation with methanol to give the methyl ester, and finally acid-mediated condensation to forge the [2.2.2]bicyclic system.9
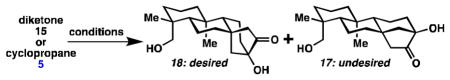
Reductive cyclopropanation of 15 would generate an overbred cyclopropanediol (16), which could undergo divergent fragmentation pathways: C12–C13 cleavage or C12–C16 cleavage to give 17 or 18 respectively. Mori et al. treated a similar system with Zn(Hg) amalgam in 6 M HCl/toluene at 110 °C for 1 hour to get a 2:1 ratio in favor of the analogous desired isomer in 41% yield.4c Treatment of diketone 15 with these conditions for 45 minutes gave only the undesired isomer 17 in 26% yield (see Table 1, entry 1). Encouragingly, when the reaction was stopped after 5 minutes, a 2.2:1 ratio in favor of the desired isomer 18 was observed (entry 3). Moreover, the desired product 18 was found to rearrange to the undesired isomer 17 under acidic conditions (see Scheme 2A). It seemed that Mori’s conditions were unsuitable due to the high temperatures, which caused the desired kinetic isomer (18) to rearrange to the thermodynamic isomer (17). Under the reaction conditions, the ketones in products 17 and 18 likely undergo further reduction. Attempts to run this reaction at lower temperature with activated zinc led to decomposition with trace formation of 17 (entry 4).
Table 1.
Attempts at conversion of 15 or 5 to 18
| ||||
|---|---|---|---|---|
| Entry | Conditions | Yield% 18 | Yield % 17 | ratio 18:17 |
| 1 | 15, Zn(Hg), 6M HCl, PhMe, 110 °C, 45 min | 0% | 26% | 0:1 |
| 2 | 15, Zn(Hg), 6M HCl, PhMe, 110 °C, 30 min | 13% | 9% | 1.4:1 |
| 3 | 15, Zn(Hg), 6M HCl, PhMe, 110 °C, 5 min | 24% | 11% | 2.2:1 |
| 4 | 15, act. Zn°, HCl in Et2O, 0 °C, 15 m | trace | -- | -- |
| 5 | 5, AcCl, MeOH, 0–6 °C, 12 h | 79% | 11% | 7.2:1 |
Scheme 2.

Reagents and Conditions: (a) 12 M HCl, PhMe, 110 °C, 30 min (79%); (b) CAN (3 equiv), MeCN, 0 °C, 10 min; (c) PPA, AcOH, 110 °C, 12 h (72% over 2 steps); (d) HBr (48% aq.) Et2O, rt, 15 h (90%); (e) HBr (48% aq.) Et2O, rt, 18 h (87%); (f) CrO3 (10 equiv), acetone, 0 °C to rt, 3 h (81%) (CAN = cerium ammonium nitrate)
To overcome these issues, cyclopropane diol 16 was trapped as diacetate 5. 15 This would allow for fragmentation at low temperatures thereby avoiding isomerization of kinetic product 18 to thermodynamic product 17. It would also avoid over-reduction. Treatment of 5 with methanolic HCl at 0–6 °C gave a greater than 7:1 ratio of 18:17 in 79% and 11% yields respectively (entry 5). Undesired isomer 17 can also be recycled to 15 (Scheme 2B).
With suitable quantities of 18 in hand, installation of the methylene was attempted (see Scheme 1). While the Wittig olefination of a similar substrate has been reported,4a,c this procedure as well as salt-free variations either yielded rearranged material or gave no reaction respectively. A modified Wittig procedure proceeded to give olefin 19,16 which was oxidized to give (±)-steviol (1) in 17 steps from geranyl acetate. 17 Acid-induced rearrangement of steviol (1) provided isosteviol (2) (see Scheme 2C). Alternatively, compound 19 could first be rearranged to the beyerane skeleton followed by Jones oxidation to provide isosteviol (2) in 17 steps (see Scheme 2D).
To summarize, a synthetic route that offers efficient access to minimally oxidized members of the ent-kaurane and beyerane class of terpenes has been developed. This route could feasibly be rendered enantioselective.18 The challenging axial C19 oxidation and [3.2.1]bicyclic motifs prompted a reevaluation and strategic modification of literature precedent. The first challenge was addressed with a unique polycyclization precursor (8) while the second necessitated the use of overbred intermediates (6 and 5) and their controlled fragmentations. Such strained intermediates enabled and simplified the overall synthetic route. The completion of this cyclase phase sets the stage for an in-depth study of the oxidation chemistry of these complex terpenes.
