

Abstract
Reactive astrocytes are traditionally thought to impede brain plasticity after stroke. However, we previously showed that reactive astrocytes may also contribute to stroke recovery, partly via the release of a nuclear protein called high-mobility group box 1 (HMGB1). Here, we investigate the mechanisms that allow stimulated astrocytes to release HMGB1. Exposure of rat primary astrocytes to IL-1b for 24 hrs elicited a dose-dependent HMGB1 response. Immunostaining and western blots of cell lysates showed increased intracellular levels of HMGB1. Western blots confirmed that IL-1b induced a release of HMGB1 into astrocyte conditioned media. MAP kinase signaling was involved. Levels of phospho-ERK were increased by IL-1b, and the MEK/ERK inhibitor U0126 decreased HMGB1 upregulation in the stimulated astrocytes. Since HMGB1 is a nuclear protein, the role of the nuclear protein exporter, chromosome region maintenance 1 (CRM1), was assessed as a candidate mechanism for linking MAP kinase signaling to HMGB1 release. IL-1b increased CRM1 expression in concert with a translocation of HMGB1 from nucleus into cytoplasm. Blockade of IL-1b -stimulated HMGB1 release with the ERK inhibitor U0126 was accompanied by a downregulation of CRM1. Our findings reveal that IL-1b stimulates the release of HMGB1 from activated astrocytes via ERK MAP kinase and CRM1 signaling. These data suggest a novel pathway by which inflammatory cytokines may enhance the ability of reactive astrocytes to release pro-recovery mediators after stroke.
Keywords: Reactive astrocytes, high-mobility group box1, IL-1beta, ERK signaling, CRM1/Exportin1
Introduction
Astrocytes comprise the most numerous non-neuronal cell type in mammalian brain (Tower et al., [1973]). Within a few hours of virtually any type of brain injury, surviving astrocytes in the affected region become hypertrophic and proliferate, a process termed reactive astrogliosis (Ridet et al., [1997]). Traditionally, it was assumed that reactive astrocytes after stroke or brain injury contribute to glial scarring that impedes neuronal remodeling and recovery (Silver and Miller, [2004]). However, it is now recognized that reactive astrocytes may also release many trophic factors, such as nerve growth factor, basic fibroblast growth factor, platelet-derived growth factor, brain-derived neurotrophic factor, ciliary neurotrophic factor, Neuropilin-1, vascular endothelial growth factor, and others (Strauss et al., [1994]; Mocchetti and Wrathall, [1995]; Ridet et al., [1997]; Tokita et al., [2001]; Zhang and Chopp, [2002]; Chen and Swanson, [2003]). Many of these trophic factors may in fact be beneficial by promoting neuronal survival and synaptogenesis, neurogenesis, and angiogenesis after stroke or brain injury (Panickar and Norenberg, [2005]; Shibuya et al., [2009]).
In addition to trophic factors, it was recently discovered that reactive astrocytes may also secrete a nuclear protein called high-mobility group box 1 (HMGB1) (Passalacqua et al., [1998]; Kim et al., [2008]). HMGB1, a non-histone DNA-binding protein, is widely expressed in various tissues, including mammalian brain. HMGB1 is a multifunctional molecule that acts as an extracellular trigger and/or modulator of critical cell processes such as inflammation, proliferation, migration and survival (Yang et al., [2005]; Ullora et al., [2006]). In neurons, HMGB1 can promote neurite outgrowth, upregulate symaptic proteins and sustain cell survival (Huttunen et al., [2000], [2002]). In endothelial cells, HMGB1 can induce proliferation (Treutiger et al., [2003]) and sprouting (Schlueter et al., [2005]). Hence, in the context of stroke and brain injury, HMGB1 released from reactive astrocytes would be a candidate mediator for enabling neurovascular remodeling during the recovery phase.
To date, the mechanisms that regulate the expression and secretion of HMGB1 in astrocytes remain unclear. In this study, we investigated HMGB1 expression in rat cortical astrocytes stimulated with IL-1b. Our findings showed that IL-1b-stimulated astrocytes upregulate and release HMGB1 via specific signaling pathways involving ERK MAP kinase and the nuclear protein exporter, chromosome region maintenance 1 (CRM1).
Methods and Materials
Reagents
Rat recombinant IL-1b and human recombinant HMGB1 were purchased from Sigma-aldrich (St Louis, MO), U0126, SB203580, SP600125 were purchased from Calbiochem (La Jolla, CA).
Cell culture
Primary astrocyte cultures were prepared from cerebral cortices of 2-day-old neonatal Sprague-Dawley rats (Arai et al, [2003]). Briefly, dissociated cortical cells were suspended in Dulbecco's modified Eagle medium (NBM, Life Technology) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 10% fetal bovine serum and plated on uncoated 25 cm2 flasks at a density of 6×105 cells/cm2. Monolayers of type 1 astrocytes were obtained 12-14 days after plating. Nonastrocytic cells such as microglia and neurons were detached from the flasks by shaking and removed by changing the medium. Astrocytes were dissociated by trypsinization and then reseeded on uncoated 6-, 12-, and 24-well plates or slide chambers at a density of 2×105 cells/cm2. After the cells reached confluence (7-8 days after seeding), cultures were switched to serum-free Dulbecco's modified Eagle medium and experiments were initiated 1 h and 24 h later. In this system, more than 95% of the cells were identified as type 1 astrocytes by GFAP staining and their flattened, polygonal morphology.
Immunohistochemistry
Cells on slide chambers were used for immunohistochemistry. After the cells reached confluence, they were washed with ice-cold PBS (pH 7.4), followed by 4% paraformaldehyde for 30 min. After being further washed three times in PBS containing 0.1% Triton X-100, they were incubated with 3% bovine serum albumin (BSA) in PBS for 1 h. Then cells were incubated with primary antibodies against the astrocytic marker glial fibrillary acidic protein (GFAP; 1:100; Chemicon) or HMGB1 (1:200; Abcam) or CRM1 (1:100; BD Transduction Laboratories) at 4°C overnight. After washing with PBS, they were incubated with goat anti rabbit IgG against HMGB1 conjugated with TRITC (1:150; Jackson ImmunoResearch) and donkey antimouse IgG against GFAP or CRM1 conjugated with fluorescein isothiocyanate (1:150; Jackson ImmunoResearch) for 1 h at room temperature. Finally, nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI), and coverslips were placed. Immunostaining was analyzed with a fluorescence microscope (Olympus BX51) interfaced with a digital charge-coupled device camera and an image analysis system.
Preparation of cellular extracts
At 24h after IL-1b (100pg/ml) treatment, astroglial cells were harvested and washed twice with cold PBS, and nuclear and cytoplasmic extracts were prepared by using NucBusterTM Protein Extraction Kit purchased from Novagen (Madison, WI). We prepared a single cell suspension 4×105 cells/25 μl and resuspend the cell pellet using 75 μl NucBuster Reagent 1 following mixing 15 sec at high speed vortex, and incubating on ice 5 min, and vortex again 15 sec at high speed. After then, cytoplasmic fraction was extracted by centrifuge at 16,000 × g for 5 min at 4°C. After that, the pellet was resuspend in 1 μl of 100X Protease Inhibitor Cocktail, 1 μl of 100 mM DTT, and 75 μl NucBuster Extraction Reagent 2 following mixing 15 sec at high speed vortex, and incubating on ice 5 min, and vortex again 15 sec at high speed. Nuclear extract was prepared from supernatant after centrifuge at 16,000 × g 5 min at 4°C.
Western blot analysis
Cultures were rinsed twice with ice-cold phosphate-buffered saline and the cells were collected into Pro-PREPTM Protein Extraction Solusion (BOCA SCIENTIFIC). Cell lysates were then homogenized and centrifuged at 14,000 rpm for 10 min at 4°C and protein concentration in the supernatant was determined with the Bradford assay (Bio-Rad). Samples were heated with equal volumes of SDS sample buffer (Novex) and 10% 2-mercaptoethanol at 95°C for 5 min, then each sample (20 μg per lane) was loaded onto 4-20% Tris-glycine gels. After electorophoresis and transferring to polyvinylidene difluoride membranes (Novex), the membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 0.2% I-block (Tropix) for 90 min at room temperature. Membranes were then incubated overnight at 4°C with monoclonal anti-p-ERK1/2 antibody (1:2,000, Cell Signaling Technology, New England Biolabs) or anti-ERK1/2 antibody (1:2,000, promega) or monoclonal anti-HMGB1 (1:10000, Abcam) or anti-CRM1 (1:2000, BD Transduction Laboratories) after incubation with peroxidase-conjugated secondary antibodies and visualization by enhanced chemiluminescence (GE Healthcare). Optical density was assessed using the NIH Image analysis software.
Cell number and viability assays
Cell proliferation/survival was assessed by WST reduction assay (Dojindo), which detects dehydrogenase activity of viable cells. The cells were incubated with 10% WST solution for 1 h at 37°C. Then the absorbance of the culture medium was measured with a microplate reader at a test wavelength of 450 nm and a reference wavelength of 630 nm.To ensure that our secreted HMGB1 measurements were not confounded by nonspecific protein release due to cell damage, cytotoxicity was quantified using a standard measurement of lactate dehydrogenase (Boehringer-Mannheim LDH kit). Measurement of the reduction of 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide (MTT) was performed to assess the integrity of mitochondrial function as a measure of cell viability.
Statistical analysis
Results were expressed as mean±SEM. Multiple comparisons were evaluated ANOVA followed by Tukey-Kramer's tests for pair-wise comparisons between all groups. Differences that reached P<0.05 were considered to be statistically significant.
Results
IL-1b upregulates and induces HMGB1 secretion in astrocytes
Immunohistochemical staining showed that IL-1b increased HMGB1 expression in astrocytes (Figure 1A). Western blots demonstrated that IL-1b (10-1000 pg/ml) upregulated HMGB1 in astrocytes following a concentration-dependent manner [F(4,16)=17.417, P<0.0001, Figure 1B]. Western blots were also used to confirm that this response to IL-1b involved an active release of HMGB1 into the culture media [F(2,8)=8.706, P=0.0098, Figure 2A]. Proliferation (WST), cytotoxicity (LDH), and cell viability (MTT) assays showed that the range of IL-1b stimulation used here did not increase cell numbers, cause cell death, or alter basic cell viability respectively (Figure 2B-D). Thus, the IL-1b-induced release of HMGB1 from astrocytes was not due to nonspecific cell lysis or injury.