Supplementary Material
Footnotes
Financial support for this work was provided by an unrestricted grant from TEVA, the NIH (GM097444-03) and NSF (a predoctoral fellowship to E.C.C). We are grateful to Prof. A. Rheingold and Dr. C.E. Moore (UCSD) for X-ray crystallographic analysis.
This work is dedicated to R. W. Hoffmann on the occasion of his 80th birthday.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Sun HD, Huang SX, Han QB. Nat Prod Rep. 2006;23:673. doi: 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]
- 2.a) Ishihara Y, Baran PS. Synlett. 2010;12:1733. [Google Scholar]; b) Chen K, Baran PS. Nature. 2009;459:824. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 3.Ruddat M, Heftmann E, Lang A. Arch Biochem Biophys. 1965;110:496. doi: 10.1016/0003-9861(65)90441-8. [DOI] [PubMed] [Google Scholar]
- 4.a) Mori K, Nakahara Y, Matsui M. Tetrahedron Lett. 1970;11:2411. [Google Scholar]; b) Nakahara Y, Mori K, Matsui M. Agr Biol Chem. 1971;35:918. [Google Scholar]; c) Mori K, Nakahara Y, Matsui M. Tetrahedron. 1972;28:3217. [Google Scholar]; d) Ziegler FE, Kloek JA. Tetrahedron. 1977;33:373. [Google Scholar]; e) Cook IF, Knox JR. Tetrahedron Lett. 1970;11:4091. [Google Scholar]
- 5.Snider BB, Kiselgof JY, Foxman BM. J Org Chem. 1998;63:7945. [Google Scholar]
- 6.Hoffmann RW. Elements of Synthesis Planning; Springer-Verlag; Berlin Heidelburg: 2009. pp. 106–117. (and references there-in) [Google Scholar]
- 7.Abad A, Agullo C, Alfonso Marzal I, Navarro I, Gris A. Tetrahedron. 2006;62:3266. [Google Scholar]
- 8.a) Crimmins MT, Reinhold TL. Org React. 1994;44:297. [Google Scholar]; b) Weisner K. Tetrahedron. 1975;31:1655. [Google Scholar]
- 9.a) Nagaoka H, Shimano M, Yamada Y. Tetrahedron Lett. 1989;30:971. [Google Scholar]; b) Poupart MA, Paquette L. Tetrahedron Lett. 1988;29:269. [Google Scholar]
- 10.a) Snider BB, Mohan R, Kates SA. J Org Chem. 1985;50:3659. [Google Scholar]; b) Zoretic PA, Weng X, Caspar ML, Davis DG. Tetrahedron Lett. 1991;32:4819. [Google Scholar]; c) Chen L, Gill GB, Pattenden G. Tetrahedron Lett. 1994;35:2593–6. [Google Scholar]; d) Rendler S, MacMillan DWCJ. Am Chem Soc. 2010;132:5027. doi: 10.1021/ja100185p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Baldwin JE, Jones RH, Najera C, Yus M. Tetrahedron. 1985;41:699. [Google Scholar]; b) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; c) Simmons EM, Hartwig JF. Nature. 2012;483:70. doi: 10.1038/nature10785. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Granados A, Lopez PE, Melguizo E, Parra A, Simeo Y. J Org Chem. 2007;72:3500. doi: 10.1021/jo070116e. [DOI] [PubMed] [Google Scholar]
- 13.a) Van Tamelen EE, Zawacky SR, Russell RK, Carlson JG. J Am Chem Soc. 1983;105:142. [Google Scholar]; b) Corey EJ, Liu K. J Am Chem Soc. 1997;119:9929. [Google Scholar]
- 14.a) Stork G, Burgstahler AW. J Am Chem Soc. 1955;77:5068. [Google Scholar]; b) Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Helv Chim Acta. 1955;38:1890. [Google Scholar]
- 15.Toda M, Hayashi M, Hirata Y, Yamamura S. Bull Chem Soc Japan. 1972;45:264. [Google Scholar]
- 16.Lebel H, Guay D, Paquet V, Huard K. Org Lett. 2004;6:3047. doi: 10.1021/ol049085p. [DOI] [PubMed] [Google Scholar]
- 17.Corey EJ, Schmidt G. Tetrahedron Lett. 1979;5:399. [Google Scholar]
- 18.Corey EJ, Noe MC, Shieh WC. Tetrahedron Lett. 1993;34:5995. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.