Figure 1.
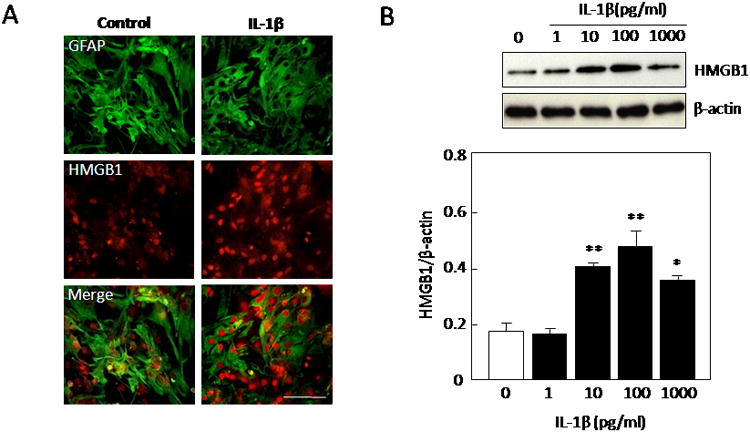
(A) Immunohistochemical staining demonstrated that HMGB1 expression is enhanced after IL-1b treatment in our purified astroglia culture system. Cells were stained for the astrocytic marker glial fibrillary acidic protein (GFAP; green), anti-HMGB1 antibody (red). Scale bar indicates 100 μm. (B) IL-1b (10-1000 pg/ml) significantly enhanced the expression of HMGB1 in astrocytes after 24 hrs. Values are means ± SEM of four independent experiments. *P<0.05, **P<0.01 compared with 0 pg/ml treatment controls.
Figure 2.
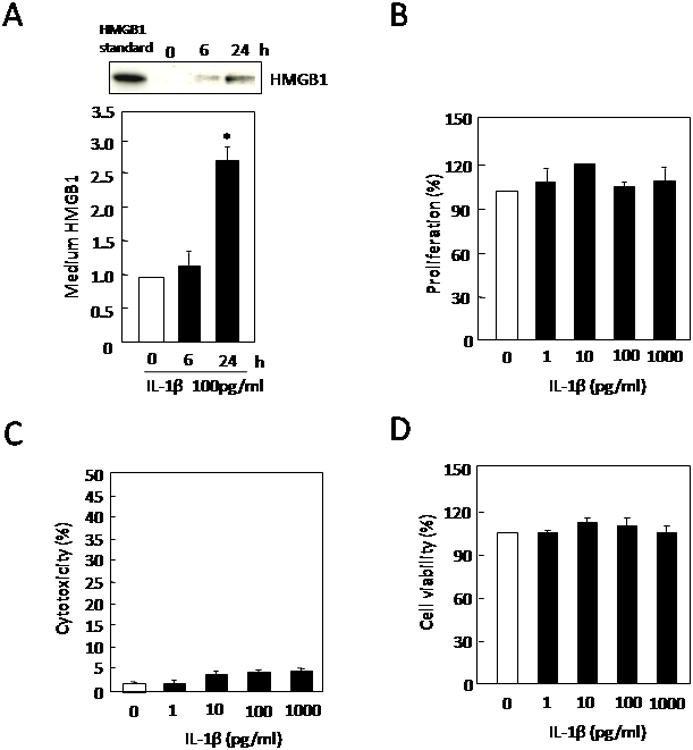
(A) Treatment with IL-1b (100 pg/ml) for 24 hrs significantly induced the release of HMGB1 into extracellular media. Values are means ± SEM of six independent experiments. *P<0.05 compared with 0 pg/ml treatment controls. IL-1b (100 pg/ml) did not affect (B) WST cell proliferation, (C) LDH release or (D) MTT cell viability after 24 hrs. Values are means ± SEM of four independent experiments.
IL-1b upregulates HMGB1 via ERK signaling
IL-1b (10-1000 pg/ml) increased phosphorylation in ERK1/2 without affecting total ERK1/2 levels (Figure 3A). The MEK/ERK inhibitor, U0126 (0.1-10 μM) effectively reduced IL-1b-induced ERK phosphorylation in a dose-dependent manner (Figure 3B). Concomitantly, U0126 also blocked the ability of IL-1b (100 pg/ml) to upregulate HMGB1 [F(4,19)=11.633, P<0.0001, Figure 3C], showing that the ERK MAP kinase signaling pathway is involved in this phenomenon. In contrast, inhibitors of the other two major MAP kinases, SB203580 for p38 and SP600125 for JNK, did not have any detectable effects at all (data not shown).
Figure 3.
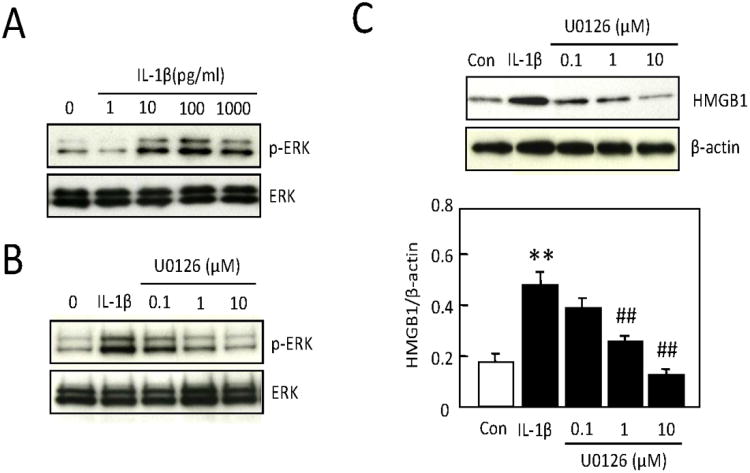
(A) IL-1b (10-1000 pg/ml) induced phosphorylation in ERK1/2 without affecting total ERK1/2 levels after 1 hr in astrocytes. (B) The MEK inhibitor U0126 (0.1-10 μM) effectively reduced IL-1b -induced ERK phosphorylation after 1 hr in a dose-dependent manner. (C) The IL-1b (100 pg/ml) stimulated elevation of HMGB1 after 24 hrs in astrocytes was attenuated after inhibition of ERK signaling with U0126 (1, 10 μM). Values are means ± SEM of four independent experiments. **P<0.01 compared with no-treated control, ##P<0.01 compared with IL-1b (100 pg/ml)-treated group.
IL-1b enhances expression of CRM1 via ERK phosphorylation
HMGB1 is a nuclear protein, so we hypothesized that the nuclear protein exporter, chromosome region maintenance 1 (CRM1), may be a candidate mechanism for IL-1b-stimulated HMGB1 release from astrocytes. IL-1b (100 pg/ml) significantly enhanced the expression of CRM1 in astrocytes. And the elevation of CRM1 was attenuated by blockade of ERK MAP kinase signaling with U0126 (10 μM) [F(2,13)=13.875, P<0.001, Figure 4].
Figure 4.
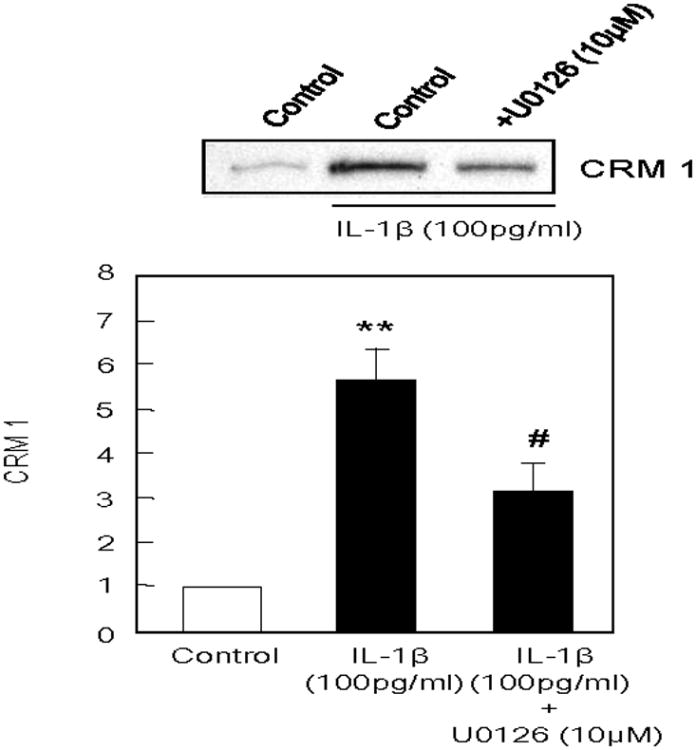
IL-1b (100 pg/ml) significantly enhanced the expression of CRM1 in astrocytes after 24 hrs, and the elevation of CRM1 in astrocytes was attenuated by U0126 (10 μM). Values are means ± SEM of six independent experiments. *P<0.05 compared with no-treated control, #P<0.05 compared with IL-1b (100 pg/ml)-treated group.
Translocation and release of HMGB1 occurs via CRM1 activation
In stimulated astrocytes, immunostaining showed that IL-1b (100 pg/ml) upregulated CRM1 expression (Figure 5). CRM1-positive astrocytes showed evidence of a translocation of HMGB1 from a mostly nuclear localization to include a diffuse staining in the cytoplasm (Figure 5). This phenomenon appeared to be dependent on upstream ERK MAP kinase signaling. U0126 (10 μM) effectively blocked CRM1 upregulation and HMGB1 translocation (Figure 5).
Figure 5.
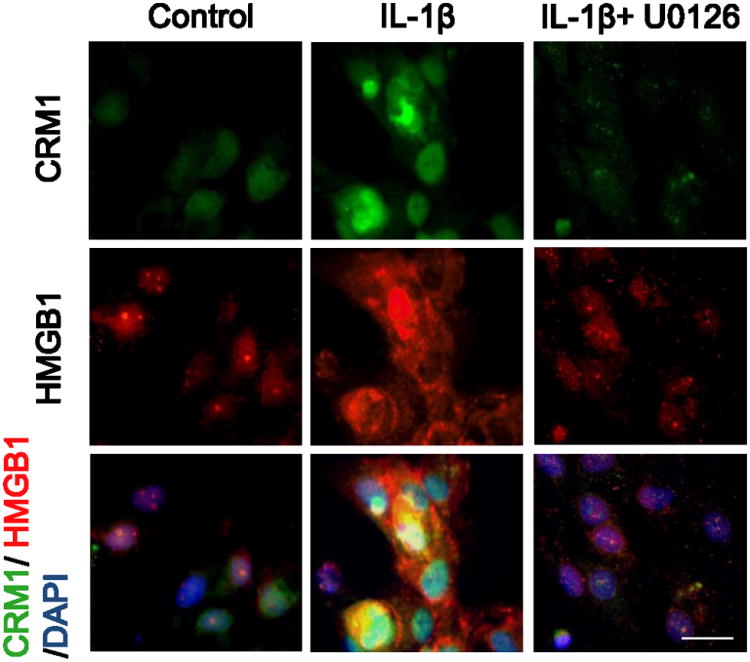
IL-1b (100 pg/ml) increased the immunostaining of CRM1 in astrocytes after 24 hrs. HMGB1 staining also showed evidence of translocation from nucleus into cytoplasm. Both CRM1 upregulation and HMGB1 translocation was inhibited by U0126 (10 μM). Cells were stained for the anti-CRM1 antibody (green), anti-HMGB1 antibody (red), and a nuclear stain DAPI (blue). Scale bar indicates 20 μm.
To quantify these findings, western blots of cell lysates were performed on IL-1b-stimulated astrocytes. Stimulation of astrocytes with IL-1b upregulated total amounts of HMGB1 and also resulted in a translocation from nuclear to cytoplasmic fractions (Figure 6A-B). Blockade of ERK MAP kinase signaling with U0126 prevented the translocation of HMGB1 into cytoplasm [F(2,9)=16.625, P<0.001, Figure 6B]. As a positive control, leptomycin B (LMB, 20 nM) was used to block the binding of CRM1 to proteins containing the nuclear export signal. Leptomycin B also effectively prevented IL-1b from trigerring the translocation of HMGB1 into cytoplasm [F(2,9)=25.364, P<0.001, Figure 6B].
Figure 6.
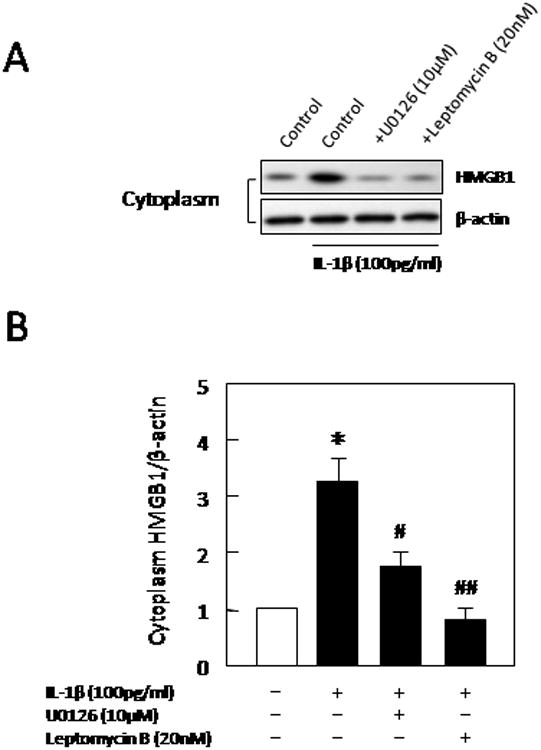
(A) Western blots showed that overall expression levels of HMGB1 were increased at 24 hrs after IL-1b stimulation. Cytosolic fractions showed evidence of HMGB1 translocation. (B) Densitometry quantitation of western blots demonstrated that cytosolic levels of HMGB1 were significantly increases by IL-1b. This response was significantly blocked by either inhibition of ERK signaling with U0126 (10 μM) or leptomycin B (LMB, 20 nM) which blocks the binding of CRM1 to proteins containing the nuclear export signal. Values are means ± SEM of four independent experiments. *P<0.05 compared with non-treated controls, #P<0.05, ##P<0.01 compared with IL-1b (100 pg/ml)-treated group.
Ultimately, CRM1 and ERK MAP kinase-mediated translocation of HMGB1 was consistent with measurable effects on total HMGB1 release. Western blot analysis of conditioned media demonstrated that IL-1b-stimulated release of HMGB1 was significantly attenuated by either blocking ERK MAP kinase signaling with U0126 (10 μM) [F(2,12)=15.022, P<0.001], or blocking CRM1 signaling with leptomycin B (20 nM) [F(2,11)=12.408, P<0.01] (Figure 7).
Figure 7.
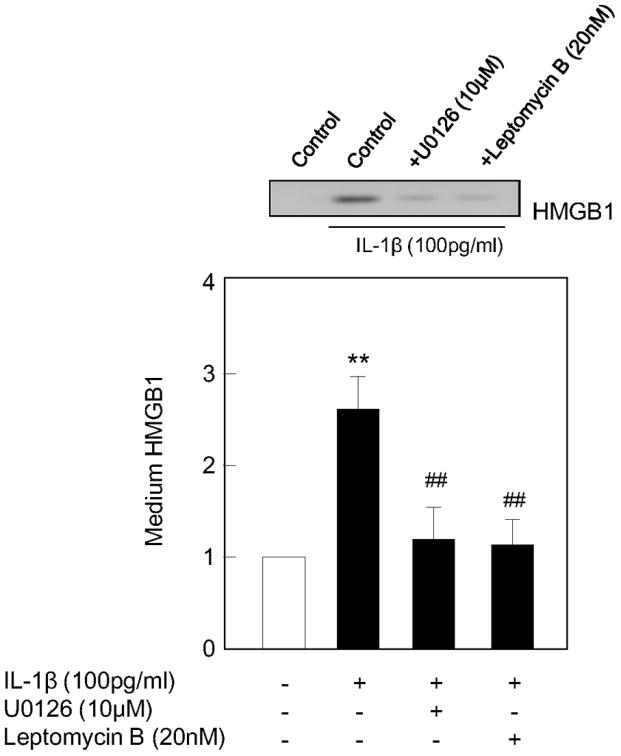
IL-1b -stimulated HMGB1 release from astrocytes was attenuated by U0126 (10 μM) and LMB (20 nM). Values are means ± SEM of four independent experiments. *P<0.05, **P<0.01 compared with non-treated control, ##P<0.01 compared with IL-1b (100 pg/ml)-treated group.
Discussion
It is increasingly recognized that reactive astrocytes play complex and potentially biphasic roles after stroke and brain injury. Glial scarring should be deleterious in terms of producing inhibitory signals and substrates that inhibit dendritic and axonal remodeling in neuronal circuits (Horner and Gage, [2000]; Rossi et al., [2007]). But some subsets of reactive astrocytes may also comprise key sources of beneficial mediators that encourage neuronal and vascular plasticity (Liberto et al, [2004]). We have previously shown that reactive astrocytes release HMGB1 that promotes neurovascular recovery within peri-infarct cortex after focal cerebral ischemia in mice (Hayakawa et al, [2009]). In the present study, we used primary cortical astrocyte cultures to dissect the mechanisms that control HMGB1 regulation. Our data showed that the inflammatory cytokine IL-1b potently upregulates HMGB1 in astrocytes. The prototypical MAP kinase ERK provides the upstream signal. And the nuclear export protein CRM1 enables nuclear HMGB1 to translocate from nucleus to cytoplasm before release. These data provide support for a novel signaling pathway that allows reactive astrocytes to release HMGB1 within the context of an inflammatory milieu.
HMGB1 is an architectural non-histone DNA-binding factor known to be released extracellularly during inflammatory responses in diverse tissues (Abraham et al., [2000]; Wang et al., [1999]). Traditionally, it was thought that release of HMGB1 occurred via passive nonspecific leakage from necrotic cells (Scaffidi et al., [2002]; Qiu et al., [2008]). However, emerging data now suggest that under some conditions, HMGB1 can also be secreted via specific signaling mechanisms. Upon inflammatory stimulation, HMGB1 can be actively secreted from monocytes and macrophages (Bonaldi et al, [2003]), endothelial cells (Kawahara et al, [2008]), fibroblasts (Feghali et al, [2009]), and brain glial cells (Passalacqua et al, [1998]). However, the mechanisms that mediate HMGB1 release are complex and still remain elusive. For example, HMGB1 is secreted by macrophages/monocytes via a non-classical, vesicle-mediated, secretory pathway (Bonaldi et al., [2003]; Garadella et al., [2002]), but the subcellular identity of these HMGB1-containing vesicles is not yet fully identified. In our study, we showed that in response to IL-1b stimulation, astrocytes upregulated HMGB1 levels and then proceeded to release it into extracellular space. There were no changes in astrocyte cell number or viability. Hence, this phenomenon is not the result of nonspecific leakage from dying cells.
In this study, we found that blockade of the ERK pathway with U0126 almost completely inhibited IL-1b-induced HMGB1 release. But inhibitors of other MAP kinases such as p38 and JNK, had no detectable effect. Hence, ERK MAP kinase may be specifically required for IL-1b-stimulated HMGB1 responses in astrocytes. This is consistent with the known ability of the MAP kinase pathways to play important roles in physiological and pathological events, including cell-cycle regulation, proliferation, apoptosis, and inflammation (Pouyssegur, [2000]; Deng et al., [2003]).
The ability of IL-1b to trigger ERK MAP kinase signaling in astrocytes is consistent with our previous findings (Arai et al, [2003]). But how would ERK signaling lead to the release of HMGB1? HMGB1 is a nuclear protein, so movement from nucleus into cytoplasm should be required before release into extracellular space occurs. CRM1 is a well-known factor that mediates the nuclear export of proteins bearing a leucine-rich nuclear export signal. Hence, we hypothesized that CRM1 may be involved in our model system. Our data supported this idea. CRM1 levels were clearly increased upon IL-1b stimulation and ERK activation. We further confirmed this pathway using leptomycin B, a drug that inhibits the function of CRM1 protein in yeast (Ossareh-Nazari et al., [1997]). Leptomycin B potently decreased IL-1b-stimulated HMGB1 release from astrocytes. Thus, these data support the existence of a signaling pathway whereby IL-1b triggers upstream ERK MAP kinase that activates CRM1, allowing HMGB1 to translocate from nucleus into cytoplasm before release into extracellular space. Our results are consistent with previous reports describing the connection between CRM1-mediated nuclear export and ERK signaling in various cancer cell lines (Qiu et al., [2003]; Le Gallic et al., [2004]). However, it must be noted that U0126 did not completely suppress CRM1 levels, suggesting that beyond ERK, other signaling pathways may also be involved in our astrocyte model system.
In the context of stroke and brain injury, HMGB1 may be released from ischemic neurons (Qiu et al., [2008]) and activated monocytes/macrophages (Wang et al., [1999]). Thereafter, HMGB1 would mediate potentially deleterious inflammatory responses by activating monocytes/macrophage (Andersson et al., [2000]; Park et al., [2004]), neutrophils (Abraham et al., [2000]; Park et al., [2004]), microvascular endothelial cells (Fiuza et al., [2003]), and microglia (Kim et al., [2006]). In one study, suppression of HMGB1-containing microglia improved functional outcomes in focal cerebral ischemia (Hayakawa et al, [2008]). More recently, however, it is beginning to be recognized that many mediators in stroke pathophysiology play biphasic roles (Lo, [2008]). Early in the stroke process, critical mediators contribute to brain cell damage and death. But later on, the same mediators contribute to neurovascular remodeling. In this regard, HMGB1 might also play biphasic roles after stroke. In contrast to the damaging actions early on, delayed HMGB1 signaling in reactive astrocytes may promote neurovascular remodeling. HMGB1 promotes neurite outgrowth (Huttunen et al., [2000], [2002]) and may also to play an essential role in endothelial cell activation that is needed for angiogenesis and vascular recovery (Treutiger et al., [2003]). In this study, we showed that ERK MAP kinase and CRM1 signaling mediates the ability of reactive astrocytes to upregulate and release pro-recovery HMGB1. These pathways may be important, because they may provide targets for future attempts to modify and augment pro-recovery mechanisms in astrocytes after stroke and brain injury.
Taken together, our findings support a novel signaling pathway involving ERK MAP kinase and CRM1 signaling that allows astrocytes to upregulate and release HMGB1 in response to inflammatory IL-1b stimulation. However, there are several caveats to keep in mind. First, these data are based on primary astrocytes cultures. The data here are consistent with our previous paper that described the potentially beneficial actions of HMGB1-positive astrocytes in peri-infarct brain (Hayakawa et al, [2009]). But whether these specific ERK and CRM1 signaling mechanisms operate in vivo remains to be confirmed. Second, we only tested a single stimulus, i.e. IL-1b. It has been previously reported that astrocytes can release HMGB1 in response to diverse stimuli, including cAMP, dexamethasone and forskolin (Passalacqua et al., [1998]). After stroke or brain injury, multiple inflammatory cytokines and chemokines would co-exist in vivo. How HMGB1 is regulated in a complex and multifactorial situation in vivo is unclear. Third, our data suggest the ERK and not p38 or JNK MAP kinases were involved. But of course, there are many other kinase mechanisms that we have not examined. For example, protein kinase C seems to be a central regulator for HMGB1 regulation in monocytes (Oh et al, [2009]), whereas Janus kinases are involved in IFN-mediated HMGB1 release from macrophages (Renden-Mitchell et al, [2003]). How ERK signaling in our astrocyte model system may interact with other kinase pathways remains to be determined. Fourth, our focus here was on astrocytes because we previously showed that subsets of HMGB1-positive astrocytes played a role in peri-infarct recovery after focal cerebral ischemia. But of course, many other cell types will contribute to stroke recovery. How HMGB1 is co-regulated in microglia and actively remodeling neurons and vasculature would be an important direction to pursue.
In conclusion, we demonstrated that ERK MAP kinase and CRM1 signaling are required for the upregulation and secretion of HMGB1 from IL-1b-stimulated astrocytes. These findings suggest a novel pathway by which inflammatory cytokines may enhance the ability of reactive astrocytes to contribute to neurovascular recovery after stroke and brain injury.
Acknowledgments
Supported in part by NIH grants P01-NS55104, R37-NS37074, R01-NS48422, R01-NS53560, the American Heart Association, the Deane Institute, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and a Postdoctoral Fellowship for Research Abroad from the Japan Society for the Promotion of Science.
References
- Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lee SR, Lo EH. Essential role for ERK mitogen-activated protein kinase in matrix metalloproteinase-9 regulation in rat cortical astrocytes. Glia. 2003;43:254–264. doi: 10.1002/glia.10255. [DOI] [PubMed] [Google Scholar]
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNF-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- Feghali K, Iwasaki K, Tanaka K, Komaki M, Machigashira M, Ishikawa I, Izumi Y. Human gingival fibroblasts release high-mobility group box-1 protein through active and passive pathways. Oral Microbial Immunol. 2009;24:292–298. doi: 10.1111/j.1399-302X.2009.00508.x. [DOI] [PubMed] [Google Scholar]
- Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, Nozako M, Hazekawa M, Mishima S, Fujioka M, Orito K, Egashira N, Iwasaki K, Fujiwara M. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke. 2008;39:951–958. doi: 10.1161/STROKEAHA.107.495820. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Nakano T, Irie K, Higuchi S, Fujioka M, Orito K, Iwasaki K, Jin G, Lo EH, Mishima K, Fujiwara M. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2009 doi: 10.1038/jcbfm.2009.257. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407:963–970. doi: 10.1038/35039559. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and s100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275:40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Kuja-Panula J, Rauvala H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J Biol Chem. 2002;41:38635–38646. doi: 10.1074/jbc.M202515200. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Hashiguchi T, Kikuchi K, Tancharoen S, Miura N, Ito T, Oyama Y, Nawa Y, Biswas KK, Meng X, Morimoto Y, Shrestha B, Sameshima H, Maruyama I. Induction of high mobility group box 1 release from serotonin-stimulated human umbilical vein endothelial cells. Int J Mol Med. 2008;22:639–44. [PubMed] [Google Scholar]
- Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26:6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res. 2008;86:1125–1131. doi: 10.1002/jnr.21555. [DOI] [PubMed] [Google Scholar]
- Le Gallic L, Virqilio L, Cohen P, Biteau B, Mavrothalassitis G. ERF nuclear shuttling, a continuous monitor of Erk activity that links it to cell cycle progression. Mol Cell Biol. 2004;24:1206–1218. doi: 10.1128/MCB.24.3.1206-1218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberto CM, Albrecht PJ, Herx LM, Yong VW, Levision SW. Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem. 2004;89:1092–1100. doi: 10.1111/j.1471-4159.2004.02420.x. [DOI] [PubMed] [Google Scholar]
- Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- Mocchetti I, Wrathall JR. Neurotrophic factors in central nervous system trauma. J Neurotrauma. 1995;12:853–870. doi: 10.1089/neu.1995.12.853. [DOI] [PubMed] [Google Scholar]
- Oh YJ, Youn JH, JiY, Lee SE, Lim KJ, Choi JE, Shin JS. HMGB1 is phosphorylated by classical protein kinase C and is secreted by a calcium-dependent mechanism. J Immunol. 2009;182:5800–5809. doi: 10.4049/jimmunol.0801873. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Panickar KS, Norenberg MD. Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia. 2005;50:287–298. doi: 10.1002/glia.20181. [DOI] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Passalacqua M, Patrone M, Picotti GB, Del Rio M, Sparatore B, Melloni E, Pontremoli S. Stimulated astrocytes release high-mobility group 1 protein, an inducer of LAN-5 neuroblastoma cell differentiation. Neuroscience. 1998;82:1021–1028. doi: 10.1016/s0306-4522(97)00352-7. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J. Signal transduction. An arresting start for MAPK. Science. 2000;290:1515–1518. doi: 10.1126/science.290.5496.1515. [DOI] [PubMed] [Google Scholar]
- Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, Salomone S, Moskowitz MA. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28:927–938. doi: 10.1038/sj.jcbfm.9600582. [DOI] [PubMed] [Google Scholar]
- Qiu M, Olsen A, Faivre E, Horwitz KB, Lange CA. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol. 2003;17:628–642. doi: 10.1210/me.2002-0378. [DOI] [PubMed] [Google Scholar]
- Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, Susarla S, Czura C, Mitchell RA, Chen G, Sama AE, Tracey KJ, Wang H. IFN- induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. 2003;170:3890–3897. doi: 10.4049/jimmunol.170.7.3890. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Brady JD, Morh C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci. 2007;10:1377–1386. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schlueter C, Weber H, Meyer B, Rogalla P, Röser K, Hauke S, Bullerdiek J. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166:1259–1263. doi: 10.1016/S0002-9440(10)62344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. Brain angiogenesis in developmental and pathological processes: therapeutic aspects of vascular endothelial growth factor. FEBS J. 2009;276:4636–4643. doi: 10.1111/j.1742-4658.2009.07175.x. [DOI] [PubMed] [Google Scholar]
- Strauss S, Otten U, Joggerst B, Plüss K, Volk B. Increased levels of nerve growth factor (NGF) protein and mRNA and reactive gliosis following kainic acid injection into the rat striatum. Neurosci Lett. 1994;168:193–196. doi: 10.1016/0304-3940(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Tokita Y, Keino H, Matsui F, Aono S, Ishiguro H, Higashiyama S, Oohira A. Regulation of neuregulin expression in the injured rat brain and cultured astrocytes. J Neurosci. 2001;21:1257–1264. doi: 10.1523/JNEUROSCI.21-04-01257.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower DB, Young OM. The activities of butyrylcholinesterase and carbonic anhydrase, the rate of anaerobic glycolysis, and the question of a constant density of glial cells in cerebral cortices of various mammalian species from mouse to whale. J Neurosci. 1973;20:269–278. doi: 10.1111/j.1471-4159.1973.tb12126.x. [DOI] [PubMed] [Google Scholar]
- Treutiger CJ, Mullins GE, Johansson AS, Rouhiainen A, Rauvala HM, Erlandsson-Harris H, Andersson U, Yang H, Tracey KJ, Andersson J, Palmblad JE. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–385. doi: 10.1046/j.1365-2796.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 2006;17:189–201. doi: 10.1016/j.cytogfr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. 2005;78:1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chopp M. Vascular endothelial growth factor and angiopoietins in focal cerebral ischemia. Trends Cardiovasc Med. 2002;12:62–66. doi: 10.1016/s1050-1738(01)00149-9. [DOI] [PubMed] [Google Scholar]